2.1. Ohmic and Voltage-Gated Channels
Leaving aside ligand-gated ion channels, the other ion channels may be either ohmic or voltage-gated. Ohmic channels exhibit a plot of the average or single channel current against
φm that is linear across the zero transmembrane potential [
13]. This linearity is maintained throughout the whole accessible
φm range if the two bathing solutions have identical composition. If the two solutions differ in their electrolyte concentration, progressive deviations from linear behavior are observed as we depart from the zero transmembrane potential, with the BLM opposing greater resistance to the flow of ions as
φm moves them from the lower to the higher concentration. Conversely, voltage-gated channels yield plots of the current against
φm that increases exponentially on one side of
φm = 0, while the current is vanishingly small on the other side [
14,
15]; in other words, they open or close in response to changes in the direction of the transmembrane potential. A typical example of ohmic channel is offered by gramicidin [
13]. This unique channel-forming peptide has a helical structure that differs from the
α-helix of common peptides and membrane proteins, in that its lumen is large enough to allow the passage of simple desolvated monovalent cations. Since its length is about one half that of a biomembrane, it spans the membrane by forming a dimeric channel, which is stabilized by the cation flow [
16].
By far the majority of channel-forming peptides and proteins have an
α-helical structure in a lipid environment and form ion channels by aggregation of monomeric units. One side of the
α-helices is relatively hydrophilic by the presence of polar or charged residues and is turned toward the lumen of the aggregate, whereas the opposite side is hydrophobic and is turned toward the surrounding lipid molecules, where it interacts attractively with them. As a rule, the aggregation of
α-helices with ion channel formation takes place by a kinetic mechanism of nucleation and growth. For these ion channels to exhibit an ohmic behavior, they must retain the same membrane-spanning structure in passing from one side of the zero transmembrane potential to the other. This situation is encountered only rarely. An example is provided by the lipodepsipeptide syringopeptin 25A (SP25A), whose single channel current vs.
φm plot is roughly linear over a narrow potential range straddling
φm = 0 [
17]. However, this plot is obtained upon adding the peptide on one side of the BLM (the
cis side), stepping
φm from positive values on the
trans side (i.e., “trans-positive” values) to equal and opposite trans-negative values, and plotting the rate of change of the current with time as measured just before and after each voltage step against
φm. While the current at trans-positive potentials increases with time, that of opposite sign flowing as a consequence of the potential steps to the corresponding trans-negative values decays with time, becoming vanishingly small in about 10 s. This denotes an unstable state at trans-negative potentials, which determines a slow conformational change of the channel, accompanied by a movement of charge (the “gating charge”). Nonetheless, the cyclic voltammograms of SP25A at a Hg-supported tBLM show a perfectly ohmic behavior [
18] (see
Section 3.4). Particular pretreatments at non-physiological trans-negative potentials also induce an ohmic behavior lasting several hours in a typical voltage-gated channel such as melittin [
19,
20].
By far the majority of
α-helical peptides yield voltage-gated ion channels, when incorporated into BLMs. Several molecular models for voltage-gated channels have been proposed, with particular emphasis on the alamethicin channel. The simplest model assumes that channel formation starts from a small aggregate of peptide monomers (the nucleus) that grows in diameter through the uptake of further monomers. The voltage dependent step is the reorientation of the monomers by the electric field from the membrane surface into its interior, where they span the whole hydrophobic region and aggregate with ion channel formation [
21]. Several other more involved models for peptide aggregation with ion channel formation have been proposed (for a review, see [
22]), but the majority of evidence seems to be in favor of the aforementioned model.
There are quite different views about the peptide orientation at zero transmembrane potential, based on different experimental data and supported by different calculations. An NMR investigation showed that alamethicin interacts primarily at the water/lipid interphase without significant insertion into the hydrocarbon tail region in the absence of voltage [
23]. On the other hand, circular dichroism data in unilamellar vesicles [
24] and infrared attenuated total reflection spectroscopy in multibilayer membranes [
25] pointed to alamethicin incorporation into the lipid film. Site-directed spin-labeling studies showed that alamethicin is in a linear form and normal to the membrane plane, with its C-terminus in the aqueous region and the N-terminus in the membrane hydrocarbon tails, even in the absence of voltage [
26]. The aforementioned uncertainty in alamethicin orientation at zero transmembrane potential is not clarified by computational approaches [
27,
28].
2.2. What Can Be Learnt from Current-Time Curves at BLMs
The response of a conventional BLM incorporating a channel-forming peptide to a potential step is usually monitored by recording current-time (
I-t) curves. This technique is referred to as
chronoamperometry in electrochemical jargon. Peptides are often added only on one side of BLMs, referred to as the
cis side. In what follows, the transmembrane potential will be defined as the electric potential on the
trans side with respect to the
cis side, taken conventionally equal to zero.
Figure 1 shows
I-t curves following transmembrane potential steps of increasing height at a BLM incorporating monazomycin [
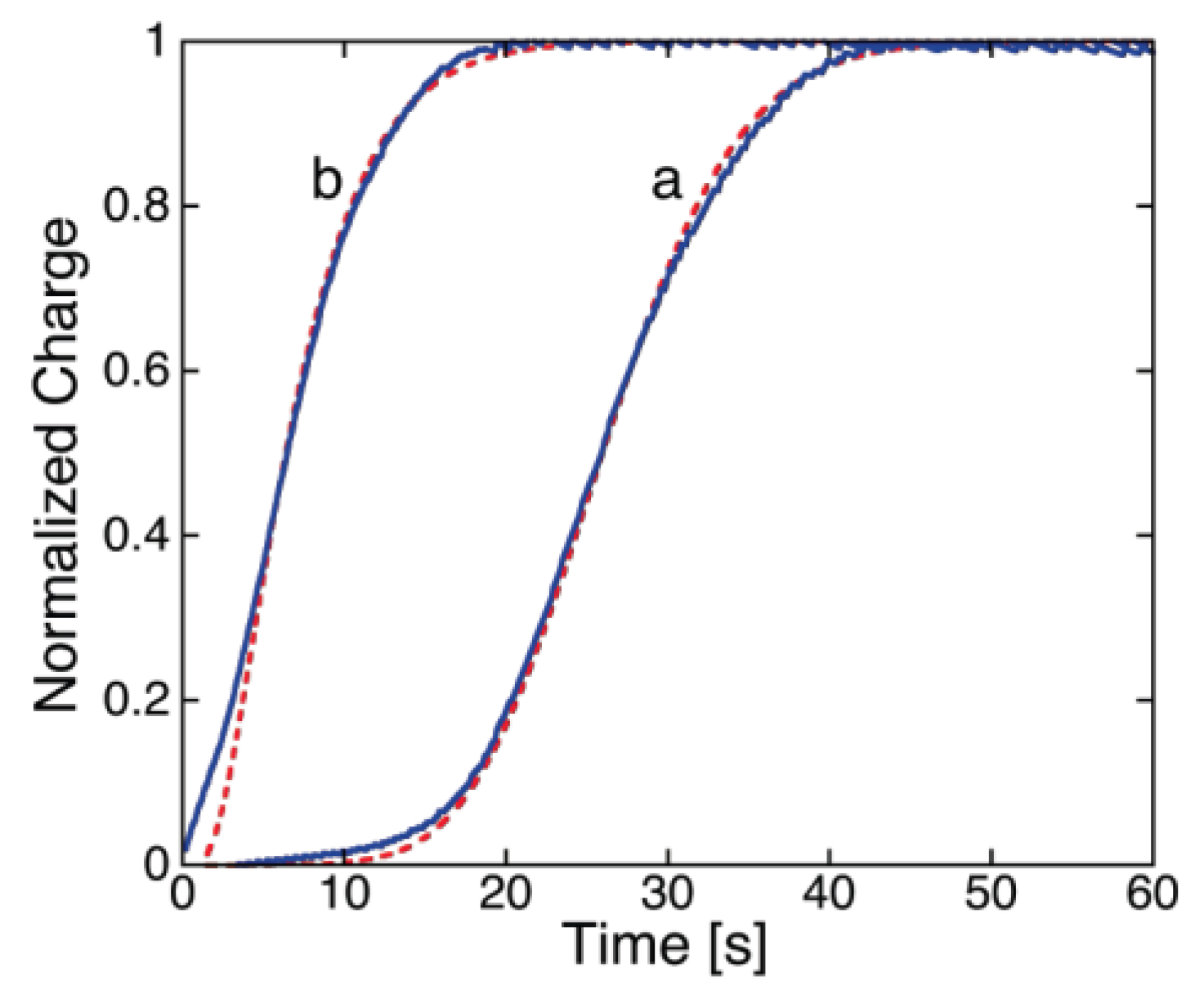
29]. The current induced by a voltage step rises in time approaching asymptotically a steady-state level, which increases with an increase in step height. This is due to an increase in the steady flow rate of permeant ions, elicited by the increase in the transmembrane potential.
