
Complex Interplay of Genes Underlies Invasiveness in Fibrosarcoma Progression Model


 , , , , , , , , , , ,
, , , , , , , , , , ,  ,
, 
Abstract
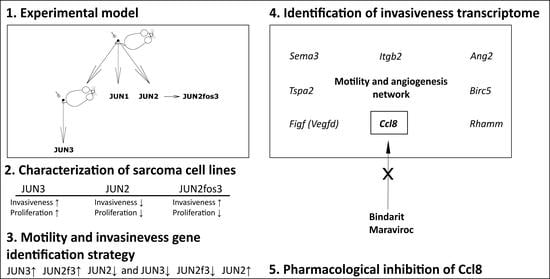
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Stable Transfection and Derivation of JUN-2fos-3 Cell Subline
2.3. Expression of Fos and Jun Genes in Fibrosarcoma Cell Lines
2.4. Indirect Immunofluorescence
2.5. Analysis of Cell Morphology
2.6. Evaluation of Growth Characteristics
2.7. Anchorage-Independent Growth
2.8. Sphere Formation Assay and Side Population Assay
2.9. Motility Assay
2.10. Assessment of Invasive Ability
2.11. Analysis of Cellular Energy Metabolism
2.12. Transcriptomic Profiling
2.13. Pharmacologic Inhibition of CCL8 Activity
2.14. Additional Statistical Analysis
3. Results
3.1. Extension of JUN-Sarcoma Progression Series for the JUN-2fos-3 Cell Line
3.2. Proliferation Characteristics
3.3. Sarcosphere Formation and Clonogenic Activity Is Not Associated with Apparent Side Population
3.4. JUN-2fos-3 and JUN-3 Cell Lines Are Highly Motile and Invasive
3.5. Invasive Cell Lines Have Different Metabolic Profiles
3.6. Distribution of Transformation Traits among JUN Fibrosarcoma Cell Lines Allows for the Straightforward Identification of Genes Potentially Responsible for Sarcoma Cell Proliferation and Motility/Invasiveness
3.7. CCL8 Represents a Druggable Target to Curtail Motility and Invasion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oda, Y.; Yamamoto, H.; Kohashi, K.; Yamada, Y.; Iura, K.; Ishii, T.; Maekawa, A.; Bekki, H. Soft Tissue Sarcomas: From a Morphological to a Molecular Biological Approach. Pathol. Internat. 2017, 67, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Quesada, J.; Amato, R. The Molecular Biology of Soft-Tissue Sarcomas and Current Trends in Therapy. Sarcoma 2012, 2012, 849456. [Google Scholar] [CrossRef] [Green Version]
- Sbaraglia, M.; Dei Tos, A.P. The Pathology of Soft Tissue Sarcomas. Radiol. Med. 2019, 124, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Skubitz, K.M.; D’Adamo, D.R. Sarcoma. Mayo Clin. Proc. 2007, 82, 1409–1432. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in Sarcoma Genomics and New Therapeutic Targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef] [Green Version]
- Riedel, R.F. Systemic Therapy for Advanced Soft Tissue Sarcomas. Cancer 2012, 118, 1474–1485. [Google Scholar] [CrossRef] [Green Version]
- Mertens, F.; Antonescu, C.R.; Mitelman, F. Gene Fusions in Soft Tissue Tumors: Recurrent and Overlapping Pathogenetic Themes. Genes Chromosomes Cancer 2016, 55, 291–310. [Google Scholar] [CrossRef] [Green Version]
- Hatina, J.; Kripnerova, M.; Houfkova, K.; Pesta, M.; Kuncova, J.; Sana, J.; Slaby, O.; Rodríguez, R. Sarcoma Stem Cell Heterogeneity. Adv. Exp. Med. Biol. 2019, 1123, 95–118. [Google Scholar] [CrossRef]
- Pennacchioli, E.; Tosti, G.; Barberis, M.; De Pas, T.M.; Verrecchia, F.; Menicanti, C.; Testori, A.; Mazzarol, G. Sarcoma Spreads Primarily through the Vascular System: Are There Biomarkers Associated with Vascular Spread? Clin. Exp. Metastasis 2012, 29, 757–773. [Google Scholar] [CrossRef]
