Fenofibrate Reduces the Severity of Neuroretinopathy in a Type 2 Model of Diabetes without Inducing Peroxisome Proliferator-Activated Receptor Alpha-Dependent Retinal Gene Expression

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Metabolic Analyses
2.3. Electroretinography
2.4. Immunohistochemistry
2.5. Gene Expression Analysis of Mice Treated with Fenofibrate-Supplemented Diet
2.6. Intravitreous Injection and Microarray Analysis
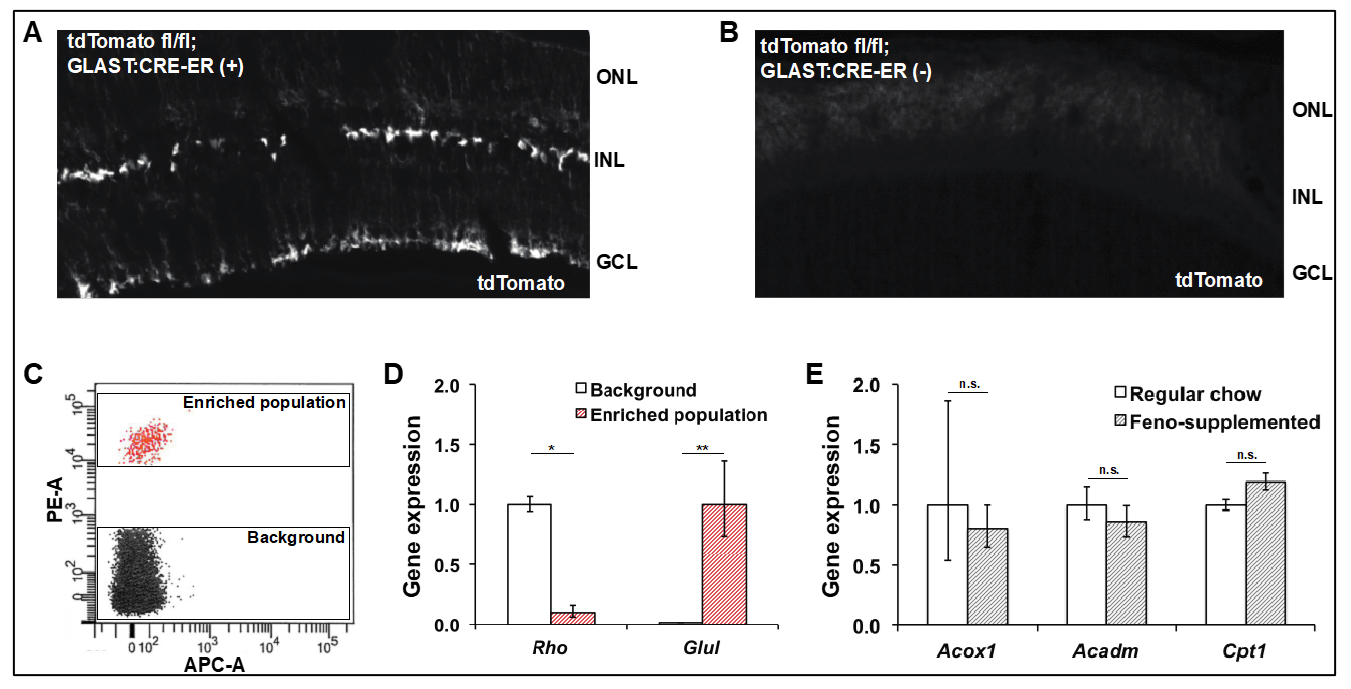
2.7. Isolation and Gene Expression Analysis of Müller Glia
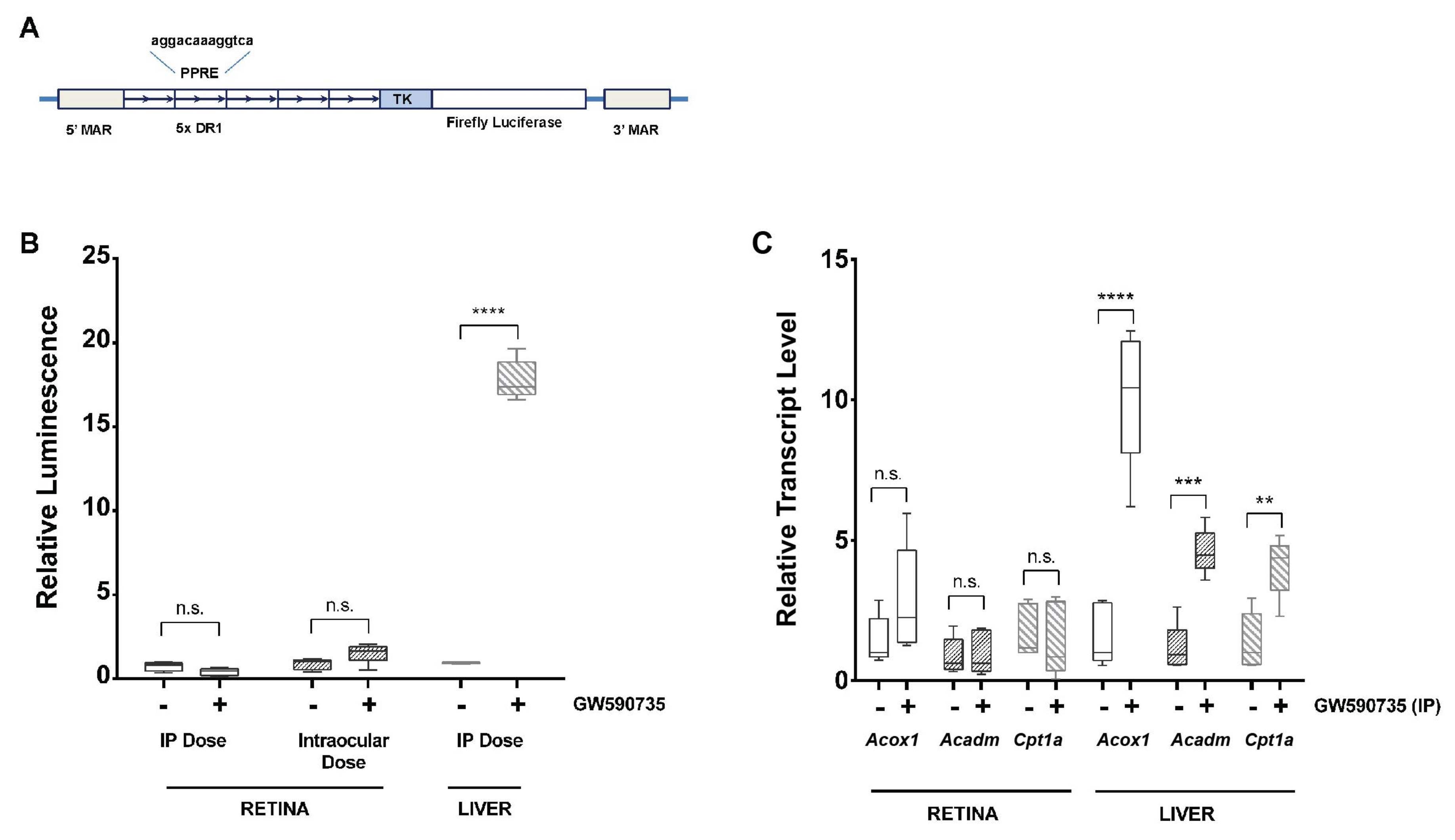
2.8. Luciferase Assay
2.9. Statistical Analyses
3. Results
3.1. Oral Fenofibrate Alters Lipid Metabolism in a Mouse Model of Type 2 Diabetes
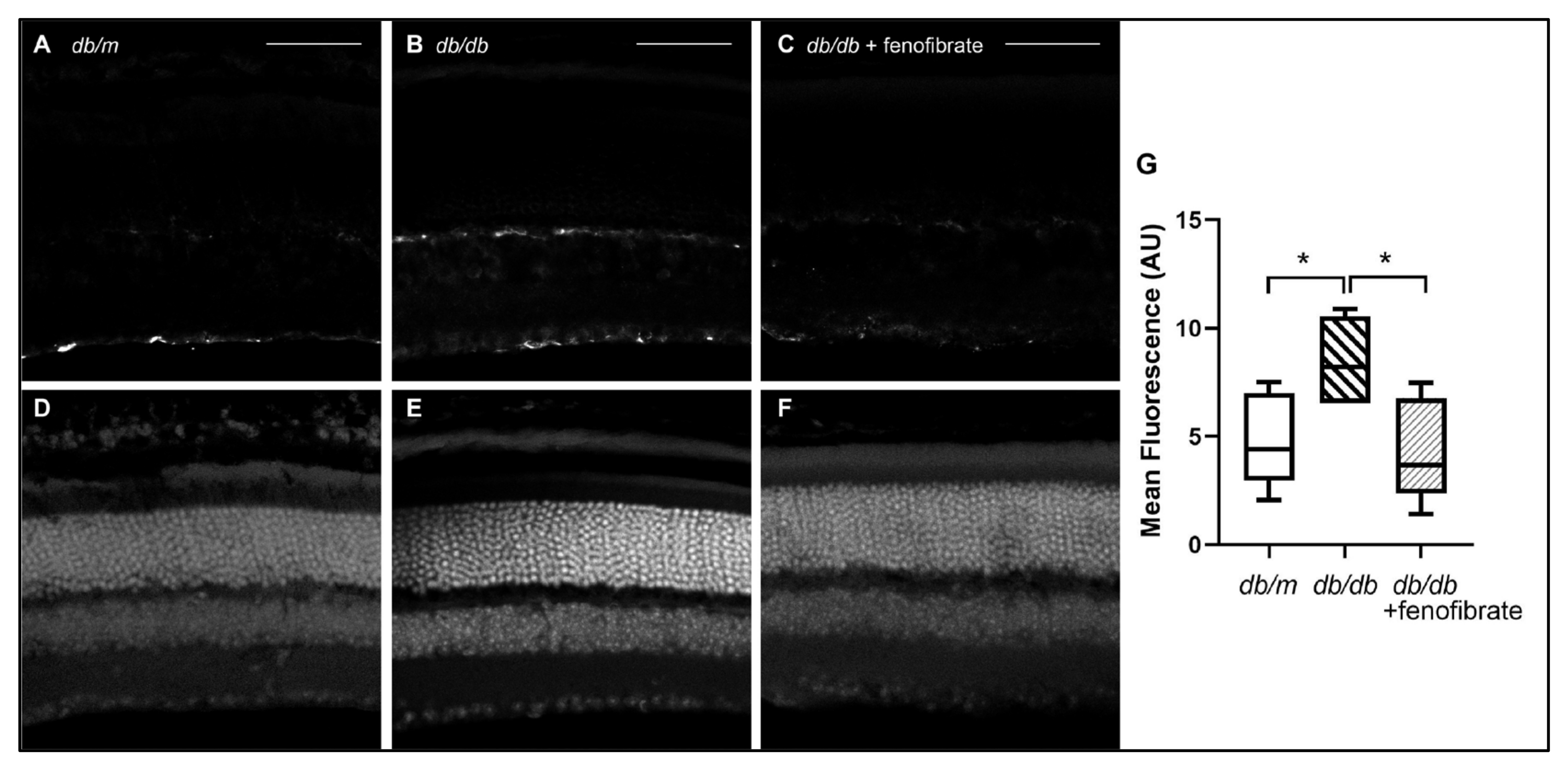
3.2. Diabetes-Associated Reactive Retinal Gliosis Is Reduced by Oral Fenofibrate
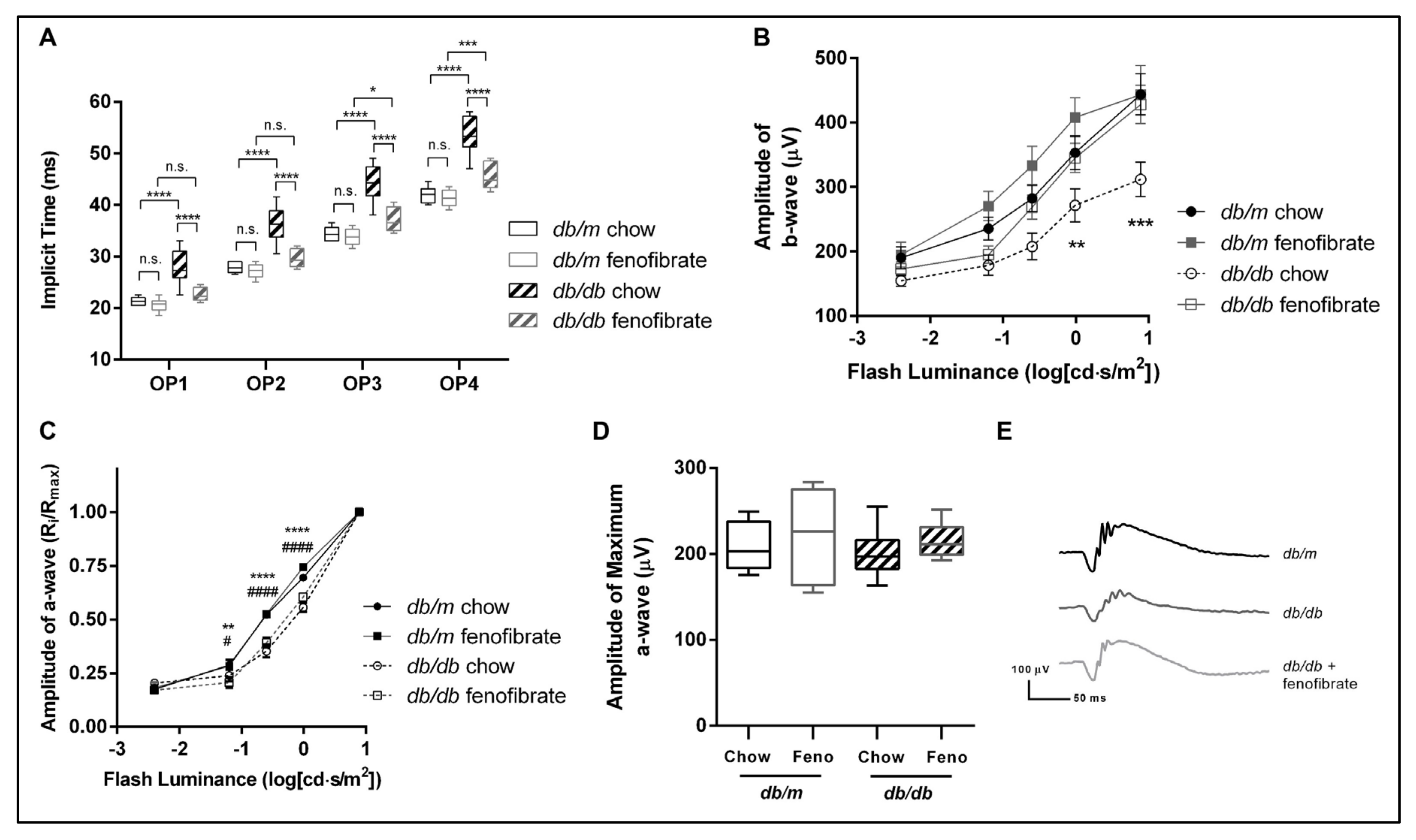
3.3. Oral Fenofibrate Attenuates Diabetes-Related Changes in Electroretinography
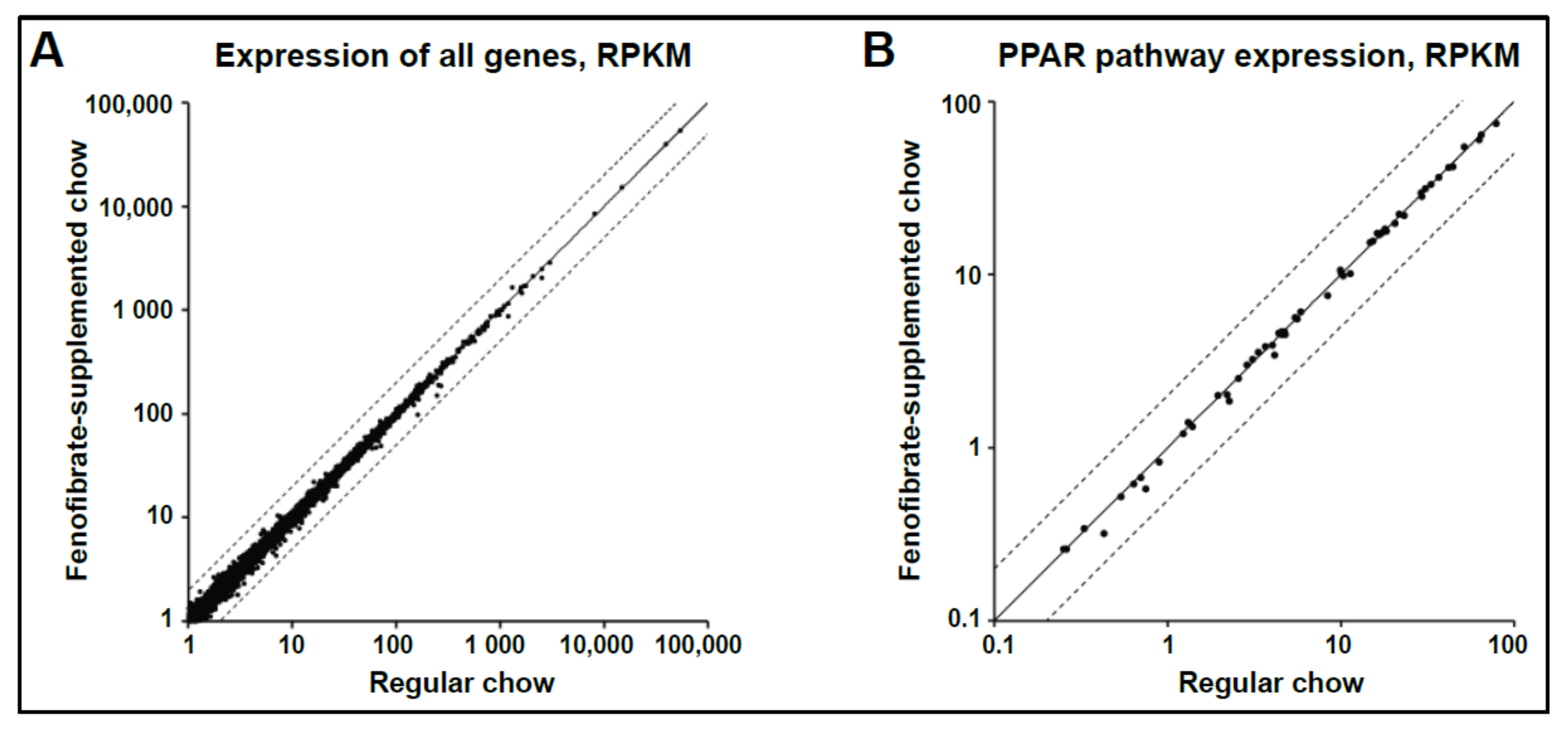
3.4. Oral Fenofibrate Does Not Alter Expression of PPARα Targets in Whole Retina of Non-Diabetic Mice
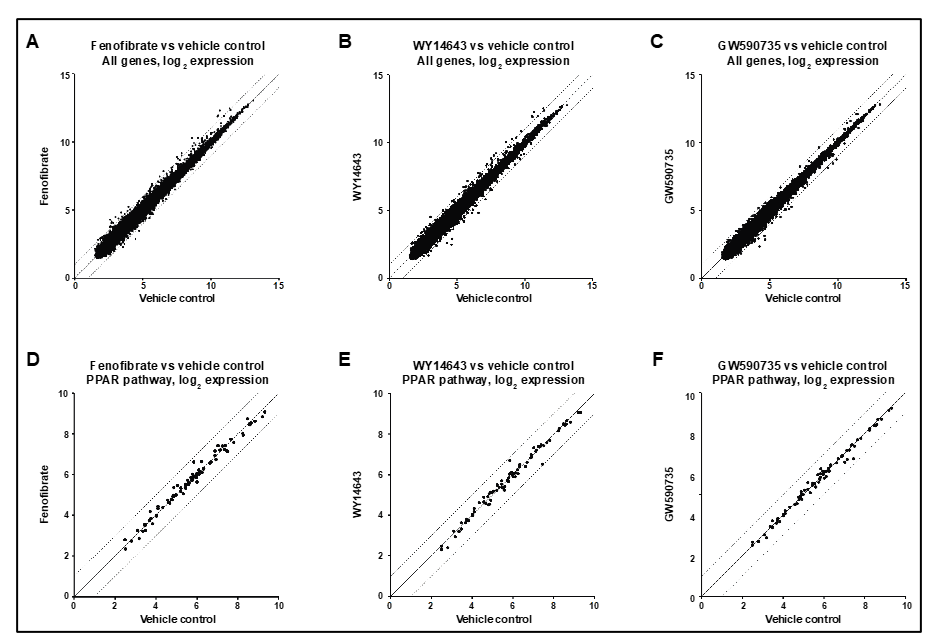
3.5. Intravitreous Fenofibrate Does Not Alter Expression of PPARα Targets in Whole Retina of Non-Diabetic Mice
3.6. Expression of PPARα Targets Is Unchanged in Müller Glia Isolated from Mice Treated with Oral Fenofibrate
3.7. Systemic or Local Delivery of a PPARα Agonist Activates a PPRE-Luciferase Reporter in Liver but Not Retina
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, R.; Wong, T.Y.; Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. 2015, 2, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Zapata, M.J.; Martí-Carvajal, A.J.; Solà, I.; Pijoán, J.; Buil-Calvo, J. Anti-vascular endothelial growth factor for proliferative diabetic retinopathy. Cochrane Database Syst. Rev. 2014, 2014, CD008721. [Google Scholar] [CrossRef] [PubMed]
