The Relative Role of Toxins A and B in the Virulence of Clotridioides difficile

 ,
,
Abstract
:1. Introduction
2. Experimental Section
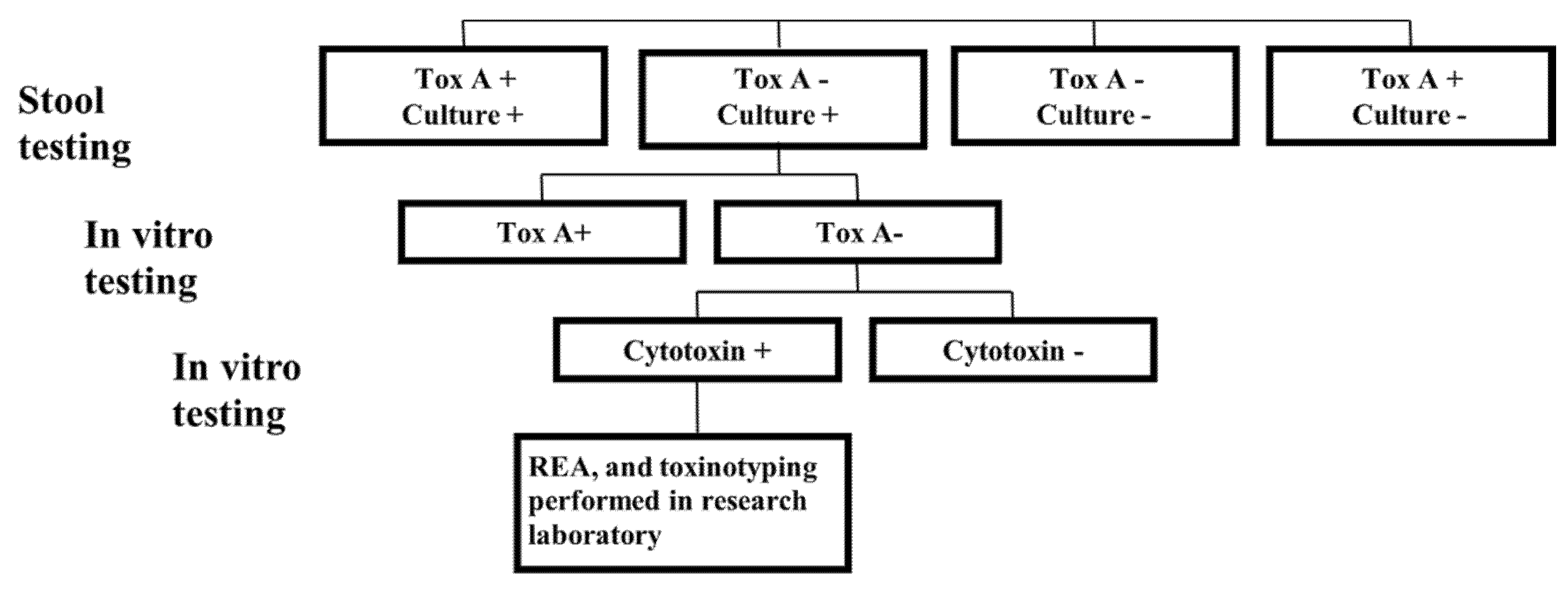
2.1. Clinical Microbiology Laboratory Testing
2.1.1. Culture
2.1.2. In Vitro Testing of Recovered C. difficile Isolates for TcdA and Cytotoxicity
2.2. Research Laboratory Testing
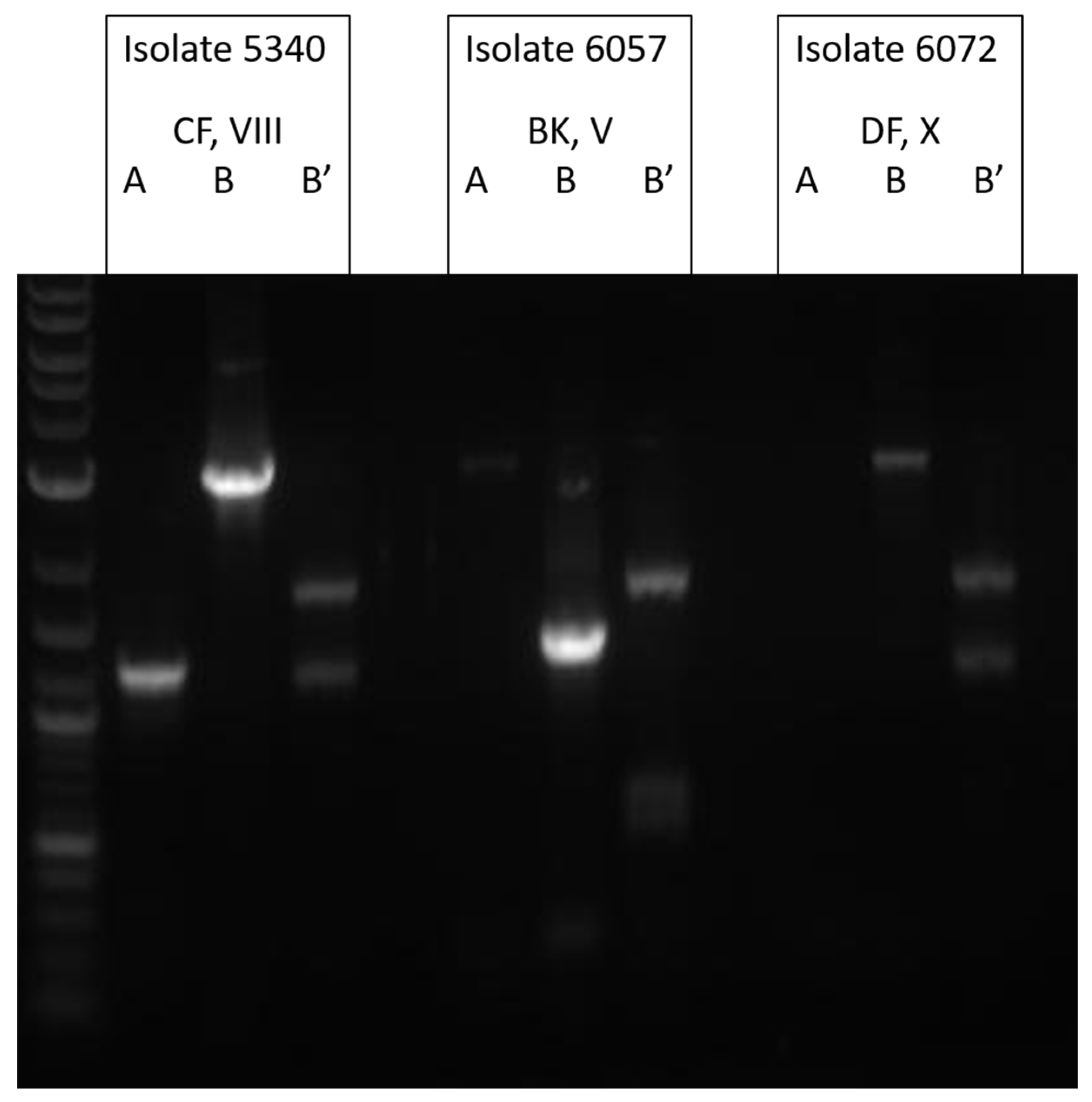
2.2.1. Restriction Endonuclease Analysis (REA) Typing
2.2.2. Toxinotyping
2.3. Clinical Data
3. Results
3.1. Surveillance by TcdA Immunoassay
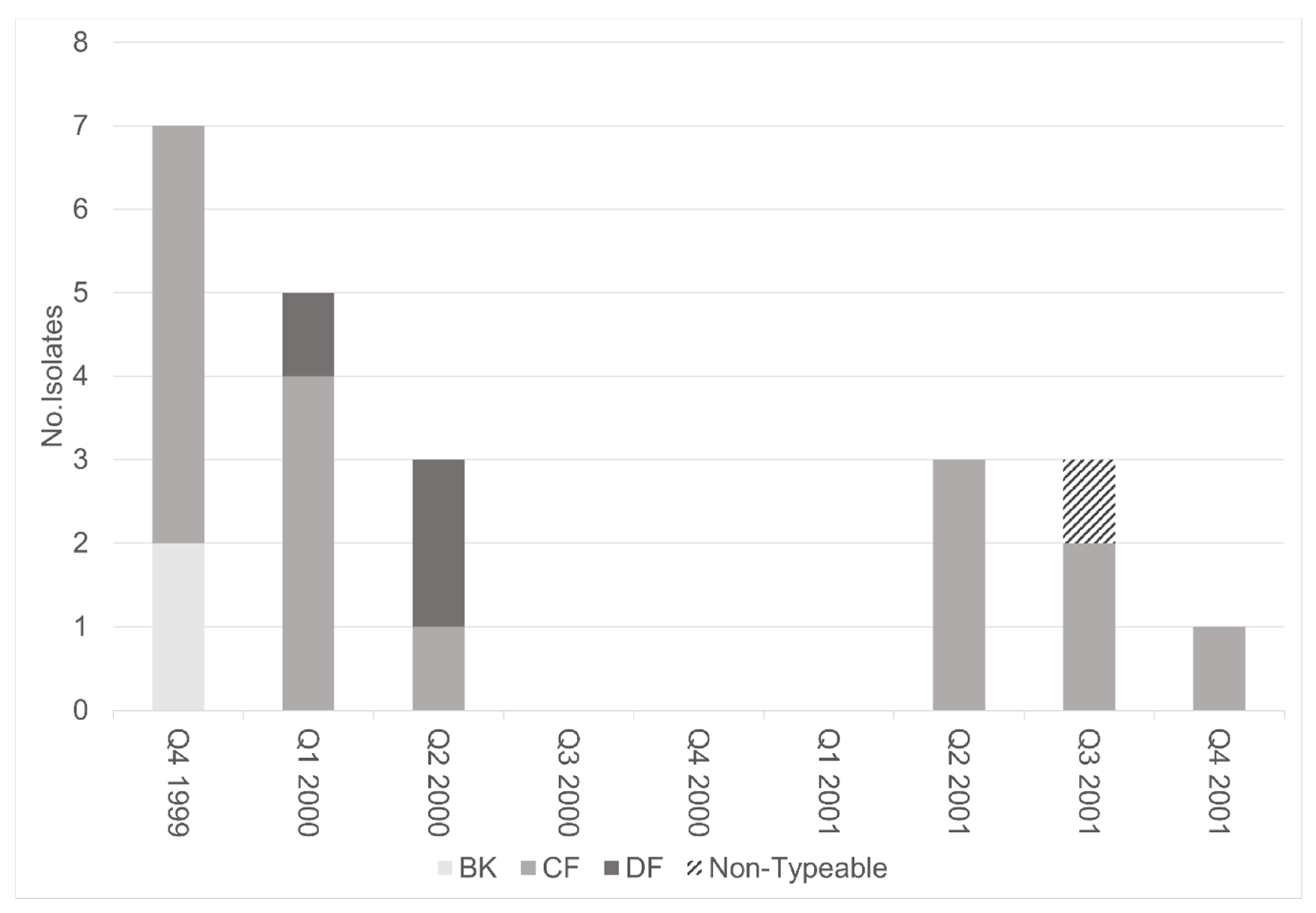
3.2. Molecular Analysis of TcdA−Negative, B-Positive Variant Isolates
3.3. Clinical Outcomes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Just, I.; Fritz, G.; Aktories, K.; Giry, M.; Popoff, M.R.; Boquet, P.; Hegenbarth, S.; von Eichel-Streiber, C. Clostridium Difficile Toxin B Acts on the GTP-Binding Protein Rho. J. Biol. Chem. 1994, 269, 10706–10712. [Google Scholar] [PubMed]
- Hundsberger, T.; Braun, V.; Weidmann, M.; Leukel, P.; Sauerborn, M.; von Eichel-Streiber, C. Transcription Analysis of the Genes TcdA-E of the Pathogenicity Locus of Clostridium Difficile. Eur. J. Biochem. 1997, 244, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Hecht, G.; Pothoulakis, C.; LaMont, J.T.; Madara, J.L. Clostridium Difficile Toxin A Perturbs Cytoskeletal Structure and Tight Junction Permeability of Cultured Human Intestinal Epithelial Monolayers. J. Clin. Investig. 1988, 82, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.A.; Johnson, J.L. The Toxigenic Element of Clostridium Difficile Strain VPI 10463. Microb. Pathog. 1995, 19, 203–213. [Google Scholar] [CrossRef]
- McDonald, L.C.; Gerding, D.N.; Johnson, S.; Bakken, J.S.; Carroll, K.C.; Coffin, S.E.; Dubberke, E.R.; Garey, K.W.; Gould, C.V.; Kelly, C.; et al. Clinical Practice Guidelines for Clostridium Difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. 2018, 66, e1–e48. [Google Scholar] [CrossRef]
- Musher, D.M.; Manhas, A.; Jain, P.; Nuila, F.; Waqar, A.; Logan, N.; Marino, B.; Graviss, E.A. Detection of Clostridium Difficile Toxin: Comparison of Enzyme Immunoassay Results with Results Obtained by Cytotoxicity Assay. J. Clin. Microbiol. 2007, 45, 2737–2739. [Google Scholar] [CrossRef] [Green Version]