- Tsukushi, S.; Nishida, Y.; Urakawa, H.; Kozawa, E.; Ishiguro, N. Prognostic Significance of Histological Invasion in High Grade Soft Tissue Sarcomas. SpringerPlus 2014, 3, 544. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.T.J.; Pollack, S.M.; Huang, P.; Jones, R.L. Phase III Soft Tissue Sarcoma Trials: Success or Failure? Curr. Treat. Options Oncol. 2017, 18, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chibon, F.; Lagarde, P.; Salas, S.; Pérot, G.; Brouste, V.; Tirode, F.; Lucchesi, C.; de Reynies, A.; Kauffmann, A.; Bui, B.; et al. Validated Prediction of Clinical Outcome in Sarcomas and Multiple Types of Cancer on the Basis of a Gene Expression Signature Related to Genome Complexity. Nat. Med. 2010, 16, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Jemaà, M.; Abdallah, S.; Lledo, G.; Perrot, G.; Lesluyes, T.; Teyssier, C.; Roux, P.; van Dijk, J.; Chibon, F.; Abrieu, A.; et al. Heterogeneity in Sarcoma Cell Lines Reveals Enhanced Motility of Tetraploid versus Diploid Cells. Oncotarget 2016, 8, 16669–16689. [Google Scholar] [CrossRef] [PubMed]
- Chibon, F.; Lesluyes, T.; Valentin, T.; Guellec, S.L. CINSARC Signature as a Prognostic Marker for Clinical Outcome in Sarcomas and Beyond. Genes Chromosomes Cancer 2019, 58, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Cavanna, T.; Pokorna, E.; Vesely, P.; Gray, C.; Zicha, D. Evidence for Protein 4.1B Acting as a Metastasis Suppressor. J. Cell Sci. 2007, 120, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosel, D.; Brabek, J.; Tolde, O.; Mierke, C.T.; Zitterbart, D.P.; Raupach, C.; Bicanova, K.; Kollmannsberger, P.; Pankova, D.; Vesely, P.; et al. Up-Regulation of Rho/ROCK Signaling in Sarcoma Cells Drives Invasion and Increased Generation of Protrusive Forces. Mol. Cancer Res. 2008, 6, 1410–1420. [Google Scholar] [CrossRef] [Green Version]
- Kainov, Y.; Favorskaya, I.; Delektorskaya, V.; Chemeris, G.; Komelkov, A.; Zhuravskaya, A.; Trukhanova, L.; Zueva, E.; Tavitian, B.; Dyakova, N.; et al. CRABP1 Provides High Malignancy of Transformed Mesenchymal Cells and Contributes to the Pathogenesis of Mesenchymal and Neuroendocrine Tumors. Cell Cycle 2014, 13, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Funes, J.M.; Quintero, M.; Henderson, S.; Martinez, D.; Qureshi, U.; Westwood, C.; Clements, M.O.; Bourboulia, D.; Pedley, R.B.; Moncada, S.; et al. Transformation of Human Mesenchymal Stem Cells Increases Their Dependency on Oxidative Phosphorylation for Energy Production. Proc. Natl. Acad. Sci. USA 2007, 104, 6223–6228. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, A.; Wang, C.; Schreiber, S.L. Perturbational Profiling of a Cell-Line Model of Tumorigenesis by Using Metabolic Measurements. Proc. Natl. Acad. Sci. USA 2005, 102, 5992–5997. [Google Scholar] [CrossRef] [Green Version]
- Hatina, J.; Hajkova, L.; Peychl, J.; Rudolf, E.; Finek, J.; Cervinka, M.; Reischig, J. Establishment and Characterization of Clonal Cell Lines Derived from a Fibrosarcoma of the H2-K/v-Jun Transgenic Mouse. Tumor Biol. 2003, 24, 176–184. [Google Scholar] [CrossRef]
- Schuh, A.C.; Keating, S.J.; Monteclaro, F.S.; Vogt, P.K.; Breitman, M.L. Obligatory Wounding Requirement for Tumorigenesis in V- Jun Transgenic Mice. Nature 1990, 346, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Takahashi, Y.; Hayashi, S.; Kondoh, H. Improved Mammalian Vectors for High Expression of G418 Resistance. Cell Struct. Funct. 1987, 12, 575–580. [Google Scholar] [CrossRef] [Green Version]