- Virgili, G.; Parravano, M.; Evans, J.R.; Gordon, I.; Lucenteforte, E. Anti-vascular endothelial growth factor for diabetic macular oedema: A network meta-analysis. Cochrane Database Syst. Rev. 2018, 10, CD007419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.R.; Michelessi, M.; Virgili, G. Laser photocoagulation for proliferative diabetic retinopathy. Cochrane Database Syst. Rev. 2014, 2014, CD011234. [Google Scholar] [CrossRef] [PubMed]
- ACCORD Study Group; ACCORD Eye Study Group; Chew, E.Y.; Ambrosius, W.T.; Davis, M.D.; Danis, R.P.; Gangaputra, S.; Greven, C.M.; Hubbard, L.; Esser, B.A.; et al. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010, 363, 233–244. [Google Scholar] [PubMed] [Green Version]
- Keech, A.; Mitchell, P.; Summanen, P.; O’Day, J.; Davis, T.M.; Moffitt, M.S.; Taskinen, M.R.; Simes, R.J.; Tse, D.; Williamson, E.; et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): A randomised controlled trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef]
- Srinivasan, S.; Hande, P.; Shetty, J.; Murali, S. Efficiency of fenofibrate in facilitating the reduction of central macular thickness in diabetic macular edema. Indian J. Ophthalmol. 2018, 66, 98–105. [Google Scholar] [CrossRef]
- Pearsall, E.A.; Cheng, R.; Matsuzaki, S.; Zhou, K.; Ding, L.; Ahn, B.; Kinter, M.; Humphries, K.M.; Farjo, R.A.; Ma, J. Neuroprotective effects of PPARalpha in retinopathy of type 1 diabetes. PLoS ONE 2019, 14, e0208399. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Chen, J.; Dong, L.; He, X.; Cheng, R.; Zhou, K.; Liu, J.; Qiu, F.; Li, X.; Ma, J. A Protective Effect of PPARalpha in Endothelial Progenitor Cells Through Regulating Metabolism. Diabetes 2019, 68, 2131–2142. [Google Scholar] [CrossRef]