Some peptides, such as alamethicin at ambient pressure [
14,
30], yield
I-t curves that constantly maintain the concavity of the curve turned toward the time axis. Some others, such as monazomycin [
29,
31] and melittin [
32], yield
I-t curves that pass from being concave upward to concave downward, thus exhibiting a clear sigmoidal shape. Even alamethicin was reported to yield a sigmoidal shape under particular experimental conditions, such as an elevated pressure of 100 MPa [
33] or during the recording of the first two or three
I-t curves at a BLM freshly formed from a dioleoylphosphatidylcholine (DOPC) solution in decane [
34]. Moreover, Mauro et al. [
31], by carefully examining the
I-t curve of alamethicin at ambient pressure in the range of a few tens of milliseconds from the start of the potential step, reported an initial S-shaped time course, followed by an asymptotic increase toward a steady-state value. It is, therefore, quite probable that the sigmoidal shape is a feature shared by all channel-forming
α-helical peptides and ascribable to a common kinetic mechanism. If this kinetics is too fast to be resolved in the accessible time scale of potential-step experiments, then only
I-t curves that are concave downward can be monitored, as in the case of the peptaibol alamethicin and of other Aib-containing peptides [
35]. A three-state molecular model predicting a sigmoidal shape of current transients in response to potential steps was proposed by Bruner [
36] by assuming that a peptide molecule moves, under the influence of an applied potential, first from a nonconducting surface state to a nonconducting precursor state and then to a conducting state. Current transients of sigmoidal shape for monazomycin [
30] and potassium channels [
37] were subsequently predicted by us on the basis a model accounting for monomer aggregation by a mechanism of nucleation and growth. Since this model also accounts for several properties of experimental current-voltage (
I-V) curves of BLMs incorporating channel-forming peptides, we will now describe in some detail the main features of this modelistic approach [
38].
Prior to a potential step inducing an I-t curve or a potential scan inducing an I-V curve, the monomers are regarded as located on the cis side of the membrane bathed by the solution where they were initially added. For the peptide dipolar molecule to span the hydrocarbon tail region of the BLM during a negative potential step or scan, the positive pole of the dipole will have to move across this region up to the attainment of the polar head region on the trans side, with the negative pole remaining on the cis side. The probability p of this charge movement depends critically upon the transmembrane potential.
Let us denote by
Ns and
Nt the number of peptide monomers located on the
cis side of the membrane surface and with a transmembrane orientation, respectively. Under equilibrium conditions, their relative electrochemical potentials,
and
, are equal for any value of the transmembrane potential
φm. These electrochemical potentials may differ not only by their electrical contribution, due to the different position of the positive pole within the membrane, but also by their chemical contribution
μθ, due to any conformational changes induced by such a charge movement. We can therefore write:
Here,
q is the charge of the positive pole, which undergoes a shift from the
cis to the
trans side of the membrane, passing from a position where the transmembrane potential equals
φs to one where it equals
φt. Rearranging terms, we obtain:
Under the simplifying assumption that the peptide monomers are only allowed either to be located on the
cis side of the membrane or to assume a transmembrane orientation, the probability
p of their assuming the latter orientation is clearly given by
p =
Nt/(
Nt +
Ns). Upon assuming for simplicity that the membrane spanned by the ion channel is homogeneous and that the electric field within it is constant, the electric potential varies linearly with distance. Denoting by
xs and
xt the distances of the charge
q from the
cis surface of the membrane in its initial
cis and final
trans locations, respectively,
φs is given by
xsφm/
d, and
φt by
xtφm/
d, where
d is the membrane thickness. Hence,
q(
φt −
φs) equals
q(
xt −
xs)
φm/
d = ∆
mφm/
d, where ∆
m is the change in the dipole moment of the peptide as a consequence of its alignment along the direction of the electric field. Carrying out this substitution into Equation (2), the probability
p is given by [
3,
38]:
∆
μθ is the difference in Gibbs energy between the two conformational states of the peptide in the absence of the electric field (i.e., for
φm = 0). Since the parameter
a is always much less that unity, it measures the probability
p at zero transmembrane potential, in view of Equation (3).
As soon as a negative potential step or scan induces the peptide monomers to span the membrane with the
φm-dependent probability
p, the resulting transmembrane monomers start nucleating, giving rise to channel-forming transmembrane clusters. Let
θ0 denote the fraction of the whole BLM surface covered by the peptide molecules, irrespective of their orientation. Upon denoting by
S the ratio of the area covered by transmembrane clusters to that covered by both transmembrane monomers and transmembrane clusters, the fraction of the whole BLM surface covered by the transmembrane monomers is given by
Θ ≡
θ0p(1 −
S), whereas
θ0pS is clearly the fraction covered by the transmembrane clusters. Clusters of transmembrane monomers resulting from a series of consecutive collisions are characterized by a critical size (the “nucleus”), below which they have a higher tendency to shrink by releasing one unit than to grow by aggregation of a further unit, and above which they have a practically irreversible tendency to increase. The formation of this critical cluster size from embedded monomeric units is called “nucleation”, whereas the irreversible increase beyond the critical size is referred to as “growth”. If the nucleus is composed of a number
n of transmembrane monomers, the elementary step yielding the nucleus consists of the incorporation of a monomer into a (
n − 1)-meric “subcritical nucleus”. This results from (
n − 1) elementary steps consisting in the incorporation of each monomeric unit into the immediately preceding subcritical nucleus, starting from an initial transmembrane monomer acting as a “nucleation center”. If all steps preceding the step yielding the nucleus are considered to be in quasi equilibrium, then the nucleation rate,
vN, will be proportional to the
nth power of the fractional surface coverage,
Θ =
θ0p(1 −
S), by the transmembrane monomers randomly distributed in the lipid bilayer, according to a nucleation rate constant
kN:
Here N is the number of nuclei per unit surface area. The irreversible aggregation of monomeric units to a nucleus, just after its formation, gives rise to a “supercritical nucleus”, whose continuous growth ultimately yields a channel-forming transmembrane cluster.
Upon assuming for simplicity that the cross-sectional area
A of a growing supercritical nucleus is a circle of radius
R, the rate of growth of
A is given by the time derivative of
πR2. This rate can be reasonably regarded as proportional to the frequency of the successful impacts of the transmembrane monomers, of surface coverage
Θ, with the circumference
2πR of the supercritical nucleus, according to a proportionality constant
kR. Hence, we can write:
It follows that the rate vR of radial growth of a supercritical nucleus is proportional to Θ according to the rate constant kR. Successful impacts require a favorable mutual orientation between the supercritical nucleus and the aggregating transmembrane monomer, and are not necessarily controlled by the two-dimensional diffusion of the transmembrane monomers within the lipid bilayer.
Let us now consider a general approach to the kinetics of nucleation and growth that allows the quantity
S to be calculated as a function of time. Upon setting equal to 0 the starting time of the nucleation and growth process, let us imagine observing it at a later time
t. Let d
N denote the infinitesimal number of nuclei that are forming in the infinitesimal time interval between
y and
y + d
y before the observation time
t. Having assumed that the resulting supercritical nuclei have a circular shape, the nuclei formed in this infinitesimal time interval make the following contribution to the area covered by the supercritical nuclei at time
t [
3,
38]:
Here,
z is an auxiliary variable that has the dimensions of time and varies between the time
y at which the nuclei form and the time
t at which the resulting supercritical nuclei are observed. If we now sum all the above infinitesimal contributions by integrating Equation (6) over time between the limits of integration
y = 0 and
y =
t, we obtain the ratio of the area covered by the supercritical nuclei (ultimately yielding the channel-forming clusters) to that originally covered by all monomers, irrespective of their orientation [
39]:
This ratio is denoted by Sx, rather than S, to emphasize that it ignores the possible overlapping of the progressively growing supercritical nuclei; it is commonly referred to as the “extended area”. However, strictly speaking, Sx is not the” extended equivalent” of S. In fact, S is the ratio of the actual area covered by the supercritical clusters to that covered by both supercritical clusters and transmembrane monomers, thus ignoring the monomers located on the cis side of the membrane. In this respect, Sx corresponds to the “extended equivalent” of the product pS, and not of S. The expression in Equation (7) includes both the rate of nucleation, (dN/dt) = vN, and the rate of radial growth, (dR/dt) = vR. Being entirely general, it can be applied to the kinetics of any nucleation-and-growth process.
The unrealistic overlapping of the progressively growing supercritical nuclei can be avoided by making use of Avrami’s formalism [
40]. Roughly speaking, Avrami’s approach relies on the consideration that the area covered by the growing supercritical nuclei is an extensive property of the system and, as such, is directly proportional to the “available area”. If,
ab absurdo, this concept is applied to the case in which the single growing supercritical nuclei are allowed to grow without being limited by the neighboring supercritical nuclei, then the available area is the total surface area,
ST, where the nucleation-and-growth process occurs, and we will write:
Sx is just the aforementioned hypothetical extended area, namely the area that would be covered by all the growing supercritical nuclei if they were free to grow without limits. This is not possible in reality, and the actual surface area available to the growing supercritical nuclei is that still uncovered, which is given by the difference,
ST −
S, between the total area,
ST, and that,
S, already covered. In this case, Equation (8) will become:
Upon eliminating the common proportionality constant between Equations (8) and (9), we obtain [
2]:
The present model of nucleation and growth is entirely general. Thus, it only assumes that the elementary steps preceding the step yielding the nucleus are in quasi equilibrium, and that the growth of the supercritical nuclei proceeds irreversibly by activated aggregation of transmembrane monomers. The expressions of Equations (4) and (5) for the rates,
vN and
vR, of nucleation and radial growth must be substituted into the expression of Equation (7) for the extended area
Sx. In doing so, we must consider that
Sx is the extended equivalent of
pS. Hence, in carrying out these substitutions, the complement of
S to unity, (1 −
S), must be multiplied by
p in the quantity
Θ =
θ0p(1 −
S), for consistency [
41], yielding:
By repeatedly differentiating Equation (11) with respect to time via the generalized Leibnitz formula, three differential equations are obtained. Combining these equations with the differential Equation (10) relating the extended area Sx to the corresponding actual area S, a set of four differential equations is obtained, which can be readily solved numerically by the fourth-order Runge-Kutta method, yielding Sx and S as a function of time. It is worth noting that the kinetics of nucleation and growth depends exclusively on the product, kNkR2, of the rate constant of nucleation by the square of the corresponding rate constant of radial growth, thus reducing the number of adjustable parameters to only four, i.e., p, θ0, n, and kNkR2.