- Chomczynski, P.; Sacchi, N. The Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction: Twenty-Something Years On. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Ausubel, F.M.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidman, J.G.; Smith, J.A.; Struhl, K. Current Protocols in Molecular Biology; John Wiley & Sons: Brooklyn, NY, USA, 1995; ISBN 978-0-471-50338-5. [Google Scholar]
- Maki, Y.; Bos, T.J.; Davis, C.; Starbuck, M.; Vogt, P.K. Avian Sarcoma Virus 17 Carries the Jun Oncogene. Proc. Natl. Acad. Sci. USA 1987, 84, 2848–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holubova, M.; Leba, M.; Sedmikova, M.; Vannucci, L.; Horak, V. Characterization of Three Newly Established Rat Sarcoma Cell Clones. In Vitro Cell Dev. Biol. Anim. 2012, 48, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Ke, N.; Wang, X.; Xu, X.; Abassi, Y.A. The XCELLigence System for Real-Time and Label-Free Monitoring of Cell Viability. Methods Mol. Biol. 2011, 740, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-Independent Cell Growth Signature Identifies Tumors with Metastatic Potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Honoki, K.; Tsujiuchi, T.; Kido, A.; Yoshitani, K.; Takakura, Y. Sphere-Forming Stem-like Cell Populations with Drug Resistance in Human Sarcoma Cell Lines. Int. J. Oncol. 2009, 34, 1381–1386. [Google Scholar]
- Liu, W.-D.; Zhang, T.; Wang, C.-L.; Meng, H.-M.; Song, Y.-W.; Zhao, Z.; Li, Z.-M.; Liu, J.-K.; Pan, S.-H.; Wang, W.-B. Sphere-Forming Tumor Cells Possess Stem-like Properties in Human Fibrosarcoma Primary Tumors and Cell Lines. Oncol. Lett. 2012, 4, 1315–1320. [Google Scholar] [CrossRef]
- Boesch, M.; Reimer, D.; Rumpold, H.; Zeimet, A.G.; Sopper, S.; Wolf, D. DyeCycle Violet Used for Side Population Detection Is a Substrate of P-Glycoprotein. Cytometry A 2012, 81, 517–522. [Google Scholar] [CrossRef]
- Quail, D.F.; Maciel, T.J.; Rogers, K.; Postovit, L.M. A Unique 3D in Vitro Cellular Invasion Assay. J. Biomol. Screen. 2012, 17, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Santini, M.T.; Rainaldi, G.; Indovina, P.L. Multicellular Tumour Spheroids in Radiation Biology. Int. J. Radiat. Biol. 1999, 75, 787–799. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-Resolution Respirometry: OXPHOS Protocols for Human Cells and Permeabilized Fibers from Small Biopsies of Human Muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Strobl, D.; Ruttmann, E.; Königsrainer, A.; Margreiter, R.; Gnaiger, E. Evaluation of Mitochondrial Respiratory Function in Small Biopsies of Liver. Anal. Biochem. 2002, 305, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of Mitochondrial Content in Skeletal Muscle of Healthy Young Human Subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Chen, J.-R.; Lazarenko, O.P.; Blackburn, M.L.; Rose, S.; Frye, R.E.; Badger, T.M.; Andres, A.; Shankar, K. Maternal Obesity Programs Senescence Signaling and Glucose Metabolism in Osteo-Progenitors From Rat and Human. Endocrinology 2016, 157, 4172–4183. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Chan, D.K.; Haugrud, A.B.; Miskimins, W.K. Mechanisms by Which Low Glucose Enhances the Cytotoxicity of Metformin to Cancer Cells Both in Vitro and in Vivo. PLoS ONE 2014, 9, e108444. [Google Scholar] [CrossRef] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating High-Throughput Genomic Analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Mirolo, M.; Fabbri, M.; Sironi, M.; Vecchi, A.; Guglielmotti, A.; Mangano, G.; Biondi, G.; Locati, M.; Mantovani, A. Impact of the Anti-Inflammatory Agent Bindarit on the Chemokinome: Selective Inhibition of the Monocyte Chemotactic Proteins. Eur. Cytokine Netw. 2008, 19, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Paccosi, S.; Giachi, M.; Di Gennaro, P.; Guglielmotti, A.; Parenti, A. The Chemokine (C-C Motif) Ligand Protein Synthesis Inhibitor Bindarit Prevents Cytoskeletal Rearrangement and Contraction of Human Mesangial Cells. Cytokine 2016, 85, 92–100. [Google Scholar] [CrossRef]
- Halvorsen, E.C.; Hamilton, M.J.; Young, A.; Wadsworth, B.J.; LePard, N.E.; Lee, H.N.; Firmino, N.; Collier, J.L.; Bennewith, K.L. Maraviroc Decreases CCL8-Mediated Migration of CCR5 + Regulatory T Cells and Reduces Metastatic Tumor Growth in the Lungs. OncoImmunology 2016, 5, e1150398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicoli, D.; Jiao, X.; Ju, X.; Velasco-Velazquez, M.; Ertel, A.; Addya, S.; Li, Z.; Ando, S.; Fatatis, A.; Paudyal, B.; et al. CCR5 Receptor Antagonists Block Metastasis to Bone of V-Src Oncogene-Transformed Metastatic Prostate Cancer Cell Lines. Cancer Res. 2014, 74, 7103–7114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scrace, S.; O’Neill, E.; Hammond, E.M.; Pires, I.M. Use of the xCELLigence System for Real-Time Analysis of Changes in Cellular Motility and Adhesion in Physiological Conditions. In Adhesion Protein Protocols; Coutts, A.S., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; pp. 295–306. ISBN 978-1-62703-538-5. [Google Scholar]
- R Core Team (2020)—European Environment Agency. Available online: https://www.eea.europa.eu/data-and-maps/indicators/oxygen-consuming-substances-in-rivers/r-development-core-team-2006 (accessed on 9 November 2020).
- DiCiccio, T.J.; Efron, B. Bootstrap Confidence Intervals. Statist. Sci. 1996, 11, 189–228. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Luo, D.; Koolaard, S.G.J. Predictmeans: Calculate Predicted Means for Linear Models. Available online: https://cran.r-project.org/web/packages/predictmeans/predictmeans.pdf (accessed on 20 November 2020).
- Tichanek, F.; Salomova, M.; Jedlicka, J.; Kuncova, J.; Pitule, P.; Macanova, T.; Petrankova, Z.; Tuma, Z.; Cendelin, J. Hippocampal Mitochondrial Dysfunction and Psychiatric-Relevant Behavioral Deficits in Spinocerebellar Ataxia 1 Mouse Model. Sci. Rep. 2020, 10, 5418. [Google Scholar] [CrossRef] [PubMed]
- Cendelin, J.; Tichanek, F. Cerebellar Degeneration Averts Blindness-Induced Despaired Behavior during Spatial Task in Mice. Neurosci. Lett. 2020, 722, 134854. [Google Scholar] [CrossRef]
- Eklund, A. Beeswarm: The Bee Swarm Plot, an Alternative to Stripchart. Available online: https://rdrr.io/cran/beeswarm/ (accessed on 25 November 2020).
- Kelly, T. TomKellyGenetics/Vioplot. Available online: https://cran.r-project.org/web/packages/vioplot/vioplot.pdf (accessed on 11 November 2020).