- Rupnik, M.; Avesani, V.; Janc, M.; von Eichel-Streiber, C.; Delmée, M. A Novel Toxinotyping Scheme and Correlation of Toxinotypes with Serogroups of Clostridium DifficileIsolates. J. Clin. Microbiol. 1998, 36, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Borek, A.; Kini, A.; Kelly, P.; Wunderlickh-Weiorka, L.; Peterson, L. Prospective Comparison of Clearview C. DIFF A (CDA) with Meridian Immunocard (IMM), Stool Culture (StC) and Cytotoxin Detection (CyD) for Diagnosis of Clostridium Difficile Associated Diarrhea (CDAD). In Proceedings of the Ninety-Eighth Annual Meeting of the American Society for Microbiology, Atlanta, GA, USA, 17–21 May 1998. [Google Scholar]
- Lyerly, D.M.; Neville, L.M.; Evans, D.T.; Fill, J.; Allen, S.; Greene, W.; Sautter, R.; Hnatuck, P.; Torpey, D.J.; Schwalbe, R. Multicenter Evaluation of the Clostridium DifficileTOX A/B TEST. J. Clin. Microbiol. 1998, 36, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Lyerly, D.M.; Lockwood, D.E.; Richardson, S.H.; Wilkins, T.D. Biological Activities of Toxins A and B of Clostridium Difficile. Infect. Immun. 1982. [Google Scholar] [CrossRef] [Green Version]
- Lyras, D.; O’Connor, J.R.; Howarth, P.M.; Sambol, S.P.; Carter, G.P.; Phumoonna, T.; Poon, R.; Adams, V.; Vedantam, G.; Johnson, S.; et al. Toxin B Is Essential for Virulence of Clostridium Difficile. Nature 2009, 458, 1176–1179. [Google Scholar] [CrossRef]
- Kuehne, S.A.; Cartman, S.T.; Heap, J.T.; Kelly, M.L.; Cockayne, A.; Minton, N.P. The Role of Toxin A and Toxin B in Clostridium Difficile Infection. Nature 2010, 467, 711–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambol, S.P.; Merrigan, M.M.; Lyerly, D.; Gerding, D.N.; Johnson, S. Toxin Gene Analysis of a Variant Strain of Clostridium Difficile That Causes Human Clinical Disease. Infect. Immun. 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Eichel-Streiber, C. A Nonsense Mutation Abrogates Production of a Functional Enterotoxin A in Clostridium Difficile Toxinotype VIII Strains of Serogroups F and X. FEMS Microbiol. Lett. 1999, 178, 163–168. [Google Scholar] [CrossRef]