- Bogdanov, P.; Hernández, C.; Corraliza, L.; Carvalho, A.R.; Simó, R. Effect of fenofibrate on retinal neurodegeneration in an experimental model of type 2 diabetes. Acta Diabetol. 2015, 52, 113–122. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, Y.; Ding, L.; He, X.; Takahashi, Y.; Gao, Y.; Shen, W.; Cheng, R.; Chen, Q.; Qi, X.; et al. Pathogenic role of diabetes-induced PPAR-α down-regulation in microvascular dysfunction. Proc. Natl. Acad. Sci. USA 2013, 110, 15401–15406. [Google Scholar] [CrossRef] [Green Version]
- Pearsall, E.A.; Cheng, R.; Zhou, K.; Takahashi, Y.; Matlock, H.G.; Vadvalkar, S.S.; Shin, Y.; Fredrick, T.W.; Gantner, M.L.; Meng, S.; et al. PPARα is essential for retinal lipid metabolism and neuronal survival. BMC Biol. 2017, 15, 113. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Cheng, R.; Hu, Y.; Takahashi, Y.; Jenkins, A.J.; Keech, A.C.; Humphries, K.M.; Gu, X.; Elliott, M.H.; Xia, X.; et al. Peroxisome Proliferator-Activated Receptor α Protects Capillary Pericytes in the Retina. Am. J. Pathol. 2014, 184, 2709–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, E.; Ding, L.; Wang, Z.; Cheng, R.; Chen, Q.; Moore, R.; Takahashi, Y.; Ma, J. Protective and antioxidant effects of PPARalpha in the ischemic retina. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4568–4576. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Lin, M.; Jenkins, A.J.; Keech, C.A.; Mott, R.; Lyons, T.J.; Ma, J. Therapeutic effects of PPARalpha agonists on diabetic retinopathy in type 1 diabetes models. Diabetes 2013, 62, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Puro, D.G. Diabetes-induced dysfunction of the glutamate transporter in retinal Muller cells. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3109–3116. [Google Scholar]
- Liu, Q.; Zhang, F.; Zhang, X.; Cheng, R.; Ma, J.; Yi, J.; Li, J.M. Fenofibrate ameliorates diabetic retinopathy by modulating Nrf2 signaling and NLRP3 inflammasome activation. Mol. Cell. Biochem. 2018, 445, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Muller cell changes in human diabetic retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef]
- Zong, H.; Ward, M.; Madden, A.; Yong, P.H.; Limb, G.A.; Curtis, T.M.; Stitt, A.W. Hyperglycaemia-induced pro-inflammatory responses by retinal Müller glia are regulated by the receptor for advanced glycation end-products (RAGE). Diabetologia 2010, 53, 2656–2666. [Google Scholar] [CrossRef] [Green Version]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Tanaka, K.; Buffo, A.; Wurst, W.; Kühn, R.; Götz, M. Inducible gene deletion in astroglia and radial glia—A valuable tool for functional and lineage analysis. Glia 2006, 54, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Charlat, O.; Tartaglia, L.; Woolf, E.; Weng, X.; Ellis, S.J.; Lakey, N.D.; Culpepper, J.; More, K.J.; Breitbart, R.; et al. Evidence That the Diabetes Gene Encodes the Leptin Receptor: Identification of a Mutation in the Leptin Receptor Gene in db/db Mice. Cell 1996, 84, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Biserni, A.; Giannessi, F.; Sciarroni, A.F.; Milazzo, F.M.; Maggi, A.; Ciana, P. In Vivo Imaging Reveals Selective Peroxisome Proliferator Activated Receptor Modulator Activity of the Synthetic Ligand 3-(1-(4-Chlorobenzyl)-3-t-butylthio-5-isopropylindol-2-yl)-2,2-dimethylpropanoic acid (MK-886). Mol. Pharmacol. 