- Hatina, J.; Fernandes, M.I.; Hoffmann, M.J.; Zeimet, A.G. Cancer Stem Cells—Basic Biological Properties and Experimental Approaches. In eLS; John Wiley & Sons: Chichester, UK, 2013. [Google Scholar] [CrossRef]
- Trucco, M.; Loeb, D. Sarcoma Stem Cells: Do We Know What We Are Looking For? Sarcoma 2012, 291702. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Basavarajaiah, S.; Limbruno, U.; Picchi, A.; Lettieri Valgimigli, M.; Sciahbasi, A.; Prati, F.; Calabresi, M.; Pierucci, D.; Guglielmotti, A. A Double-Blind Randomised Study to Evaluate the Efficacy and Safety of Bindarit in Preventing Coronary Stent Restenosis. EuroIntervention 2015, 11, 20140918-01. [Google Scholar] [CrossRef] [PubMed]
- Parra, J.; Portilla, J.; Pulido, F.; Sánchez-de la Rosa, R.; Alonso-Villaverde, C.; Berenguer, J.; Blanco, J.L.; Domingo, P.; Dronda, F.; Galera, C.; et al. Clinical Utility of Maraviroc. Clin. Drug Investig. 2011, 31, 527–542. [Google Scholar] [CrossRef]
- Ozanne, B.W.; McGarry, L.; Spence, H.J.; Johnston, I.; Winnie, J.; Meagher, L.; Stapleton, G. Transcriptional Regulation of Cell Invasion: AP-1 Regulation of a Multigenic Invasion Programme. Eur. J. Cancer 2000, 36, 1640–1648. [Google Scholar] [CrossRef]
- Ozanne, B.W.; Spence, H.J.; McGarry, L.C.; Hennigan, R.F. Transcription Factors Control Invasion: AP-1 the First among Equals. Oncogene 2007, 26, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-Q.; Liang, J.; Schellander, K.; Wagner, E.F.; Grigoriadis, A.E. C-Fos-Induced Osteosarcoma Formation in Transgenic Mice: Cooperativity with c-Jun and the Role of Endogenous c-Fos. Cancer Res. 1995, 55, 6244–6251. [Google Scholar] [PubMed]
- Mariani, O.; Brennetot, C.; Coindre, J.-M.; Gruel, N.; Ganem, C.; Delattre, O.; Stern, M.-H.; Aurias, A. JUN Oncogene Amplification and Overexpression Block Adipocytic Differentiation in Highly Aggressive Sarcomas. Cancer Cell 2007, 11, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Snyder, E.L.; Sandstrom, D.J.; Law, K.; Fiore, C.; Sicinska, E.; Brito, J.; Bailey, D.; Fletcher, J.A.; Loda, M.; Rodig, S.J.; et al. C-Jun Amplification and Overexpression Are Oncogenic in Liposarcoma but Not Always Sufficient to Inhibit the Adipocytic Differentiation Programme. J. Pathol. 2009, 218, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Ivorra, C.; Kubicek, M.; González, J.M.; Sanz-González, S.M.; Alvarez-Barrientos, A.; O’Connor, J.-E.; Burke, B.; Andrés, V. A Mechanism of AP-1 Suppression through Interaction of c-Fos with Lamin A/C. Genes Dev. 2006, 20, 307–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamood, A.S.; Gyles, P.; Balan, K.V.; Hollis, V.W.; Eckberg, W.R.; Asseffa, A.; Han, Z.; Wyche, J.H.; Anderson, W.A. Estrogen Receptor, Growth Factor Receptor and Protooncogene Protein Activities and Possible Signal Transduction Crosstalk in Estrogen Dependent and Independent Breast Cancer Cell Lines. J. Submicrosc. Cytol. Pathol. 1997, 29, 1–17. [Google Scholar] [PubMed]
- La Vecchia, S.; Sebastián, C. Metabolic Pathways Regulating Colorectal Cancer Initiation and Progression. Semin. Cell Dev. Biol. 2020, 98, 63–70. [Google Scholar] [CrossRef]
- Caneba, C.A.; Bellance, N.; Yang, L.; Pabst, L.; Nagrath, D. Pyruvate Uptake Is Increased in Highly Invasive Ovarian Cancer Cells under Anoikis Conditions for Anaplerosis, Mitochondrial Function, and Migration. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1036–E1052. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Su, Y.; Song, Q.; Tung, B.; Oyinlade, O.; Liu, S.; Ying, M.; Ming, G.; Song, H.; Qian, J.; et al. Methylated Cis-Regulatory Elements Mediate KLF4-Dependent Gene Transactivation and Cell Migration. eLife 2017, 6, e20068. [Google Scholar] [CrossRef]