- Kato, H.; Kato, N.; Watanabe, K.; Iwai, N.; Nakamura, H.; Yamamoto, T.; Suzuki, K.; Kim, S.-M.; Chong, Y.; Wasito, E.B. Identification of Toxin A-Negative, Toxin B-Positive Clostridium Difficile by PCR. J. Clin. Microbiol. 1998, 36, 2178–2182. [Google Scholar] [CrossRef] [Green Version]
- Barbut, F.; Lalande, V.; Burghoffer, B.; Thien, H.V.; Grimprel, E.; Petit, J.-C. Prevalence and Genetic Characterization of Toxin A Variant Strains of Clostridium Difficile among Adults and Children with Diarrhea in France. J. Clin. Microbiol. 2002, 40, 2079–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limaye, A.P.; Turgeon, D.K.; Cookson, B.T.; Fritsche, T.R. Pseudomembranous Colitis Caused by a Toxin A(-) B(+) Strain of Clostridium Difficile. J. Clin. Microbiol. 2000, 38, 1696–1697. [Google Scholar] [CrossRef] [Green Version]
- Rupnik, M.; Kato, N.; Grabnar, M.; Kato, H. New Types of Toxin A-Negative, Toxin B-Positive Strains among Clostridium Difficile Isolates from Asia. J. Clin. Microbiol. 2003, 41, 1118–1125. [Google Scholar] [CrossRef] [Green Version]
- Goorhuis, A.; Legaria, M.C.; van den Berg, R.J.; Harmanus, C.; Klaassen, C.H.W.; Brazier, J.S.; Lumelsky, G.; Kuijper, E.J. Application of Multiple-Locus Variable-Number Tandem-Repeat Analysis to Determine Clonal Spread of Toxin A-Negative Clostridium Difficile in a General Hospital in Buenos Aires, Argentina. Clin. Microbiol. Infect. 2009, 15, 1080–1086. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Riley, T.V.; Kim, M.; Kim, C.K.; Yong, D.; Lee, K.; Chong, Y.; Park, J.-W. Increasing Prevalence of Toxin A-Negative, Toxin B-Positive Isolates of Clostridium Difficile in Korea: Impact on Laboratory Diagnosis. J. Clin. Microbiol. 2008, 46, 1116–1117. [Google Scholar] [CrossRef] [Green Version]
- Alfa, M.J.; Kabani, A.; Lyerly, D.; Moncrief, S.; Neville, L.M.; Al-Barrak, A.; Harding, G.K.H.; Dyck, B.; Olekson, K.; Embil, J.M. Characterization of a Toxin A-Negative, Toxin B-Positive Strain of Clostridium Difficile Responsible for a Nosocomial Outbreak of Clostridium Difficile-Associated Diarrhea. J. Clin. Microbiol. 2000, 38, 2706–2714. [Google Scholar] [CrossRef] [Green Version]