Finally, the current density
j across the BLM induced by the transmembrane-potential jump is obtained by setting it proportional to the fractional surface coverage by the channel-forming transmembrane clusters,
θ0pS, since each newly formed channel makes a contribution to this current:
The limiting value attained by j is clearly given by θ0p, since S will ultimately tend to unity.
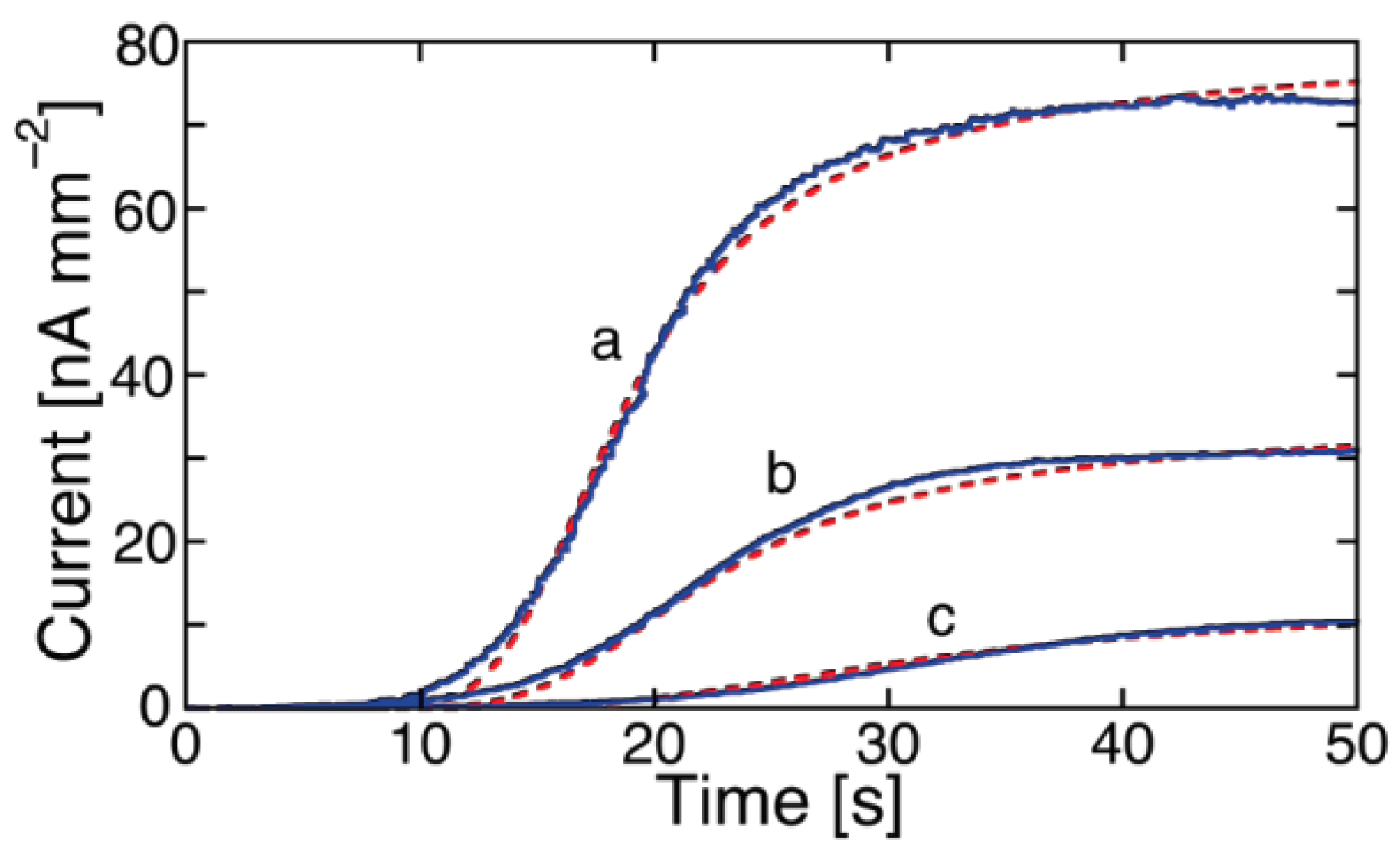
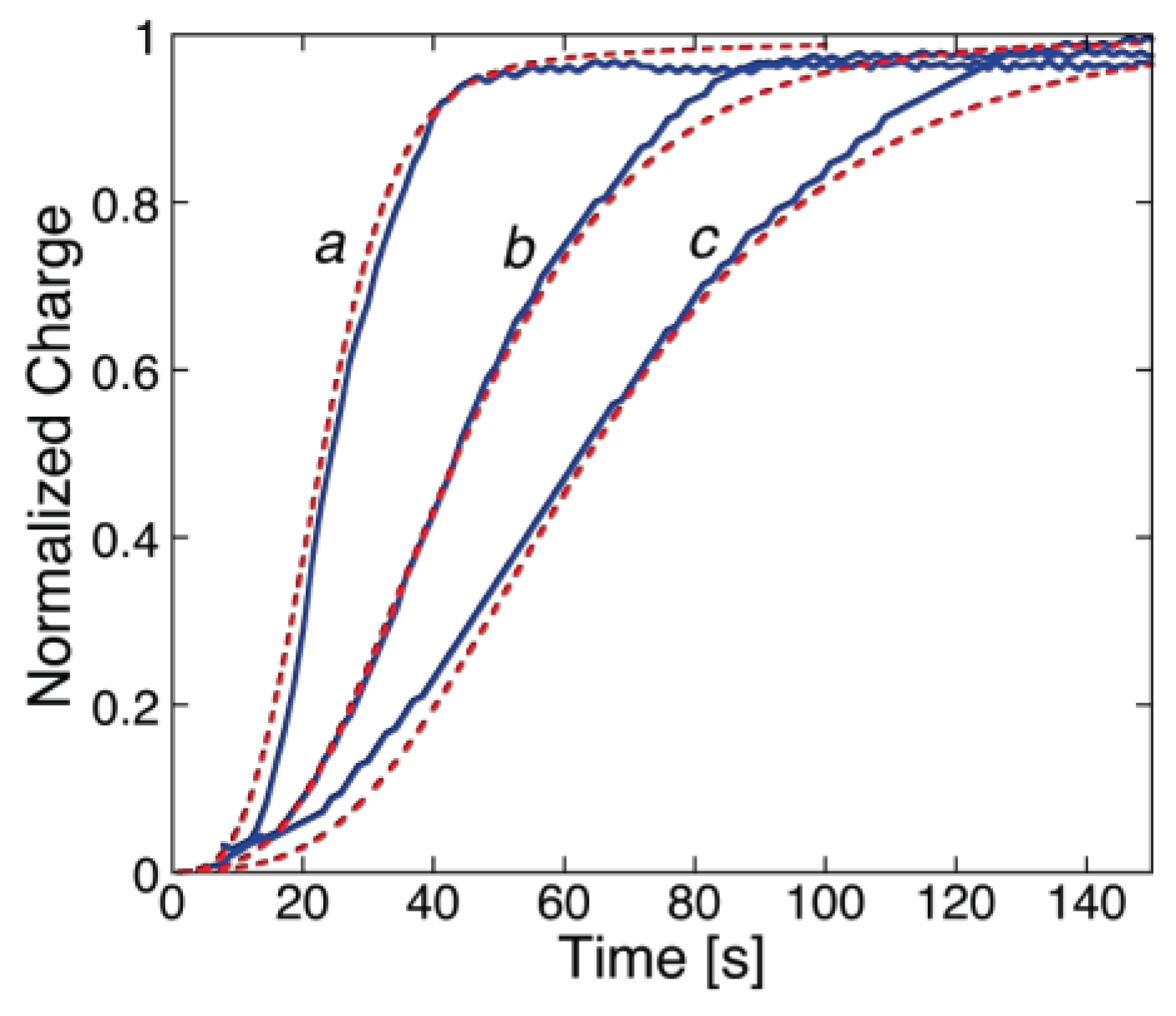
The polyene-like antibiotic monazomycin incorporated in a BLM yields sigmoidal
I-t curves [
29], as shown in
Figure 1. However, the typical sigmoidal current rise is preceded by a relatively long period of time during which no ion flow across the BLM occurs. The resulting long “foot” of the current transients is to be ascribed to the presence of peptide aggregates flatly adsorbed on the BLM surface at the initial potential. These flat clusters have the polar/charged side chains turned toward the aqueous phase and the hydrophobic side chains turned toward the interior of the clusters, with an arrangement opposite to that in the ion channels. Hence, their incorporation into the BLM following the potential step requires their previous disruption into flat monomers, to be converted into transmembrane monomers. The kinetic process of disruption of flat clusters can be formally treated as a nucleation of holes and growth of hole aggregates within the clusters. By nucleation of holes, we mean the quasi-reversible detachment of an initial number of flat monomers from a flat cluster and their random intercalation with the water molecules on top of the BLM. In other words, a hole is just a flat monomer that, upon detachment from a flat cluster, leaves behind a hole in the cluster. These flat monomers are considered to detach from a flat cluster and to re-aggregate into it in a quasi-reversible manner, until the number of nearest-neighboring holes in the cluster attains a critical value (
n) beyond which this number increases irreversibly up to complete disruption of the flat cluster. This critical number of nearest-neighboring holes can again be loosely regarded as a nucleus. Nucleation is followed by the irreversible disruption of the flat clusters, which can be viewed as an irreversible “growth of hole aggregates”, according to the nucleation-and-growth terminology. This disruption is triggered by the potential step, which starts stripping the flat monomers (the holes) from the flat clusters by dragging them into the BLM as transmembrane monomers. The rate constant of nucleation of holes,
kh,N, and the rate of radial growth of hole aggregates,
vh,R, are not directly affected by the electric field. However, they depend indirectly upon the electric potential via the penetration probability
p of the flat monomers. In
Figure 1, the experimental
I-t curves of monazomycin are fitted by a model that combines the nucleation of holes and growth of hole aggregates with the nucleation of transmembrane monomers and growth of transmembrane supercritical nuclei [
30].
