
Membrane and Protein Interactions of the Pleckstrin Homology Domain Superfamily

Abstract
:
1. Introduction
2. Experimental Section
2.1. Alignment and Conservation
2.2. Predication of Membrane Binding Sites
3. Results
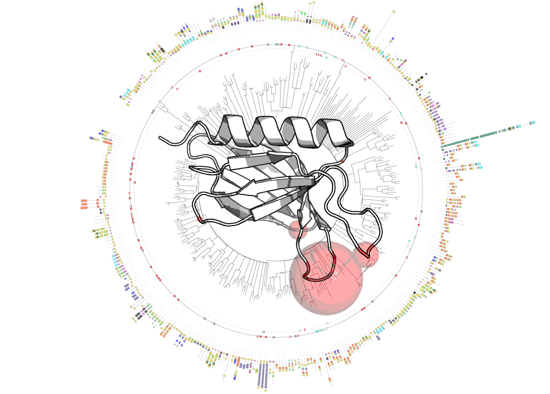
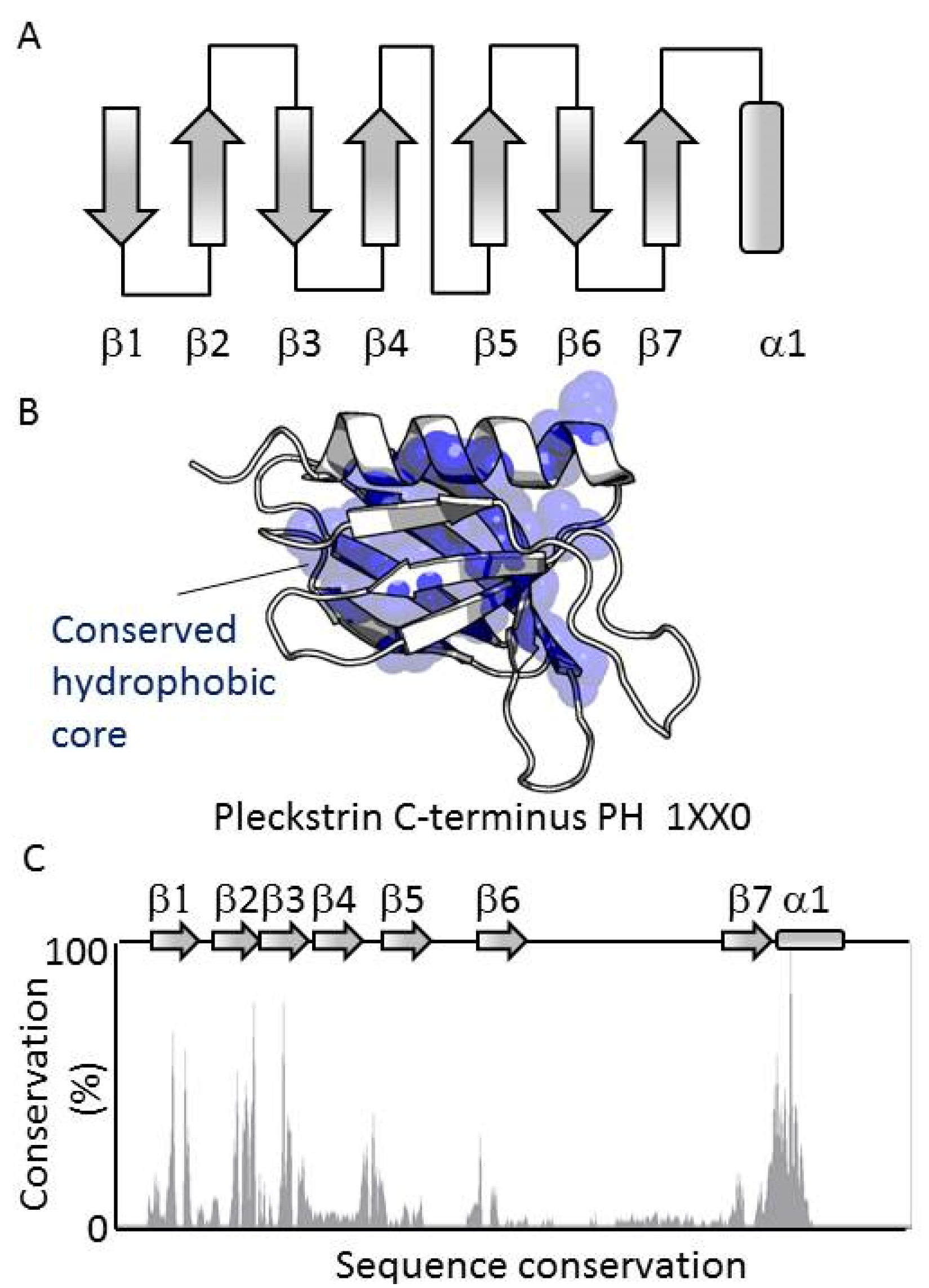
3.1. PH Domains, Similarities and Differences


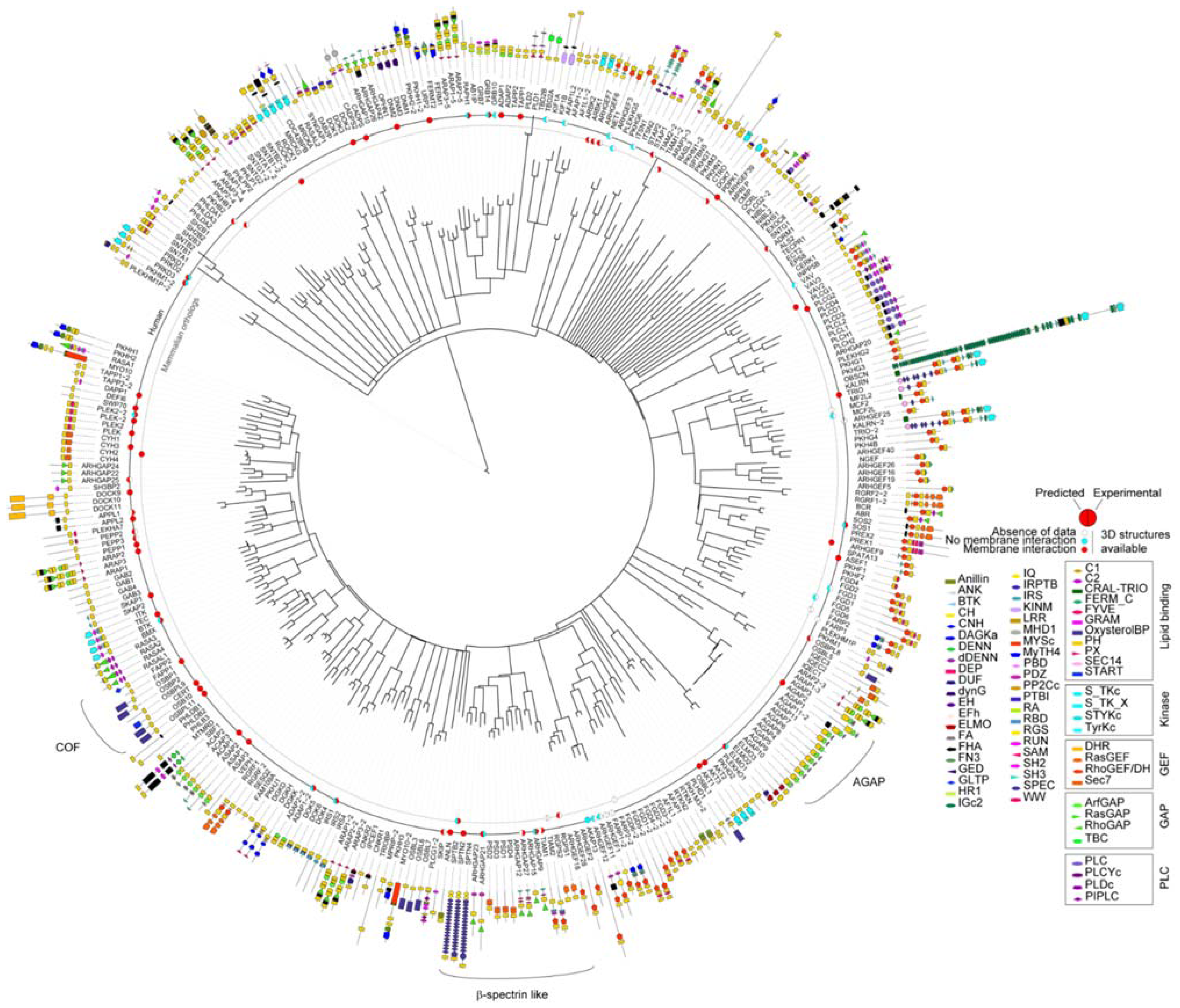
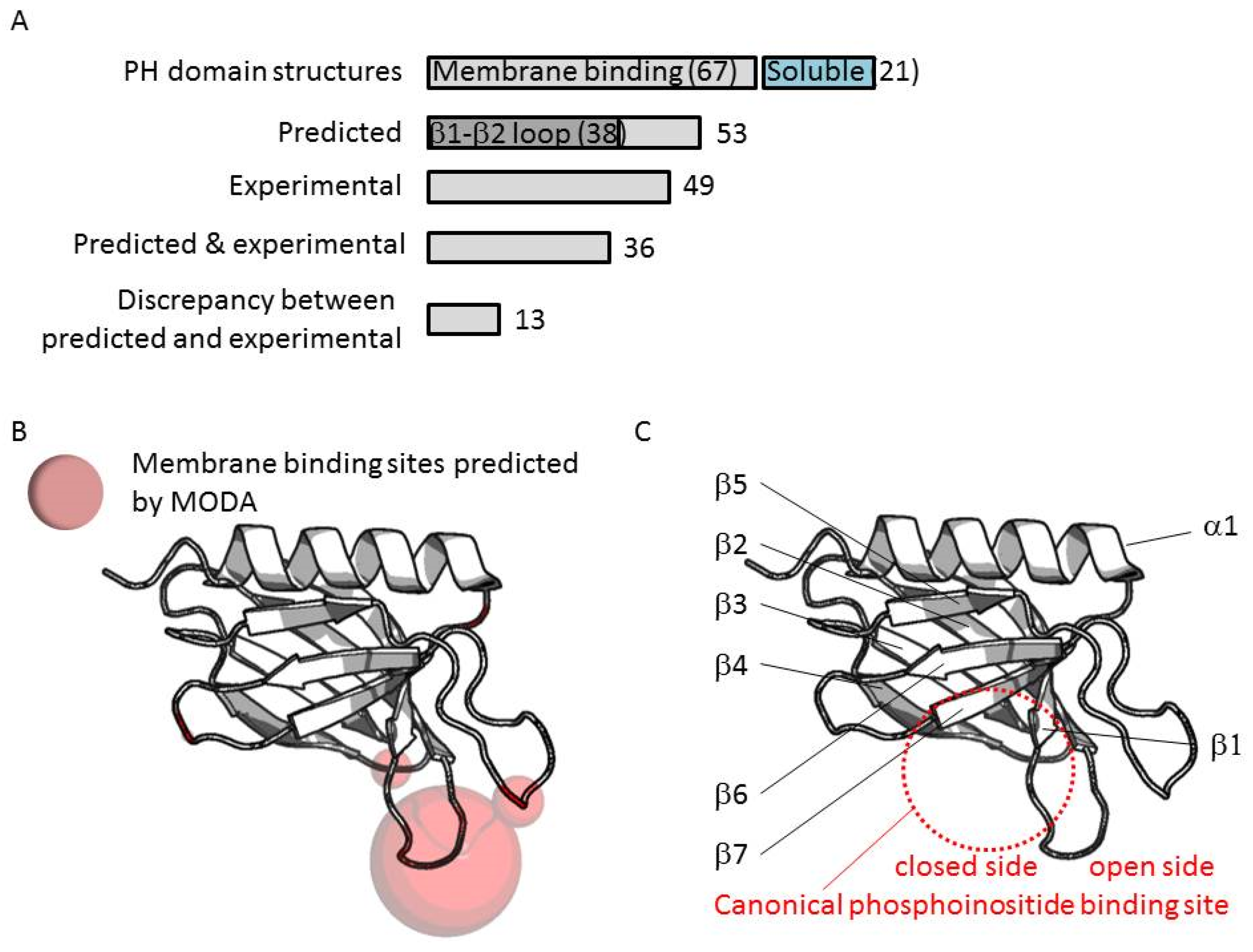
3.2. Genome-Wide Analysis of Membrane Binding of PH Domains

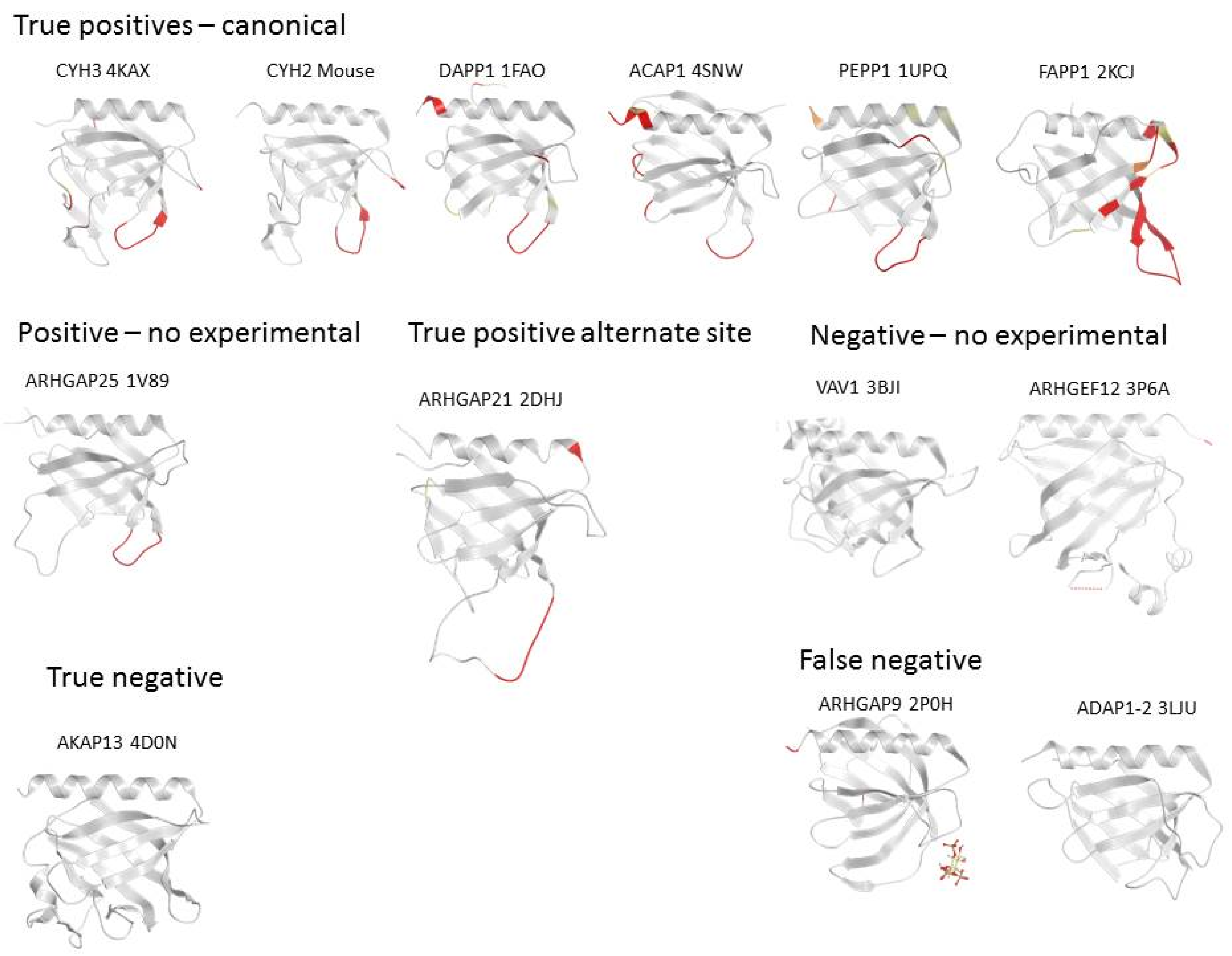
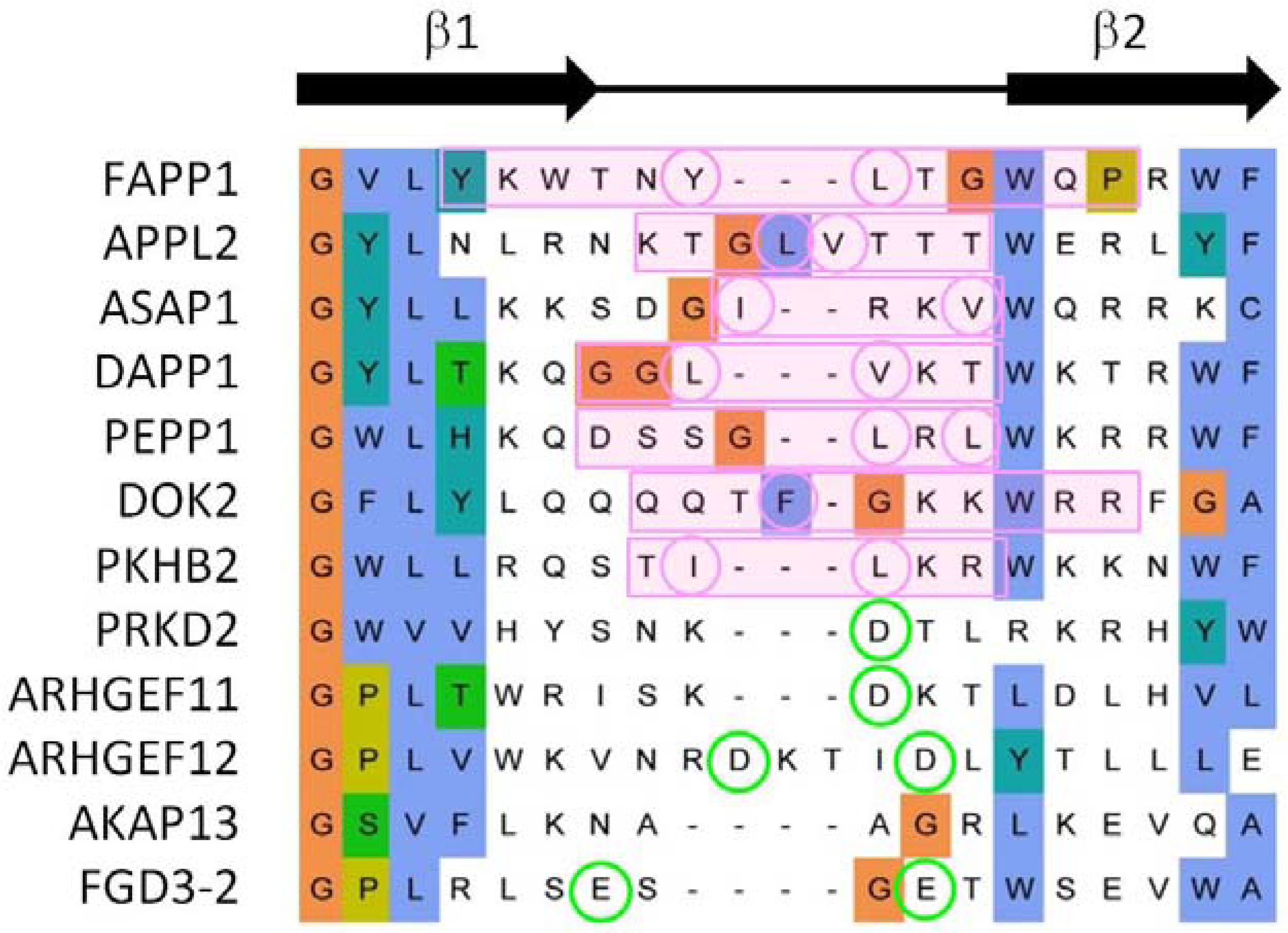
3.3. Comparison of Membrane Interactions Designates the β1-β2 Loop as the Canonical Binding Element


3.4. Extrapolation of Membrane Interaction Predictions
3.5. Four Phosphate Adaptor Protein 1 (FAPP1) as a Paradigm of Canonical Membrane Binding
3.6. ARHGAP9 and Discrepancies between Predictions and Experimental Validations
3.7. Evidence for an Alternate Binding Site in ARHGEF9
3.8. A-Kinase Anchoring Protein 13 (AKAP13) and the Absence of Membrane Interaction
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Name |
|---|---|
| ANK | Ankyrin repeats |
| BTK | Bruton’s tyrosine kinase Cys-rich motif (Zn2+) |
| C1 | Protein kinase C conserved region 1 (C1) domains (Cysteine-rich domains) |
| C2 | Protein kinase C conserved region 2 (CalB) |
| CH | Calponin homology domain |
| CNH | Domain found in NIK1-like kinases, mouse citron and yeast ROM1, ROM2 |
| CRAL-TRIO | cellular retinaldehyde-binding protein and TRIO guanine exchange factor domain |
| DAGKa | Diacylglycerol kinase accessory domain (presumed) |
| DENN | Differentially expressed in neoplastic versus normal cells |
| DEP | Domain found in Dishevelled, Egl-10, and Pleckstrin |
| DUF | Domains of unknown function |
| dynG | Dynamin-type guanine nucleotide-binding (G) |
| EFh | EF-hand, calcium binding motif (Ca2+) |
| EH | Eps15 homology domain |
| ELMO | EnguLfment and cell Motility |
| FA | FERM adjacent |
| FERM | Ezrin/radixin/moesin |
| FERM_C | FERM C-terminal PH-like domain |
| FHA | Forkhead associated domain |
| FN3 | Fibronectin type 3 domain |
| FYVE | Protein present in Fab1, YOTB, Vac1, and EEA1 |
| GED | Dynamin GTPase effector domain |
| GLTP | Glycolipid transfer protein |
| GRAM | Domain in glucosyltransferases, myotubularins and other putative membrane-associated proteins |
| HR1 | Rho effector or protein kinase C-related kinase homology region 1 homologues |
| IGc2 | Immunoglobulin C-2 Type |