- Kuijper, E.; Weerdt, J.; Kato, H.; Kato, N.; Dam, A.; Vorm, E.; Weel, J.; Rheenen, C.; Dankert, J. Nosocomial Outbreak of Clostridium Difficile-Associated Diarrhoea Due to a Clindamycin-Resistant Enterotoxin A-Negative Strain. Eur. J. Clin. Microbiol. Infect. Dis. 2001, 20, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Drudy, D.; Harnedy, N.; Fanning, S.; Hannan, M.; Kyne, L. Emergence and Control of Fluoroquinolone-Resistant, Toxin A–Negative, Toxin B–Positive Clostridium Difficile. Infect. Control Hosp. Epidemiol. 2007, 28, 932–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.; Kent, S.A.; O’Leary, K.J.; Merrigan, M.M.; Sambol, S.P.; Peterson, L.R.; Gerding, D.N. Fatal Pseudomembranous Colitis Associated with a Variant Clostridium Difficile Strain Not Detected by Toxin A Immunoassay. Ann. Intern. Med. 2001, 135, 434. [Google Scholar] [CrossRef] [PubMed]
- Clabots, C.R.; Johnson, S.; Bettin, K.M.; Mathie, P.A.; Mulligan, M.E.; Schaberg, D.R.; Peterson, L.R.; Gerding, D.N. Development of a Rapid and Efficient Restriction Endonuclease Analysis Typing System for Clostridium Difficile and Correlation with Other Typing Systems. J. Clin. Microbiol. 1993, 31, 1870–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzo, C.E.; Merrigan, M.M.; Johnson, S.; Gerding, D.N.; Riley, T.V.; Silva, J.; Brazier, J.S. International Typing Study of Clostridium Difficile. Anaerobe 2014, 28, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Kociolek, L.K.; Perdue, E.R.; Fawley, W.N.; Wilcox, M.H.; Gerding, D.N.; Johnson, S. Correlation between Restriction Endonuclease Analysis and PCR Ribotyping for the Identification of Clostridioides (Clostridium) Difficile Clinical Strains. Anaerobe 2018, 54, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.; Sambol, S.P.; Brazier, J.S.; Delmee, M.; Avesani, V.; Merrigan, M.M.; Gerding, D.N. International Typing Study of Toxin A-Negative, Toxin B-Positive Clostridium Difficile Variants. J. Clin. Microbiol. 2003, 41, 1543–1547. [Google Scholar] [CrossRef] [Green Version]
- Rupnik, M.; Brazier, J.S.; Duerden, B.I.; Grabnar, M.; Stubbs, S.L.J. Comparison of Toxinotyping and PCR Ribotyping of Clostridium Difficile Strains and Description of Novel Toxinotypes. Microbiology 2001, 147, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Tenover, F.C.; Åkerlund, T.; Gerding, D.N.; Goering, R.V.; Boström, T.; Jonsson, A.M.; Wong, E.; Wortman, A.T.; Persing, D.H. Comparison of Strain Typing Results for Clostridium Difficile Isolates from North America. J. Clin. Microbiol. 