2008, 73, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Ciana, P.; Di Luccio, G.; Belcredito, S.; Polllio, G.; Vegeto, E.; Tatangelo, L.; Tiveron, C.; Maggi, A. Engineering of a mouse for the in vivo profiling of estrogen receptor activity. Mol. Endocrinol. 2001, 15, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; Grefhorst, A.; Van Dijk, T.H.; Havinga, R.; Staels, B.; Kuipers, F.; Groen, A.K.; Reijngoud, D.-J. Fenofibrate Simultaneously Induces Hepatic Fatty Acid Oxidation, Synthesis, and Elongation in Mice. J. Biol. Chem. 2009, 284, 34036–34044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopal, R.; Bligard, G.W.; Zhang, S.; Yin, L.; Lukasiewicz, P.D.; Semenkovich, C.F. Functional Deficits Precede Structural Lesions in Mice with High-Fat Diet–Induced Diabetic Retinopathy. Diabetes 2016, 65, 1072–1084. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Rajaratnam, V.S.; Kai, S.E.; Penn, J.S. Expression of Peroxisome Proliferator Activated Receptors (PPAR) Alpha, Beta and Gamma and Their Angiostatic Effects in a Rat Model of Retinopathy of Prematurity. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3717. [Google Scholar]
- Enright, J.M.; Lawrence, K.; Hadžić, T.; Corbo, J.C. Transcriptome profiling of developing photoreceptor subtypes reveals candidate genes involved in avian photoreceptor diversification. J. Comp. Neurol. 2015, 523, 649–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Forte, T.M.; Taniguchi, S.; Ishida, B.Y.; Oka, K.; Chan, L. The db/db mouse, a model for diabetic dyslipidemia: Molecular characterization and effects of western diet feeding. Metabolism 2000, 49, 22–31. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, X.; Cheng, R.; Ma, J.; Yi, J.; Li, J. Salutary effect of fenofibrate on type 1 diabetic retinopathy via inhibiting oxidative stress-mediated Wnt/beta-catenin pathway activation. Cell Tissue Res. 2019, 376, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zou, C.; Zhao, S.; Wang, Y.; Han, C.; Zheng, Z. Fenofibrate Exerts Protective Effects in Diabetic Retinopathy via Inhibition of the ANGPTL3 Pathway. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4210–4217. [Google Scholar] [CrossRef]
- Tzekov, R.; Arden, G.B. The Electroretinogram in Diabetic Retinopathy. Surv. Ophthalmol. 1999, 44, 53–60. [Google Scholar] [CrossRef]
- Tomita, Y.; Ozawa, N.; Miwa, Y.; Ishida, A.; Ohta, M.; Tsubota, K.; Kurihara, T. Pemafibrate Prevents Retinal Pathological Neovascularization by Increasing FGF21 Level in a Murine Oxygen-Induced Retinopathy Model. Int. J. Mol. Sci. 2019, 20, 5878. [Google Scholar] [CrossRef] [Green Version]
- Ju, H.-B.; Zhang, F.-X.; Wang, S.; Song, J.; Cui, T.; Li, L.-F.; Zhang, H.-Y. Effects of fenofibrate on inflammatory cytokines in diabetic retinopathy patients. Medicine 2017, 96, e7671. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, J.; Guo, W.; Li, F.; Sun, W.; Chen, J.; Zhang, C.; Lu, X.; Tan, Y.; Feng, W.; et al. Up-regulation of Nrf2 is involved in FGF21-mediated fenofibrate protection against type 1 diabetic nephropathy. Free. Radic. Biol. Med. 2016, 93, 94–109. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| Mm_rho_f1 | CTCTGCCAGCTTTCTTTGCT |
| Mm_rho_r1 | GTCGTCATCTCCCAGTGGAT |