| IQ | Short calmodulin-binding motif containing conserved Ile and Gln residues. |
| IRPTB | IRS-type PTB |
| IRS | Phosphotyrosine-binding domain |
| KINM | Kinesin motor domain |
| LRR | Leucine-rich repeats, outliers |
| MHD1 | Munc13 homology domain |
| MORN | Membrane Occupation and Recognition Nexus |
| MYSc | Myosin. Large ATPases |
| MyTH4 | Domain in Myosin and Kinesin Tails |
| Oxysterol_BP | Oxysterol binding protein |
| PBD | P21-Rho-binding domain |
| PDZ | Domain present in PSD-95, Dlg, and ZO-1/2 |
| PH | Pleckstrin homology |
| PIPLC | Phosphatidylinositol-specific phospholipase C Y domain |
| PP2Cc | Serine/threonine phosphatases, family 2C, catalytic domain |
| PTBI | Phosphotyrosine-binding domain (IRS1-like) |
| PX | Phox homology |
| RA | Ras association (RalGDS/AF-6) domain |
| RAS | Ras subfamily of RAS small GTPases |
| RBD | Raf-like Ras-binding domain |
| RGS | Regulator of G protein signalling domain |
| RUN | Domain involved in Ras-like GTPase signalling |
| SAM | Sterile alpha motif |
| SEC14 | Domain in homologues of a S. cerevisiae phosphatidylinositol transfer protein |
| SH2 | Src homology 2 domains |
| SH3 | Src homology 3 domains |
| SPEC | Spectrin repeats |
| START | StAR-related lipid-transfer |
| TBC | Domain in Tre-2, BUB2p, and Cdc16p—probable Rab-GAP |
| uDENN/dDENN | Upstream/downstream DENN |
| WW | Domain with 2 conserved Trp (W) residues |
References
- Mayer, B.J.; Ren, R.; Clark, K.L. A putative modular domain present in diverse signaling proteins. Cell 1993, 73, 629–630. [Google Scholar] [CrossRef]
- Haslam, R.J.; Koide, H.B.; Hemmings, B.A. Pleckstrin domain homology. Nature 1993, 363, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.S.; Hajduk, P.J.; Petros, A.M. Solution structure of a pleckstrin-homology domain. Nature 1994, 369, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Harlan, J.E.; Hajduk, P.J.; Yoon, H.S. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature 1994, 371, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Ferguson, K.M. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem. J. 2000, 350, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Mendrola, J.M.; Audhya, A. Genome-wide analysis of membrane targeting by S. cerevisiae pleckstrin homology domains. Mol. Cell 2004, 13, 677–688. [Google Scholar] [CrossRef]