2011, 49, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, D.; Fawley, W.; Kachrimanidou, M.; Bowden, R.; Crook, D.W.; Fung, R.; Golubchik, T.; Harding, R.M.; Jeffery, K.J.M.; Jolley, K.A.; et al. Multilocus Sequence Typing of Clostridium Difficile. J. Clin. Microbiol. 2010, 48, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Imwattana, K.; Knight, D.R.; Kullin, B.; Collins, D.A.; Putsathit, P.; Kiratisin, P.; Riley, T.V. Clostridium Difficile Ribotype 017—Characterization, Evolution and Epidemiology of the Dominant Strain in Asia. Emerg. Microbes Infect. 2019, 8, 796–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, Y.; Pai, H. Clinical Characteristics and Treatment Outcomes of Clostridium Difficile Infections by PCR Ribotype 017 and 018 Strains. PLoS ONE 2016, 11, e0168849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Fang, H.; Weintraub, A.; Nord, C.E. Distinct Ribotypes and Rates of Antimicrobial Drug Resistance in Clostridium Difficile from Shanghai and Stockholm. Clin. Microbiol. Infect. 2009, 15, 1170–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Jeong, S.H.; Roh, K.H.; Hong, S.G.; Kim, J.W.; Shin, M.-G.; Kim, M.-N.; Shin, H.B.; Uh, Y.; Lee, H.; et al. Investigation of Toxin Gene Diversity, Molecular Epidemiology, and Antimicrobial Resistance of Clostridium Difficile Isolated from 12 Hospitals in South Korea. Ann. Lab. Med. 2010, 30, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Goorhuis, A.; Debast, S.B.; Dutilh, J.C.; van Kinschot, C.M.; Harmanus, C.; Cannegieter, S.C.; Hagen, E.C.; Kuijper, E.J. Type-Specific Risk Factors and Outcome in an Outbreak With 2 Different Clostridium Difficile Types Simultaneously in 1 Hospital. Clin. Infect. Dis. 2011, 53, 860–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, T.J.; Ketley, J.M.; Haslam, S.C.; Stephen, J.; Burdon, D.W.; Candy, D.C.; Daniel, R. Effect of Toxin A and B of Clostridium Difficile on Rabbit Ileum and Colon. Gut 1986, 27, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Chumbler, N.M.; Farrow, M.A.; Lapierre, L.A.; Franklin, J.L.; Lacy, D.B. Clostridium Difficile Toxins TcdA and TcdB Cause Colonic Tissue Damage by Distinct Mechanisms. Infect. Immun. 2016, 84, 2871–2877. [Google Scholar] [CrossRef] [Green Version]