2.3. What Can Be Learnt from Current-Voltage (I-V) Curves at BLMs
I-V curves at BLMs are obtained by scanning the applied electric potential between an initial and a final value at a constant scan rate v. The electric potential is usually applied between two identical reference electrodes immersed in the two aqueous solutions that bath the two sides of the BLM. This potential, which is just the transmembrane potential φm, is a thermodynamically significant quantity and is referred to as “voltage” (V) in biophysical jargon. The voltage may also be scanned back and the resulting voltage cycle may be repeated several times, a procedure referred to as “cyclic voltammetry” in electrochemical jargon; in this case the current is plotted versus the voltage back and forth to yield the cyclic voltammetry trace. When the peptide is added on only one side of the BLM, identified with the cis side, the voltage V will be referred to the trans side with respect to the cis side, taken as zero. This definition, which is the opposite of that adopted in the biophysical literature, complies with that adopted at metal-supported tBLMs, where the applied potential E is always referred to the metal with respect to the solution. Accordingly, the current will be taken as negative when cations move from the cis to the trans side, at variance with the biophysical usage.
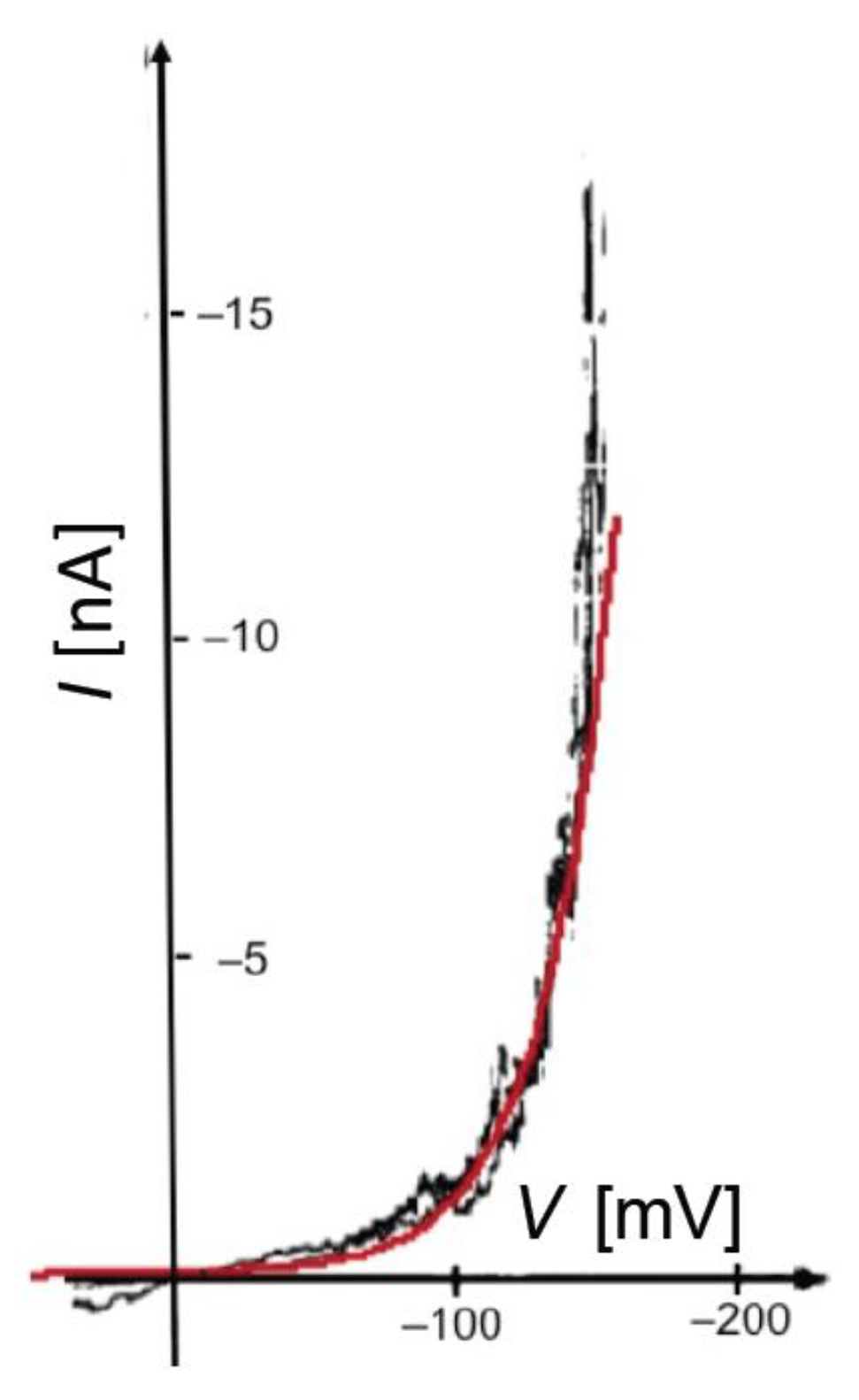
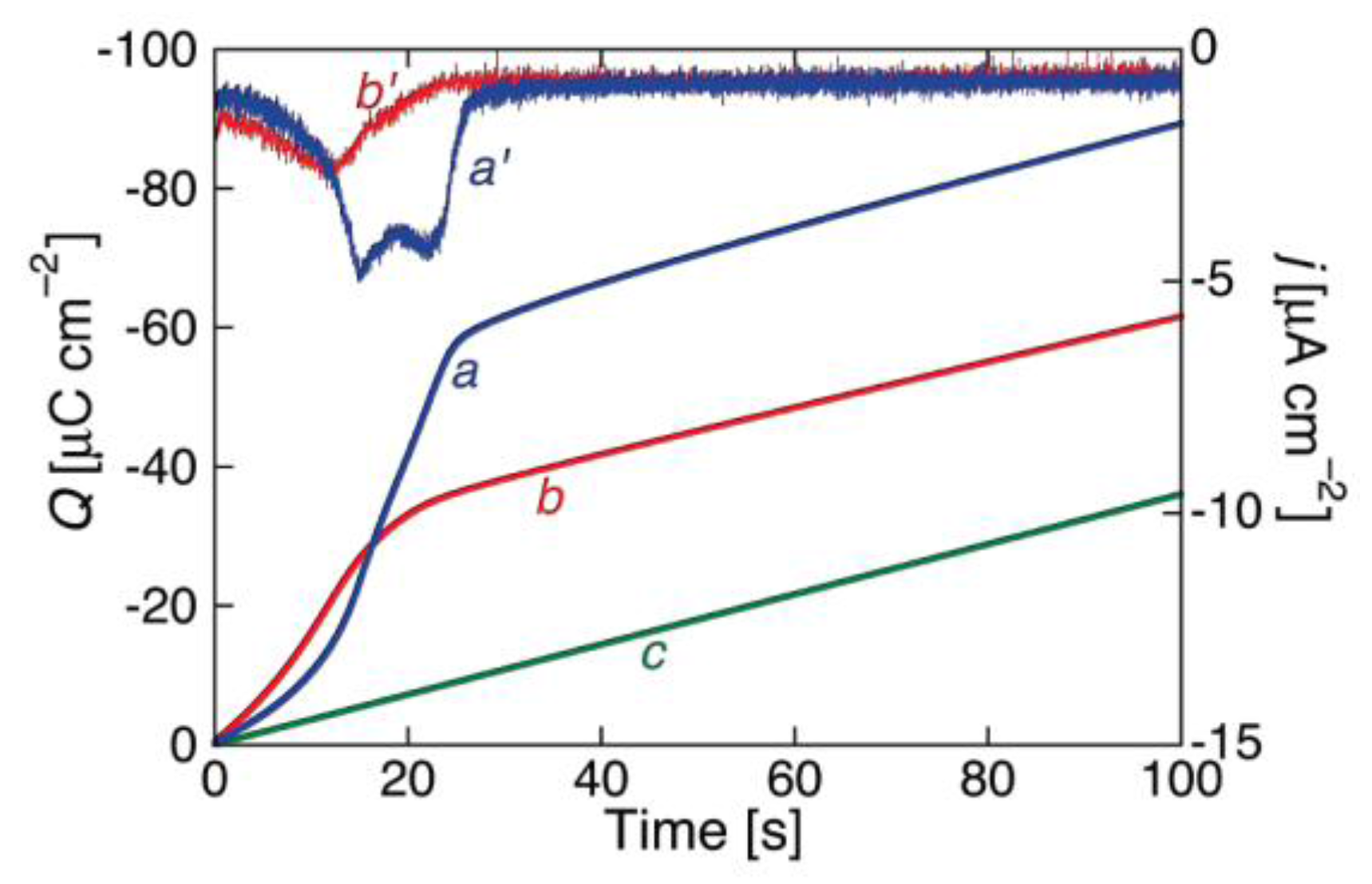
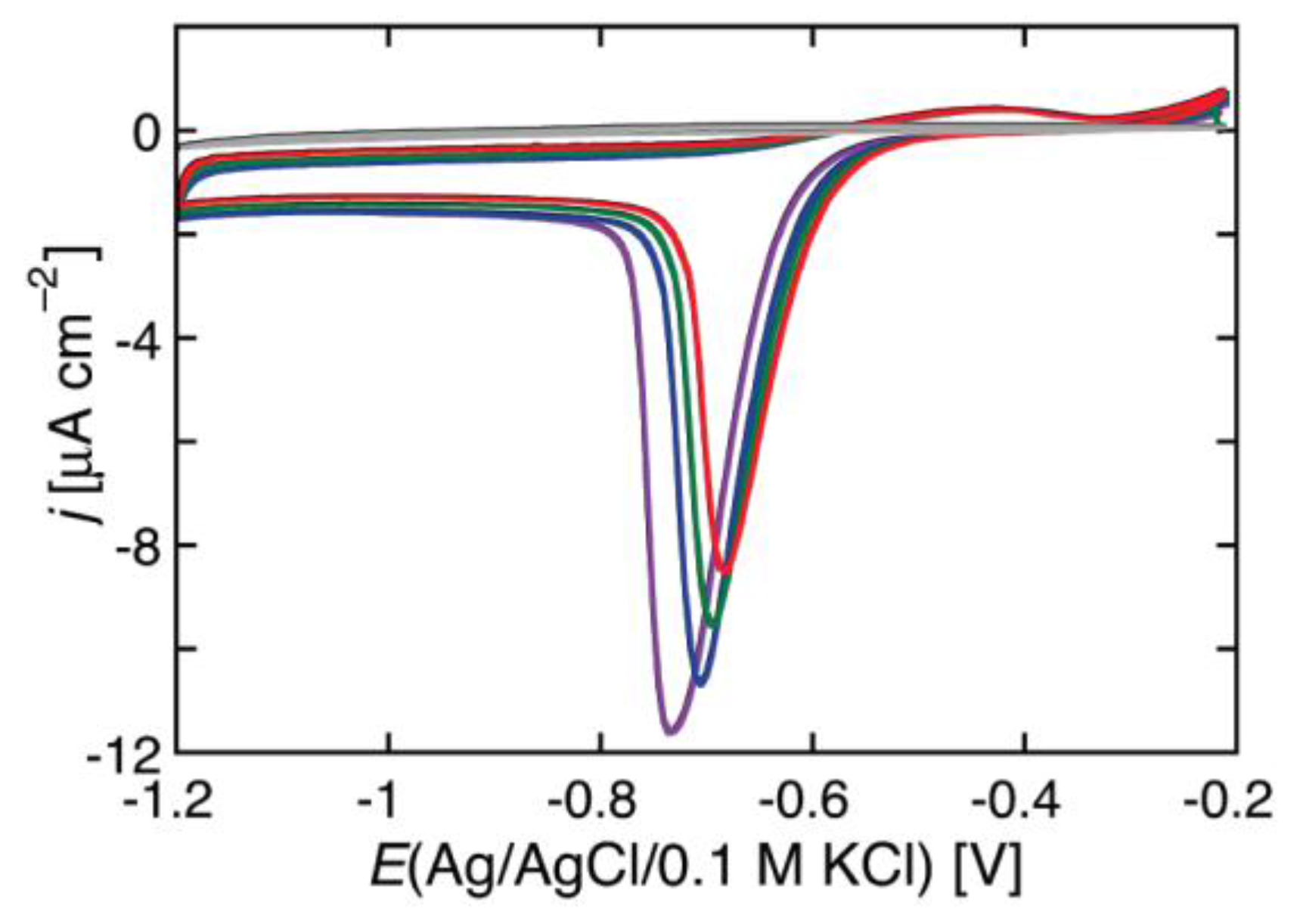
In BLMs incorporating channel-forming peptides from only the
cis side, a negative voltage scan induces an exponential increase in the negative current, as shown in
Figure 2 for a BLM incorporating melittin. Thus, the slope of the resulting
I-V curve, which measures the conductance
G, is such that its logarithm increases linearly with an increase in the absolute value, |
V|, of the voltage. An increase in the concentration of a channel-forming peptide in the bathing solution shifts the
I-V curve toward less negative
V values. At constant conductance, |
V| decreases linearly with the logarithm of the peptide concentration.
Alamethicin and melittin are the two most thoroughly investigated voltage-gated channel-forming peptides in BLMs. In spite of their structural differences, they have several functional features in common. Thus, under suitable experimental conditions, they both exhibit two distinct relaxation processes, a fast relaxation process with weakly voltage-dependent conductance, and a slow relaxation one with strongly voltage-dependent conductance. In particular, this situation is encountered when the peptide is added to only the