- Lutz, S.; Shankaranarayanan, A.; Coco, C. Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 2007, 318, 1923–1927. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, N.; Gabdoulline, R.R.; Nilges, M. Classification of protein sequences by homology modeling and quantitative analysis of electrostatic similarity. Proteins 1999, 37, 379–387. [Google Scholar] [CrossRef]
- Vetter, I.R.; Nowak, C.; Nishimoto, T. Structure of a Ran-binding domain complexed with Ran bound to a GTP analogue: Implications for nuclear transport. Nature 1999, 398, 39–46. [Google Scholar] [PubMed]
- Carpten, J.D.; Faber, A.L.; Horn, C. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007, 448, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Lenoir, M.; Dancea, F. Discovery of novel membrane binding structures and functions. Biochem. Cell Biol. 2014, 92, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Kuo, Y.-C.; Rosche, T.J.; Zhang, X. Structural basis for autoinhibition of the guanine nucleotide exchange factor FARP2. Structure 2013, 21, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Bach, T.L.; Kerr, W.T.; Wang, Y. PI3K regulates pleckstrin-2 in T-cell cytoskeletal reorganization. Blood 2007, 109, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, D.F.; Blasutig, I.M.; Goudreault, M. Non-canonical interaction of phosphoinositides with pleckstrin homology domains of Tiam1 and ArhGAP9. J. Biol. Chem. 2007, 282, 13864–13874. [Google Scholar] [CrossRef] [PubMed]
- Che, M.M.; Boja, E.S.; Yoon, H.Y. Regulation of ASAP1 by phospholipids is dependent on the interface between the PH and Arf GAP domains. Cell. Signal. 2005, 17, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, L.; Biederer, T. The novel synaptogenic protein Farp1 links postsynaptic cytoskeletal dynamics and transsynaptic organization. J. Cell Biol. 2012, 199, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, L.; Biederer, T. Activity-dependent regulation of dendritic complexity by semaphorin 3A through Farp1. J. Neurosci. 2014, 34, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.H.; Corbalan-Garcia, S.; Bar-Sagi, D. The role of the PH domain in the signal-dependent membrane targeting of Sos. EMBO J. 1997, 16, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.A.; Honda, A.; Varnai, P. Active Arf6 recruits ARNO/cytohesin GEFs to the PM by binding their PH domains. Mol. Biol. Cell 2007, 18, 2244–2253. [Google Scholar] [CrossRef] [PubMed]