- Carter, G.P.; Chakravorty, A.; Pham Nguyen, T.A.; Mileto, S.; Schreiber, F.; Li, L.; Howarth, P.; Clare, S.; Cunningham, B.; Sambol, S.P.; et al. Defining the Roles of TcdA and TcdB in Localized Gastrointestinal Disease, Systemic Organ Damage, and the Host Response during Clostridium Difficile Infections. MBio 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, M.H.; Gerding, D.N.; Poxton, I.R.; Kelly, C.; Nathan, R.; Birch, T.; Cornely, O.A.; Rahav, G.; Bouza, E.; Lee, C.; et al. Bezlotoxumab for Prevention of Recurrent Clostridium Difficile Infection. N. Engl. J. Med. 2017, 376, 305–317. [Google Scholar] [CrossRef]
- Gerding, D.N.; Kelly, C.P.; Rahav, G.; Lee, C.; Dubberke, E.R.; Kumar, P.N.; Yacyshyn, B.; Kao, D.; Eves, K.; Ellison, M.C.; et al. Bezlotoxumab for Prevention of Recurrent Clostridium Difficile Infection in Patients at Increased Risk for Recurrence. Clin. Infect. Dis. 2018, 67, 649–656. [Google Scholar] [CrossRef]
- Moncrief, J.S.; Lyerly, D.; Wilkins, T. Molecular biology of the clostridium difficile toxins. In The Clostridia: Molecular Biology and Pathogenesis; Rood, J.I., McClane, B.A., Songer, J.G., Titball, R.W., Eds.; Academic Press: London, UK, 1997; pp. 369–392. [Google Scholar]
- Voth, D.E.; Ballard, J.D. Clostridium Difficile Toxins: Mechanism of Action and Role in Disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambol, S.P.; Tang, J.K.; Merrigan, M.M.; Johnson, S.; Gerding, D.N. Infection of Hamsters with Epidemiologically Important Strains of Clostridium Difficile. J. Infect. Dis. 2001, 183, 1760–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goorhuis, A.; Bakker, D.; Corver, J.; Debast, S.B.; Harmanus, C.; Notermans, D.W.; Bergwerff, A.A.; Dekker, F.W.; Kuijper, E.J. Emergence of Clostridium Difficile Infection Due to a New Hypervirulent Strain, Polymerase Chain Reaction Ribotype 078. Clin. Infect. Dis. 2008, 47, 1162–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Pérez, S.; Blanco, J.L.; Harmanus, C.; Kuijper, E.; García, M.E. Subtyping and Antimicrobial Susceptibility of Clostridium Difficile PCR Ribotype 078/126 Isolates of Human and Animal Origin. Vet. Microbiol. 2017, 199, 15–22. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Testing Modality | All Stools (n = 10,668) | 95% Confidence Interval |