| Mm_GS_f1 | CTGCCATACCAACTTCAGCA |
| Mm_GS_r1 | TGTGGTACTGGTGCCTCTTG |
| Mm_Cpt1a_f1 | TGGGAGAGAATTTCATCCACTT |
| Mm_Cpt1a_r1 | TCCATCATGGCTTGTCTCAA |
| Mm_Acox1_f1 | GATGTGACCCTTGGCTCTGT |
| Mm_Acox1_r1 | GACTGCAGGGGCTTCAAGT |
| Mm_Acadm_f1 | AGCTCTAGACGAAGCCACGA |
| Mm_Acadm_r1 | GCGAGCAGAAATGAAACTCC |
| Mm_GAPDH_f1 | TGCACCACCAACTGCTTAGC |
| Mm_GAPDH_r1 | GGCATGGACTGTGGTCATGAG |
| Mm_L32_f1 | TTCCTGGTCCACAATGTCAA |
| Mm_L32_r1 | GGCTTTTCGGTTCTTAGAGGA |
| Non-Diabetic db/m | Diabetic db/db | |||
|---|---|---|---|---|
| Vehicle | Fenofibrate | Vehicle | Fenofibrate | |
| n | 10 | 8 | 10 | 10 |
| Body Weight, g, 3 months (at randomization) | 23.0 (1.2) | 24.8 (1.4) | 49.7 (1.1) **** | 51.2 (2.1) **** |
| Body Weight, g, 6 months (at assay) | 28.3 (1.3) | 25.5 (0.7) | 57.0 (2.4) **** | 62.6 (0.9) **** |
| Plasma Glucose (mg/dL) 3 months | 121.0 (6.8) | 121.8 (8.8) | 421.4 (29.9) **** | 445.3 (28.1) **** |
| Plasma Glucose (mg/dL) 6 months | 208.8 (9.1) | 179.5 (8.1) | 536.2 (33.8) **** | 540.1 (32.9) **** |
| Plasma Triglycerides (mg/dL) 6 months | 46.0 (1.4) | 55.3 (5.1) | 71.7 (4.5) **** | 49.2 (2.2) #### |
| Plasma Free Fatty Acids (mM) 6 months | 0.81 (0.04) | 1.06 (0.09) | 1.46 (0.04) **** | 1.34 (0.07) * |
| Plasma Cholesterol (mg/dL) 6 months | 49.6 (1.6) | 84.2 (5.7) ## | 103.2 (5.9) **** | 127.6 (9.0) ***, # |
| Non-Diabetic db/m | Diabetic db/db | |||
|---|---|---|---|---|
| Vehicle | Fenofibrate | Vehicle | Fenofibrate | |
| n | 3 | 3 | 3 | 3 |
| Liver Cholesterol Content (mg/dL/g tissue) 6 months | 0.158 (0.016) | 0.15 (0.005) | 0.238 (0.015) * | 0.255 (0.017) ** |