- Currie, R.A.; Walker, K.S.; Gray, A. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem. J. 1999, 337, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Base, C.; Manna, D. Unexpected complexity in the mechanisms that target assembly of the spectrin cytoskeleton. J. Biol. Chem. 2008, 283, 12643–12653. [Google Scholar] [CrossRef] [PubMed]
- Depetris, R.S.; Wu, J.; Hubbard, S.R. Structural and functional studies of the Ras-associating and pleckstrin-homology domains of Grb10 and Grb14. Nat. Struct. Mol. Biol. 2009, 16, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Dowler, S.; Currie, R.A.; Campbell, D.G. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem. J. 2000, 351, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Dowler, S.; Currie, R.A.; Downes, C.P. DAPP1: A dual adaptor for phosphotyrosine and 3-phosphoinositides. Biochem. J. 1999, 342, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Dowler, S.; Montalvo, L.; Cantrell, D. Phosphoinositide 3-kinase-dependent phosphorylation of the dual adaptor for phosphotyrosine and 3-phosphoinositides by the Src family of tyrosine kinase. Biochem. J. 2000, 349, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Edlich, C.; Stier, G.; Simon, B. Structure and phosphatidylinositol-(3,4)-bisphosphate binding of the C-terminal PH domain of human pleckstrin. Structure 2005, 13, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Logan, S.K.; Lehto, V.P. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 1998, 17, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Felder, B.; Radlwimmer, B.; Benner, A. FARP2, HDLBP and PASK are downregulated in a patient with autism and 2q37.3 deletion syndrome. Am. J. Med. Genet. A 2009, 149, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Guittard, G.; Gerard, A.; Dupuis-Coronas, S. Cutting edge: Dok-1 and Dok-2 adaptor molecules are regulated by phosphatidylinositol 5-phosphate production in T cells. J. Immunol. 2009, 182, 3974–3978. [Google Scholar] [CrossRef] [PubMed]
- Kabachinski, G.; Yamaga, M.; Kielar-Grevstad, D.M. CAPS and Munc13 utilize distinct PIP2-linked mechanisms to promote vesicle exocytosis. Mol. Biol. Cell 2014, 25, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Tsuji, S.; Muroya, K. The adenomatous polyposis coli-associated exchange factors Asef and Asef2 are required for adenoma formation in Apc(Min/+)mice. EMBO Rep. 2009, 10, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.E.; Lee, A.; Frank, D.W. The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J. Biol. Chem. 1998, 273, 27725–27733. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Patel, M.; Laurin, M. An alpha-helical extension of the ELMO1 pleckstrin homology domain mediates direct interaction to DOCK180 and is critical in Rac signaling. Mol. Biol. Cell 2008, 19, 4837–4851. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Leslie, J.D.; Stewart, M. Lamellipodin, an Ena/VASP ligand, is implicated in the regulation of lamellipodial dynamics. Dev. Cell 2004, 7, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Tan, D.; Hu, G. A study on HBV DNA pre C region A83 mutation in patients with hepatitis B. Hunan Yi Ke Da Xue Xue Bao, 1998; 23, 269–271. [Google Scholar] [PubMed]
- Liu, H.; Yang, Z.; Chen, T. One-stage operative treatment of atlanto-axial instability with stenosis of lower cervical level of spinal canal. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi, 1998; 12, 269–271. [Google Scholar] [PubMed]
- Liu, Q.; Frutos, A.G.; Thiel, A.J. DNA computing on surfaces: Encoding information at the single base level. J. Comput. Biol. 1998, 5, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, H.; Eberstadt, M. NMR structure and mutagenesis of the N-terminal Dbl homology domain of the nucleotide exchange factor Trio. Cell 1998, 95, 269–277. [Google Scholar] [CrossRef]
- Meller, N.; Westbrook, M.J.; Shannon, J.D. Function of the N-terminus of zizimin1: Autoinhibition and membrane targeting. Biochem. J. 2008, 409, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Menetrey, J.; Perderiset, M.; Cicolari, J. Structural basis for ARF1-mediated recruitment of ARHGAP21 to Golgi membranes. EMBO J. 2007, 26, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Komatsu, S.; Ikebe, M. Dynamics of RhoA and ROKalpha translocation in single living cells. Cell Biochem. Biophys. 2006, 45, 243–254. [Google Scholar] [CrossRef]
- Muroya, K.; Kawasaki, Y.; Hayashi, T. PH domain-mediated membrane targeting of Asef. Biochem. Biophys. Res. Commun. 2007, 355, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Heo, W.D.; Whalen, J.H. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol. Cell 2008, 30, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Smith, X.; Matthess, Y. SKAP1 protein PH domain determines RapL membrane localization and Rap1 protein complex formation for T cell receptor (TCR) activation of LFA-1. J. Biol. Chem. 2011, 286, 29663–29670. [Google Scholar] [CrossRef] [PubMed]