- Wang, S.; Shi, X.; Wei, S.; Ma, D.; Oyinlade, O.; Lv, S.-Q.; Ying, M.; Zhang, Y.A.; Claypool, S.M.; Watkins, P.; et al. Krüppel-like Factor 4 (KLF4) Induces Mitochondrial Fusion and Increases Spare Respiratory Capacity of Human Glioblastoma Cells. J. Biol. Chem. 2018, 293, 6544–6555. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial Spare Respiratory Capacity: Mechanisms, Regulation, and Significance in Non-Transformed and Cancer Cells. FASEB J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-K.; Jee, B.A.; Kwon, S.M.; Yoon, Y.-S.; Xu, W.G.; Wang, H.-J.; Wang, X.W.; Thorgeirsson, S.S.; Lee, J.-S.; Woo, H.G.; et al. Identification of a Mitochondrial Defect Gene Signature Reveals NUPR1 as a Key Regulator of Liver Cancer Progression. Hepatology 2015, 62, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Machida, K. Pluripotency Transcription Factors and Metabolic Reprogramming of Mitochondria in Tumor-Initiating Stem-like Cells. Antioxid. Redox Signal. 2018, 28, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Basu-Roy, U.; Bayin, N.S.; Rattanakorn, K.; Han, E.; Placantonakis, D.G.; Mansukhani, A.; Basilico, C. Sox2 Antagonizes the Hippo Pathway to Maintain Stemness in Cancer Cells. Nat. Commun. 2015, 6, 6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurizi, G.; Verma, N.; Gadi, A.; Mansukhani, A.; Basilico, C. Sox2 Is Required for Tumor Development and Cancer Cell Proliferation in Osteosarcoma. Oncogene 2018, 37, 4626–4632. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Taouk, G.M. A Potential Role of YAP/TAZ in the Interplay Between Metastasis and Metabolic Alterations. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Würl, P.; Kappler, M.; Meye, A.; Bartel, F.; Köhler, T.; Lautenschläger, C.; Bache, M.; Schmidt, H.; Taubert, H. Co-Expression of Survivin and TERT and Risk of Tumour-Related Death in Patients with Soft-Tissue Sarcoma. Lancet 2002, 359, 943–945. [Google Scholar] [CrossRef]
- Nikitovic, D.; Kouvidi, K.; Karamanos, N.K.; Tzanakakis, G.N. The Roles of Hyaluronan/RHAMM/CD44 and Their Respective Interactions along the Insidious Pathways of Fibrosarcoma Progression. BioMed Res. Int. 2013, 2013, 929531. [Google Scholar] [CrossRef] [PubMed]
- Farmaki, E.; Chatzistamou, I.; Kaza, V.; Kiaris, H. A CCL8 Gradient Drives Breast Cancer Cell Dissemination. Oncogene 2016, 35, 6309–6318. [Google Scholar] [CrossRef]
- Barbai, T.; Fejős, Z.; Puskas, L.G.; Tímár, J.; Rásó, E. The Importance of Microenvironment: The Role of CCL8 in Metastasis Formation of Melanoma. Oncotarget 2015, 6, 29111–29128. [Google Scholar] [CrossRef] [Green Version]
- Otsubo, C.; Otomo, R.; Miyazaki, M.; Matsushima-Hibiya, Y.; Kohno, T.; Iwakawa, R.; Takeshita, F.; Okayama, H.; Ichikawa, H.; Saya, H.; et al. TSPAN2 Is Involved in Cell Invasion and Motility during Lung Cancer Progression. Cell Rep. 2014, 7, 527–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fils-Aimé, N.; Dai, M.; Guo, J.; El-Mousawi, M.; Kahramangil, B.; Neel, J.-C.; Lebrun, J.-J. MicroRNA-584 and the Protein Phosphatase and Actin Regulator 1 (PHACTR1), a New Signaling Route through Which Transforming Growth Factor-β Mediates the Migration and Actin Dynamics of Breast Cancer Cells. J. Biol. Chem. 2013, 288, 11807–11823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagci, T.; Wu, J.K.; Pfannl, R.; Ilag, L.L.; Jay, D.G. Autocrine Semaphorin 3A Signaling Promotes Glioblastoma Dispersal. Oncogene 2009, 28, 3537–3550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Cong, H.; Wang, H.; Zhang, D.; Liu, C.; Chu, H.; Qing, Q.; Wang, K. MiR-30a-5p Inhibits Osteosarcoma Cell Proliferation and Migration by Targeting FOXD1. Biochem. Biophys. Res. Commun. 2018, 503, 1092–1097. [Google Scholar] [CrossRef]