| Retinal Cholesterol Content (mg/dL/g tissue) 6 months | 0.123 (0.004) | 0.145 (0.013) | 0.137 (0.006) | 0.127 (0.012) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enright, J.M.; Zhang, S.; Thebeau, C.; Siebert, E.; Jin, A.; Gadiraju, V.; Zhang, X.; Chen, S.; Semenkovich, C.F.; Rajagopal, R. Fenofibrate Reduces the Severity of Neuroretinopathy in a Type 2 Model of Diabetes without Inducing Peroxisome Proliferator-Activated Receptor Alpha-Dependent Retinal Gene Expression. J. Clin. Med. 2021, 10, 126. https://doi.org/10.3390/jcm10010126
Enright JM, Zhang S, Thebeau C, Siebert E, Jin A, Gadiraju V, Zhang X, Chen S, Semenkovich CF, Rajagopal R. Fenofibrate Reduces the Severity of Neuroretinopathy in a Type 2 Model of Diabetes without Inducing Peroxisome Proliferator-Activated Receptor Alpha-Dependent Retinal Gene Expression. Journal of Clinical Medicine. 2021; 10(1):126. https://doi.org/10.3390/jcm10010126
Chicago/Turabian StyleEnright, Jennifer M., Sheng Zhang, Christina Thebeau, Emily Siebert, Alexander Jin, Veda Gadiraju, Xiaodong Zhang, Shiming Chen, Clay F. Semenkovich, and Rithwick Rajagopal. 2021. "Fenofibrate Reduces the Severity of Neuroretinopathy in a Type 2 Model of Diabetes without Inducing Peroxisome Proliferator-Activated Receptor Alpha-Dependent Retinal Gene Expression" Journal of Clinical Medicine 10, no. 1: 126. https://doi.org/10.3390/jcm10010126
APA StyleEnright, J. M., Zhang, S., Thebeau, C., Siebert, E., Jin, A., Gadiraju, V., Zhang, X., Chen, S., Semenkovich, C. F., & Rajagopal, R. (2021). Fenofibrate Reduces the Severity of Neuroretinopathy in a Type 2 Model of Diabetes without Inducing Peroxisome Proliferator-Activated Receptor Alpha-Dependent Retinal Gene Expression. Journal of Clinical Medicine, 10(1), 126. https://doi.org/10.3390/jcm10010126