- Razzini, G.; Ingrosso, A.; Brancaccio, A. Different subcellular localization and phosphoinositides binding of insulin receptor substrate protein pleckstrin homology domains. Mol. Endocrinol. 2000, 14, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Seddeek, M.A. Influence of viscous dissipation and thermophoresis on Darcy-Forchheimer mixed convection in a fluid saturated porous media. J. Colloid Interface Sci. 2006, 293, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki-Narikawa, N.; Kodama, T.; Shibasaki, Y. Cooperation of phosphoinositides and BAR domain proteins in endosomal tubulation. Traffic 2006, 7, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Cox, A.; Shen, Y. Farp2 and Stk25 are candidate genes for the HDL cholesterol locus on mouse chromosome 1. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Takegahara, N.; Kang, S.; Nojima, S. Integral roles of a guanine nucleotide exchange factor, FARP2, in osteoclast podosome rearrangements. FASEB J. 2010, 24, 4782–4792. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.C.; Dowler, S.; Deak, M. Crystal structure of the phosphatidylinositol 3,4-bisphosphate-binding pleckstrin homology (PH) domain of tandem PH-domain-containing protein 1 (TAPP1): Molecular basis of lipid specificity. Biochem. J. 2001, 358, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Jais, P.; Cohen-Akenine, A. Genetics of prostate cancer. Bull. Cancer 2001, 88, 287–294. [Google Scholar] [PubMed]
- Thomas, R.H.; Lindell, B. In radiological protection, the protection quantities should be expressed in terms of measurable physical quantities. Radiat. Prot. Dosimetry 2001, 94, 287–292. [Google Scholar] [PubMed]
- Tong, Y.; Tempel, W.; Wang, H. Phosphorylation-independent dual-site binding of the FHA domain of KIF13 mediates phosphoinositide transport via centaurin alpha1. Proc. Natl. Acad. Sci. USA 2010, 107, 20346–20351. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, T.; Yoshida, J.; Sugimoto, T. FARP2 triggers signals for Sema3A-mediated axonal repulsion. Nat. Neurosci. 2005, 8, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, I.; Ihara, S.; Fukui, Y. Mutational analysis on the function of the SWAP-70 PH domain. Mol. Cell. Biochem. 2006, 293, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Wen, W.; Chan, L.N. Split pleckstrin homology domain-mediated cytoplasmic-nuclear localization of PI3-kinase enhancer GTPase. J. Mol. Biol. 2008, 378, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, B.; Su, Y.S.; Sockanathan, S. FARP1 promotes the dendritic growth of spinal motor neuron subtypes through transmembrane Semaphorin6A and PlexinA4 signaling. Neuron 2009, 61, 359–372. [Google Scholar] [CrossRef]