cis side of the membrane, and the current flows only in the direction from the
cis to the
trans side, provided the voltage is sufficiently negative. The conductance
G associated with any of the two different relaxation processes depends upon
V and the peptide concentration,
cpep, according to the equation [
22,
42]:
The fast relaxation process has a time constant two orders of magnitude smaller than that of the slow relaxation one and occurs in the millisecond range, which is of the same order of magnitude as the mean lifetime of single pore states [
34]; it appears to arise from a shift in the probability distribution of the different conductance levels of a pore as the voltage is changed. Conversely, the slow relaxation process is attributed to the fluctuation in the number of channels present in the membrane [
34,
43] and correlates well with the lifetime of single current bursts. The conductance of the fast relaxation process is from one to two orders of magnitude smaller than that of the slow relaxation process. The passage from a weakly to a strongly voltage-dependent conductance with a progressive negative shift in voltage is shown by a number of synthetic Aib-based polypeptides of different length incorporated into palmitoyloleoylphosphatidylcholine (POPC) BLMs [
35]. The critical voltage marking the passage from a weakly to a strongly voltage-dependent conductance regime is accompanied by a dramatic rise of current and shifts toward more negative values with a decrease in the polypeptide chain length and in its concentration in the bathing solution.
During the slow relaxation process, alamethicin yields
γ values ranging from 4.4 to 6 and
δ values ranging from 6 to 11, depending on the lipid composition of the BLM [
34,
42], whereas melittin yields
γ = 4.3 and
δ ≈ 4 [
32] and synthetic Aib-based peptides yield
γ = 4.7 and
δ = 8.3 [
35]. Conversely, during the fast relaxation process, alamethicin yields
γ = 0.96 and
δ ≈ 2 [
34], synthetic Aib-based peptides yield
γ values ranging from 2.8 to 3.3 and
δ ≈ 2 [
35], whereas melittin yields
γ values ranging from 1.3 to 1.6 and
δ ≈ 4 [
32], although a
δ value of 2.5 at zero voltage was also reported [
44]. The
δ parameter for melittin was found to decrease from ~2 to ~0.5 with a decrease in the chain length of the lipid molecules forming monoglyceride/squalene BLMs [
45]. Summarizing, the dependence of the current upon the peptide concentration increases by several orders of magnitude in passing from the fast to the slow relaxation process.