- Li, D.; Fan, S.; Yu, F.; Zhu, X.; Song, Y.; Ye, M.; Fan, L.; Lv, Z. FOXD1 Promotes Cell Growth and Metastasis by Activation of Vimentin in NSCLC. Cell Physiol. Biochem. 2018, 51, 2716–2731. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Larribère, L.; Sun, Q.; Novak, D.; Sachindra, S.; Granados, K.; Umansky, V.; Utikal, J. Loss of Neural Crest-Associated Gene FOXD1 Impairs Melanoma Invasion and Migration via RAC1B Downregulation. Int. J. Cancer 2018, 143, 2962–2972. [Google Scholar] [CrossRef] [Green Version]
- Ondondo, B.; Colbeck, E.; Jones, E.; Smart, K.; Lauder, S.N.; Hindley, J.; Godkin, A.; Moser, B.; Ager, A.; Gallimore, A. A Distinct Chemokine Axis Does Not Account for Enrichment of Foxp3(+) CD4(+) T Cells in Carcinogen-Induced Fibrosarcomas. Immunology 2015, 145, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A.; Mordoh, J.; Wainstok, R. Targeting Tumor-Associated Macrophages and Inhibition of MCP-1 Reduce Angiogenesis and Tumor Growth in a Human Melanoma Xenograft. J. Investig. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Gao, H.; Gao, C.; Liu, W.; Xing, D. Bindarit Attenuates Pain and Cancer-Related Inflammation by Influencing Myeloid Cells in a Model of Bone Cancer. Arch. Immunol. Ther. Exp. 2018, 66, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.T.; Li, K.K.; Hepburn, E.; Weston, C.J.; Curbishley, S.M.; Reynolds, G.M.; Hejmadi, R.K.; Bicknell, R.; Eksteen, B.; Ismail, T.; et al. The Effects of CCR5 Inhibition on Regulatory T-Cell Recruitment to Colorectal Cancer. Br. J. Cancer. 2015, 112, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, Y.; Sasaki, S.; Mukaida, N.; Baba, T. Blockade of the Chemokine Receptor, CCR5, Reduces the Growth of Orthotopically Injected Colon Cancer Cells via Limiting Cancer-Associated Fibroblast Accumulation. Oncotarget 2016, 7, 48335–48345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zollo, M.; Di Dato, V.; Spano, D.; De Martino, D.; Liguori, L.; Marino, N.; Vastolo, V.; Navas, L.; Garrone, B.; Mangano, G.; et al. Targeting Monocyte Chemotactic Protein-1 Synthesis with Bindarit Induces Tumor Regression in Prostate and Breast Cancer Animal Models. Clin. Exp. Metastasis 2012, 29, 585–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.-E.A.; Bae, S.; Lillard, J.W.; Singh, R. CCR5/CCL5 Axis Interaction Promotes Migratory and Invasiveness of Pancreatic Cancer Cells. Sci. Rep. 2018, 8, 1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervaiz, A.; Zepp, M.; Mahmood, S.; Ali, D.M.; Berger, M.R.; Adwan, H. CCR5 Blockage by Maraviroc: A Potential Therapeutic Option for Metastatic Breast Cancer. Cell. Oncol. 2019, 42, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sontag, D.; Gong, Y.; Minuk, G.Y. Alterations in Chemokine Receptor CCR5 Activity Influence Tumor Cell Biology in Human Cholangiocarcinoma Cell Lines. Ann. Hepatol. 2021, 21, 100265. [Google Scholar] [CrossRef]
- Maione, F.; Molla, F.; Meda, C.; Latini, R.; Zentilin, L.; Giacca, M.; Seano, G.; Serini, G.; Bussolino, F.; Giraudo, E. Semaphorin 3A Is an Endogenous Angiogenesis Inhibitor That Blocks Tumor Growth and Normalizes Tumor Vasculature in Transgenic Mouse Models. J. Clin. Investig. 2009, 119, 3356–3372. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Cheng, S.-Y. Angiopoietin-2: Development of Inhibitors for Cancer Therapy. Curr. Oncol. Rep. 2009, 11, 111–116. [Google Scholar] [CrossRef]
- Marconcini, L.; Marchio, S.; Morbidelli, L.; Cartocci, E.; Albini, A.; Ziche, M.; Bussolino, F.; Oliviero, S. C-Fos-Induced Growth Factor/Vascular Endothelial Growth Factor D Induces Angiogenesis in Vivo and in Vitro. Proc. Natl. Acad. Sci. USA 1999, 96, 9671–9676. [Google Scholar] [CrossRef] [Green Version]