- Zugaza, J.L.; Lopez-Lago, M.A.; Caloca, M.J. Structural determinants for the biological activity of Vav proteins. J. Biol. Chem. 2002, 277, 45377–45392. [Google Scholar] [CrossRef] [PubMed]
- Dubois, T.; Paleotti, O.; Mironov, A.A. Golgi-localized GAP for Cdc42 functions downstream of ARF1 to control Arp2/3 complex and F-actin dynamics. Nat. Cell Biol. 2005, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Recio, J.; Totrov, M.; Skorodumov, C. Optimal docking area: a new method for predicting protein-protein interaction sites. Proteins 2005, 58, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Kavran, J.M.; Sankaran, V.G. Structural basis for discrimination of 3-phosphoinositides by pleckstrin homology domains. Mol. Cell 2000, 6, 373–384. [Google Scholar] [CrossRef]
- Hanada, K.; Kumagai, K.; Yasuda, S. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Ridgway, N.D. Oxysterol-binding protein and vesicle-associated membrane protein-associated protein are required for sterol-dependent activation of the ceramide transport protein. Mol. Biol. Cell 2006, 17, 2604–2616. [Google Scholar] [CrossRef] [PubMed]
- Kam, J.L.; Miura, K.; Jackson, T.R. Phosphoinositide-dependent activation of the ADP-ribosylation factor GTPase-activating protein ASAP1. Evidence for the pleckstrin homology domain functioning as an allosteric site. J. Biol. Chem. 2000, 275, 9653–9063. [Google Scholar] [CrossRef] [PubMed]
- Manna, D.; Albanese, A.; Park, W.S. Mechanistic basis of differential cellular responses of phosphatidylinositol 3,4-bisphosphate- and phosphatidylinositol 3,4,5-trisphosphate-binding pleckstrin homology domains. J. Biol. Chem. 2007, 282, 32093–32105. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, M.; Sugawara, M.; Kaur, J. Structural insights into the activation of the RhoA GTPase by the lymphoid blast crisis (Lbc) oncoprotein. J. Biol. Chem. 2014, 289, 23992–24004. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, M.; Coskun, U.; Grzybek, M. Structural basis of wedging the Golgi membrane by FAPP pleckstrin homology domains. EMBO Rep. 2010, 11, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, M.; Grzybek, M.; Majkowski, M. Structural basis of dynamic membrane recognition by trans-Golgi network specific FAPP proteins. J. Mol. Biol. 2015, 427, 966–981. [Google Scholar] [CrossRef] [PubMed]