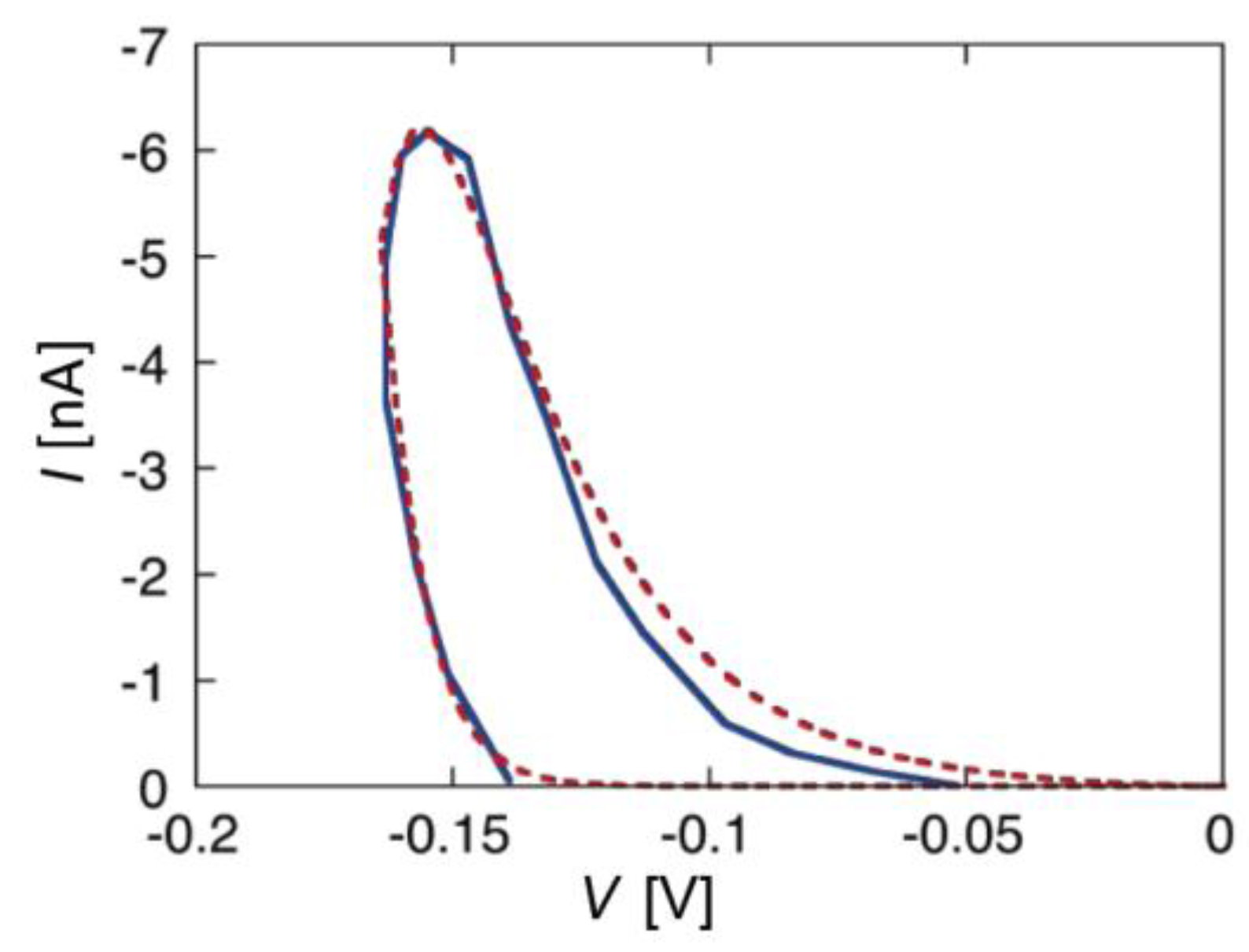
When melittin is added on the
cis side of the BLM, the steady-state
I-V curve shows both a positive and a negative branch, albeit not perfectly symmetrical with respect to
V = 0 [
20]. Adding alamethicin on the
cis side yields a single negative branch of the
I-V curve if the BLM is formed with a saturated lipid [
14,
46]. On the other hand, with unsaturated or halogenated membrane lipids, alamethicin yields both branches, with the positive branch more shifted with respect to zero voltage than the negative one [
46]. This suggests that the presence of saturated lipids in the BLM hinders the diffusion of alamethicin across the membrane.
The salient features of the experimental behavior of voltage gated channels at BLMs can be accounted for by the same general approach outlined in
Section 2.2, which treats the time-dependent formation of ion channels as a nucleation of transmembrane peptide monomers and growth of the resulting supercritical nuclei. The probability
p for the passage from a flat to a transmembrane orientation is again expressed by Equation (3). Being a function of the transmembrane potential
φm =
V,
p is time independent after the instantaneous voltage step in
I-t measurements, whereas it varies with time in
I-V measurements. Nonetheless, even in the latter case, the probability
p must be removed from under the integral signs in the expression of
Sx, as done in Equation (11). This removal is necessary, since
p is an implicit function of time
t and is operative from the starting time set equal to 0, thanks to the constant voltage scan rate
v ≡ d
V/d
t. Conversely, the quantity
Sx is a function of the integration variables
z and
y and refers to the events occurring throughout the various nucleation processes starting at different times
y and followed by the growth of the resulting supercritical nuclei, up to the final observation time
t.
The last step consists in calculating the current density
j from the absolute rate theory of ion transport across membranes [
47], as applied to ion translocation across the potential energy barrier located in the hydrocarbon tail region of the BLM. In applying this equation, the potential energy barrier will be regarded as symmetrical and nonselective toward ion flow, for simplicity, by ascribing a common value,
kt, to the forward and backward rate constants at zero voltage. Finally, the ionic current density
j across the membrane, which would be equal to zero in the absence of ion channels, is set proportional to the fractional surface coverage by the embedded channel-forming clusters,
θ0pS, since each newly formed channel makes a contribution to this current. With the above assumptions, the current density
j expressed by the absolute rate theory becomes:
Here, α is the transfer coefficient, c+cis, c+trans are the concentrations of a monovalent cation on the cis and trans sides of the membrane, and c−cis, c−trans are those of a monovalent anion. The first and second expressions between parentheses in Equation (14) measure the cation and anion currents, respectively.
Upon setting for simplicity
c+cis =
c+trans =
c−cis =
c−trans ≡
c,
j turns out to be proportional to
c, besides being proportional to
kt. The behavior of the dimensionless quantity
j/(
Fktc) will serve to show the way in which the quantities ∆
m,
θ0,
a and the kinetic parameters of nucleation and growth affect the shape of
I-V curves and the dependence of ln
G upon
V and the concentration,
cpep, of the peptide in the bathing solution. Strictly speaking, setting the current density proportional to the number density of the embedded clusters that have undergone a kinetic process of nucleation and growth with ion channel formation amounts to assuming that only a single type of aggregate develops during the whole voltage scan. However, this assumption is also valid if the single-channel state distributions are independent of voltage and the macroscopic conductance induced by the peptide exclusively arises from the increase in the number of channels as |
V| increases. These requirements are approximately satisfied by the alamethicin channel in a phosphatidylethanolamine BLM [
14] and by melittin in a DOPC BLM [
20].