|---|---|---|
| TcdA EIA positive OR Culture positive | 1661 (15.6%) | 14.9–16.3% |
| Culture- or toxin A EIA-positive stools (n = 1661) | ||
| Culture positive | 1436 (86.5%) | 84.8–88.1% |
| TcdA EIA positive | 361 (25.1%) | 22.9–27.4% |
| TcdA EIA negative | 1075 (74.9%) | 72.6–77.1% |
| TcdA EIA positive | 586 (35.3%) | 32.9–37.6% |
| Culture negative | 225 (38.4%) | 34.5–42.3% |
| Recovered C. difficile isolates from culture-positive/EIA-negative stools (n = 1075) | ||
| In vitro TcdA positive | 786 (73.1%) | 70.4–75.8% |
| In vitro TcdA negative | 289 (26.9%) | 24.3–29.6% |
| In vitro cytotoxicity assay positive | 23 (8.0%) * | 5.1–11.7% |
| Median Age (Range) | 60 (37–86) |
|---|---|
| Female (n) | 47.6% (10/21) |
| HA-CDI (n) | 71.4% (15/21) |
| Prior Antibiotics (n) | 84.2% (16/19) |
| Diarrhea (n) | 95% (19/20) |
| Treated for CDI (n) | 36.8% (7/19) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skinner, A.M.; Phillips, S.T.; Merrigan, M.M.; O’Leary, K.J.; Sambol, S.P.; Siddiqui, F.; Peterson, L.R.; Gerding, D.N.; Johnson, S. The Relative Role of Toxins A and B in the Virulence of Clotridioides difficile. J. Clin. Med. 2021, 10, 96. https://doi.org/10.3390/jcm10010096
Skinner AM, Phillips ST, Merrigan MM, O’Leary KJ, Sambol SP, Siddiqui F, Peterson LR, Gerding DN, Johnson S. The Relative Role of Toxins A and B in the Virulence of Clotridioides difficile. Journal of Clinical Medicine. 2021; 10(1):96. https://doi.org/10.3390/jcm10010096
Chicago/Turabian StyleSkinner, Andrew M., S. Tyler Phillips, Michelle M. Merrigan, Kevin J. O’Leary, Susan P. Sambol, Farida Siddiqui, Lance R. Peterson, Dale N. Gerding, and Stuart Johnson. 2021. "The Relative Role of Toxins A and B in the Virulence of Clotridioides difficile" Journal of Clinical Medicine 10, no. 1: 96. https://doi.org/10.3390/jcm10010096
APA StyleSkinner, A. M., Phillips, S. T., Merrigan, M. M., O’Leary, K. J., Sambol, S. P., Siddiqui, F., Peterson, L. R., Gerding, D. N., & Johnson, S. (2021). The Relative Role of Toxins A and B in the Virulence of Clotridioides difficile. Journal of Clinical Medicine, 10(1), 96. https://doi.org/10.3390/jcm10010096