- Soykan, T.; Schneeberger, D.; Tria, G. A conformational switch in collybistin determines the differentiation of inhibitory postsynapses. EMBO J. 2014, 33, 2113–2133. [Google Scholar] [CrossRef] [PubMed]
- Reddy-Alla, S.; Schmitt, B.; Birkenfeld, J. PH-domain-driven targeting of collybistin but not Cdc42 activation is required for synaptic gephyrin clustering. Eur. J. Neurosci. 2010, 31, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.F.; Defeo-Jones, D.; Fu, S. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 2005, 385, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Yang, H.; Trinczek, B. The Akt activation inhibitor TCN-P inhibits Akt phosphorylation by binding to the PH domain of Akt and blocking its recruitment to the plasma membrane. Cell Death Differ. 2010, 17, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Chuang, H.C.; Chou, C.C. Vitamin E facilitates the inactivation of the kinase Akt by the phosphatase PHLPP1. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Lo, P.K.; Li, Y. Deactivation of Akt by a small molecule inhibitor targeting pleckstrin homology domain and facilitating Akt ubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 6486–6491. [Google Scholar] [CrossRef] [PubMed]
- Joh, E.H.; Hollenbaugh, J.A.; Kim, B. Pleckstrin homology domain of Akt kinase: A proof of principle for highly specific and effective non-enzymatic anti-cancer target. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Blomberg, N.; Baraldi, E.; Nilges, M.; Saraste, M. The PH superfold: A structural scaffold for multiple functions. Trends Biochem. Sci. 1999, 24, 441–445. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenoir, M.; Kufareva, I.; Abagyan, R.; Overduin, M. Membrane and Protein Interactions of the Pleckstrin Homology Domain Superfamily. Membranes 2015, 5, 646-663. https://doi.org/10.3390/membranes5040646
Lenoir M, Kufareva I, Abagyan R, Overduin M. Membrane and Protein Interactions of the Pleckstrin Homology Domain Superfamily. Membranes. 2015; 5(4):646-663. https://doi.org/10.3390/membranes5040646
Chicago/Turabian StyleLenoir, Marc, Irina Kufareva, Ruben Abagyan, and Michael Overduin. 2015. "Membrane and Protein Interactions of the Pleckstrin Homology Domain Superfamily" Membranes 5, no. 4: 646-663. https://doi.org/10.3390/membranes5040646
APA StyleLenoir, M., Kufareva, I., Abagyan, R., & Overduin, M. (2015). Membrane and Protein Interactions of the Pleckstrin Homology Domain Superfamily. Membranes, 5(4), 646-663. https://doi.org/10.3390/membranes5040646