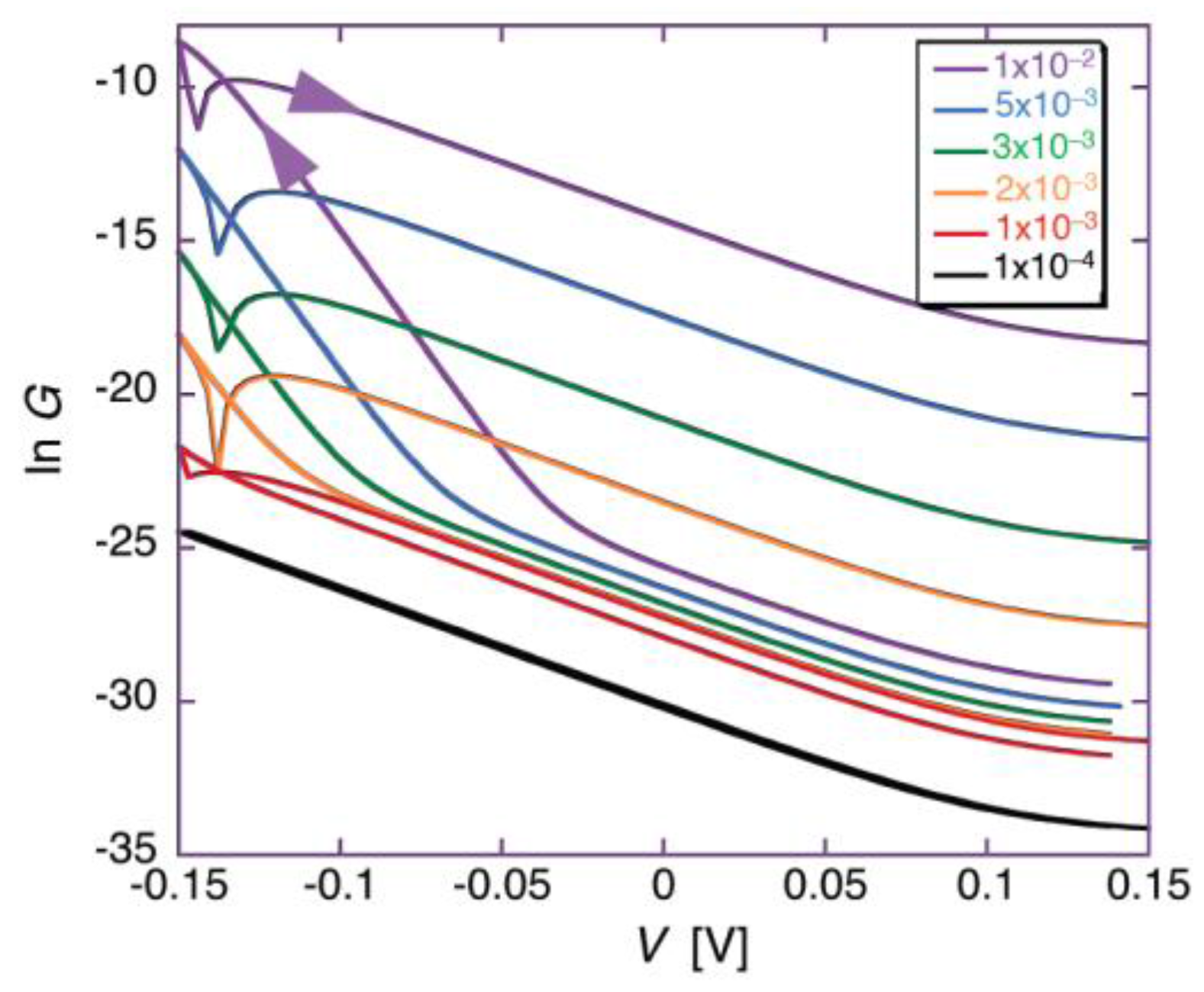
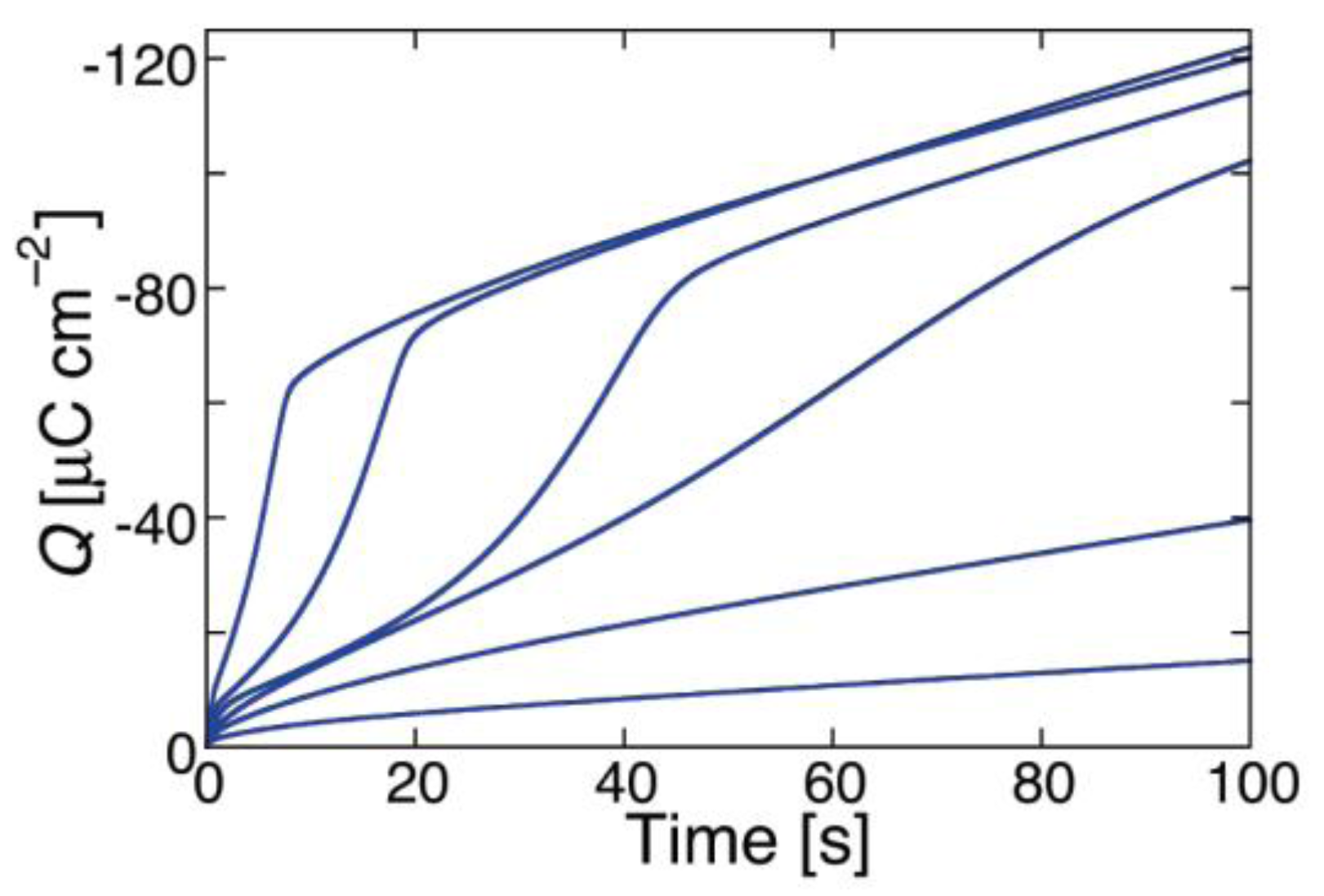
Figure 3 shows plots of ln
G against
V for different values of the probability,
a, of the peptide dipoles being in the transmembrane orientation at zero voltage, and for ∆
m = 70 D,
θ0 = 0.1,
kNkR2 = 1 × 10
6 s
−3 and
n = 1. All plots refer to a temperature of 298 K, a voltage scan rate of 10 mV/s and a membrane thickness
d = 30 Å. The plots show ln
G both along the negative-going voltage scan and along the subsequent positive-going one. The sharp dip marks the maximum negative current attained at the beginning of the reverse voltage scan.
During the negative-going voltage scan, the plots exhibit a region of weakly voltage-dependent conductance, with
γ = 0.95, at lower negative voltages, as well as a region of strongly voltage-dependent conductance, with
γ = 3.85, at higher negative voltages. At extreme positive voltages,
G tends to attain a voltage-independent minimum value. During the positive-going voltage scan, the voltage dependence of the conductance is characterized by a
γ value of 0.95, other than in the proximity of the voltage reversal. A progressive decrease of
a lowers the ln G vs.
V curve and gradually shifts its “elbow” toward more negative voltages. This causes the region of weakly voltage-dependent conductance to cover the whole negative voltage range usually spanned by experimental
I-V curves. As a result of this trend, the
I-V curves falling over this voltage range and calculated for
a values varying from 1 × 10
−2 to 5 × 10
−3 show a strongly voltage-dependent conductance and an appreciable hysteresis, whereas those with
a < 2 × 10
−3 show a weakly voltage-dependent conductance and a small or negligible hysteresis. This behavior is exemplified by the calculated
I-V curves in
Figure 2 and
Figure 4, which refer to the two different situations.
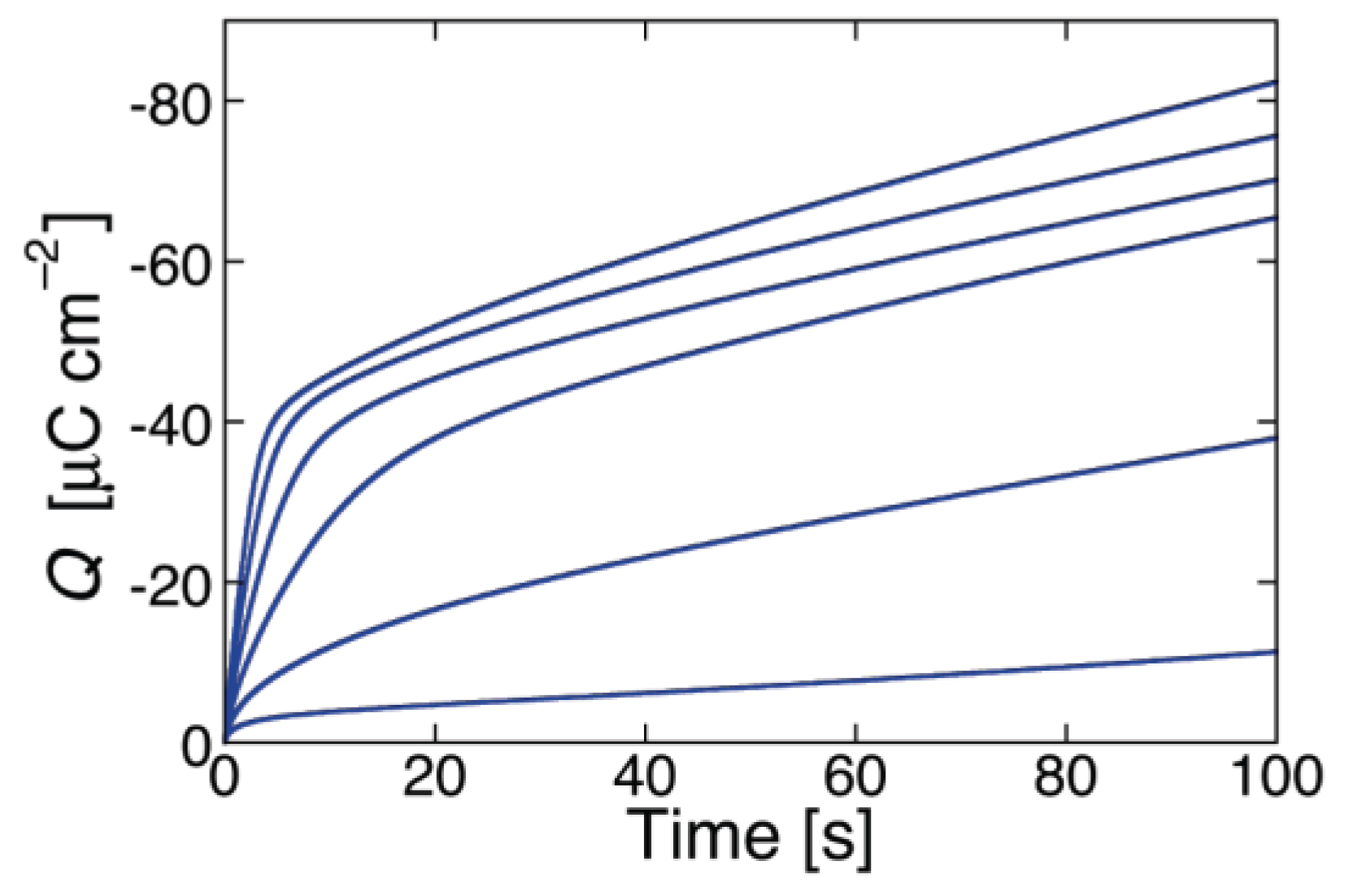
Figure 4 shows an experimental
I-V curve for 0.2 μg/mL alamethicin in a phosphatidylethanolamine BLM [
46] and a corresponding curve calculated for
a = 1 × 10
−2, ∆
m = 70 D,
θ0 = 0.1,
kNkR2 = 1 × 10
5 s
−3 and
n = 1. The shape of the calculated curve closely simulates those reported for alamethicin [
45,
46,
48] and for synthetic Aib-based polypeptides [
35]; the
γ value of 3.85 is also close to those for the synthetic polypeptides, although that for alamethicin is somewhat higher [
34,
35,
42]. Conversely,
Figure 2 shows an experimental
I-V curve for 0.4 μg/mL melittin in a DOPC BLM [
20] and a corresponding curve calculated for
a = 1 × 10
−4, ∆
m = 70 D,
θ0 = 0.1,
kNkR2 = 1 × 10
6 s
−3 and
n = 1, in which the forward and backward voltage scans practically coincide.
The experimental
I-V curves for alamethicin and melittin in
Figure 2 and
Figure 4 being simulated with
a = 1 × 10
−2 and 1 × 10
−4, respectively, is consistent with their different structures. Thus, if both peptides are incorporated in the
cis polar head region of the BLM at positive voltages, the Gibbs energy required for a negative-going voltage scan to reorient the peptide dipoles from a flat to a transmembrane stand is expected to be higher for melittin than for alamethicin. In fact, the Gibbs energy required to push the positively charged N-terminal amino group and the Lys-7 residue of the N-terminal sequence of melittin into the hydrocarbon tail region is much greater than that required to push the neutral N-terminal of the alamethicin molecule. The regions of weakly and strongly voltage-dependent conductance in the ln
G vs.
V curves of
Figure 3 are also in semiquantitative agreement with the corresponding fast and slow relaxation processes exhibited by the current-time curves in voltage step experiments, characterized by
γ ≈ 0.96 and 6.4 for alamethicin [
34], and by
γ ≈ 1.3 and 4.3 for melittin [
32].
The
γ values for the regions of weakly and strongly voltage-dependent conductance are only slightly affected by a change in the combined rate,
kNkR2, of nucleation and growth. The main effect of an increase in
kNkR2 from 10 to 1 × 10
7 s
−3, for
a = 10
−2 and the other parameters as in
Figure 3, consists in a shift of the elbow of the bent ln
G vs.
V curves toward less negative voltages [
38]. The effect of a decrease in the number of peptide monomers composing the critical nucleus from
n = 4 to
n = 1, with the other parameters as in
Figure 3, is qualitatively similar to that produced by an increase in
kNkR2. Thus, apart for the obvious changes in the absolute value of ln
G with varying the adjustable parameters of the model, the slope of ln
G against
V yields
γ values in fairly good agreement with the experimental ones over both regions of strongly and weakly voltage-dependent conductance, irrespective of the values ascribed to the adjustable parameters.
In the regime of strongly voltage-dependent conductance, the
γ value predicted by the model increases linearly with the magnitude of the dipole moment and is expressed by the equation:
γ = 0.66 + 0.044 Δ
m [
D], for
θ0 = 0.1,
a = 1 × 10
−2,
kNkR2 = 1 × 10
7 s
−3 and
n = 1 [
38]. Hence, the model predicts a
γ value of 4, close to that reported for several channel-forming
α-helical peptides, when ∆
m is close to 70 D, which is the value estimated for an
α-helical peptide spanning a hydrocarbon tail region 30 Å thick [
22]. This strongly suggests that the change, ∆
m, in the dipole moment normal component undergone by these peptides during a negative-going voltage scan is determined by the movement of their
α-helix from the
cis polar head region, with an orientation parallel to the membrane plane, to one spanning the hydrocarbon tail region (about 30 Å in thickness for a typical BLM). The above connection between the effective dipole moment of the peptide and the
γ value has contributed to regarding
γ as an “apparent gating charge”.
Along the region of weakly voltage-dependent conductance, the model predicts a
γ value about equal to unity. This is due to the fact that over this region the ratio,
S, of the number of peptide molecules aggregated into ion channels to the total number of molecules spanning the lipid bilayer is very low and almost voltage independent. Under these conditions, the voltage dependence of the current density
j is only expressed by the product of
p by the term between square brackets in Equation (14), and leads to a ln
G vs.
V plot of slope very close to unity. Since the kinetics of nucleation and growth exclusively affects the parameter
S, and this is very small and roughly constant, the current is clearly insensitive to the
kNkR2 and
n values. Moreover, for
S practically constant, the current turns out to be proportional to the fraction,
θ0, of the membrane unit area covered by the peptide. Within the limits in which
θ0 can be regarded as proportional to the peptide concentration,
cpep, in the aqueous solution, the current along the low conductance region is proportional to
cpep, and the parameter
δ in Equation (13) equals unity. This prediction is consistent with the experimental observation that the low
γ values characterizing the regime of weakly voltage-dependent conductance are often associated with
δ values ranging from 1 to 2.5 [
34,
35,
44], with the exception of melittin in asolectin BLMs [
32,
49]. It must be pointed out that the prediction of a
γ value of about 0.95 along the region of weakly voltage-dependent conductance results from a passage of the peptide dipoles from a parallel to a vertical orientation with respect to the membrane plane as voltage becomes progressively more negative, similarly to what happens along the region of strongly voltage-dependent conductance. However, in this case no aggregation of transmembrane monomers occurs. For a typical dipole moment of 70 D, the mere dipole reorientation without aggregation yields a
γ value of about 0.95. It is the aggregation of transmembrane dipoles with nucleation-and-growth kinetics that causes a significant
γ increase from about 1 to about 4, when passing from the regime of weakly to that of strongly voltage-dependent conductance. This suggests that, in the regime of weakly voltage-dependent conductance, the ion movement elicited by the electric field occurs within a bunch of transmembrane monomers, whose side chains are randomly distributed with respect to each other (i.e., without aggregation into a proper ion channel).
The dependence of the conductance upon the peptide concentration predicted by the model in the regime of strongly voltage-dependent conductance is higher than that in the regime of weakly voltage-dependent conductance, as expressed by
δ = 1. Thus, the plot of ln
G against ln
θ0 obtained from
I-V curves calculated for ∆
m = 70 D,
a = 1 × 10
−2,
kNkR2 = 1 × 10
6 s
−3 and
n = 1, i.e., under conditions of strongly voltage-dependent conductance, is linear and exhibits a slope of 3.85 over the whole
θ0 range from 0.1 to 1 [
38]. This value can be identified with
δ, if we can reasonably assume that the peptide is incorporated from the bathing solution into the
cis polar head region of the membrane according to Henry’s adsorption isotherm. This
δ value is close to those reported in the regime of strongly voltage-dependent conductance for certain Aib-based polypeptides [
35] and for melittin [
32], whereas alamethicin exhibits appreciably higher values ranging from 6 to 11 [
42]. The fact that the sole introduction of a mechanism of nucleation and growth for monomer aggregation into ion channels yields a
δ value close to 4 as a natural consequence, demonstrates the inconsistency of the frequent assumption [
20,
50] that such a value is indicative of the formation of a tetrameric ion channel. The
δ value for alamethicin being appreciably higher than predicted by the model, especially in certain BLMs, may possibly be ascribed to the failure of the assumption of voltage independence of the single-channel state distributions, on which Equation (11) relies. A higher
δ value is expected if an increase in negative voltage tends to favor higher aggregates at the expense of lower ones.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}