- Yanagawa, T.; Shinozaki, T.; Watanabe, H.; Saito, K.; Raz, A.; Takagishi, K. Vascular Endothelial Growth Factor-D Is a Key Molecule That Enhances Lymphatic Metastasis of Soft Tissue Sarcomas. Exp. Cell Res. 2012, 318, 800–808. [Google Scholar] [CrossRef] [Green Version]
- Kilvaer, T.K.; Valkov, A.; Sorbye, S.; Smeland, E.; Bremnes, R.M.; Busund, L.-T.; Donnem, T. Profiling of VEGFs and VEGFRs as Prognostic Factors in Soft Tissue Sarcoma: VEGFR-3 Is an Independent Predictor of Poor Prognosis. PLoS ONE 2010, 5, e15368. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Zhao, W.; Meng, W.; Liu, C.; Chen, Y.; Bhattacharya, S.K.; Sun, Y. Vascular Endothelial Growth Factor-D Mediates Fibrogenic Response in Myofibroblasts. Mol. Cell Biochem. 2016, 413, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chen, S.-H.; Hung, W.-C.; Paul, C.; Zhu, F.; Guan, P.-P.; Huso, D.L.; Kontrogianni-Konstantopoulos, A.; Konstantopoulos, K. Fluid Shear Promotes Chondrosarcoma Cell Invasion by Activating Matrix Metalloproteinase 12 via IGF-2 and VEGF Signaling Pathways. Oncogene 2015, 34, 4558–4569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemann, N.M.; Siemann, D.W. Angiopoietin-2 Axis Inhibitors: Current Status and Future Considerations for Cancer Therapy. Available online: https://www.eurekaselect.com/115040/article (accessed on 2 November 2020).
- Bezuidenhout, L.; Zilla, P.; Davies, N. Association of Ang-2 with Integrin Beta 2 Controls Ang-2/PDGF-BB-Dependent Upregulation of Human Peripheral Blood Monocyte Fibrinolysis. Inflammation 2009, 32, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jarzynka, M.J.; Guo, P.; Imanishi, Y.; Schlaepfer, D.D.; Cheng, S.-Y. Angiopoietin 2 Induces Glioma Cell Invasion by Stimulating Matrix Metalloprotease 2 Expression through the Alphavbeta1 Integrin and Focal Adhesion Kinase Signaling Pathway. Cancer Res. 2006, 66, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Morii, T.; Mochizuki, K.; Tajima, T.; Ichimura, S.; Satomi, K. D-Dimer Levels as a Prognostic Factor for Determining Oncological Outcomes in Musculoskeletal Sarcoma. BMC Musculoskelet. Disord. 2011, 12, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, S.D.; Zhou, X.; Bueso-Ramos, C.E.; Ravi, V.; Patel, S.; Benjamin, R.S.; Vadhan-Raj, S. Prognostic Significance of Elevated D-Dimer for Survival in Patients with Sarcoma. Am. J. Clin. Oncol. 2012, 35, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Bure, I.V.; Kuznetsova, E.B.; Zaletaev, D.V. Long Noncoding RNAs and Their Role in Oncogenesis. Mol. Biol. 2018, 52, 907–920. [Google Scholar] [CrossRef]
- Zhang, R.; Xia, T. Long Non-Coding RNA XIST Regulates PDCD4 Expression by Interacting with MiR-21-5p and Inhibits Osteosarcoma Cell Growth and Metastasis. Int. J. Oncol. 2017, 51, 1460–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, G.-Y.; Miao, J.; Zhang, X.-L. Long Noncoding RNA XIST Promotes Osteosarcoma Progression by Targeting Ras-Related Protein RAP2B via MiR-320b. Oncol. Res. 2018, 26, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA Is a Potent Suppressor of Hematologic Cancer in Mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef] [Green Version]
- Navarro, P.; Chambers, I.; Karwacki-Neisius, V.; Chureau, C.; Morey, C.; Rougeulle, C.; Avner, P. Molecular Coupling of Xist Regulation and Pluripotency. Science 2008, 321, 1693–1695. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Matsuda, M.; Kawamura, T.; Sogo, T.; Shigeno, A.; Nishida, E.; Ebisuya, M. Foxd1 Is a Mediator and Indicator of the Cell Reprogramming Process. Nat. Commun. 2014, 5, 3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics-Anti-Metastatic and Anti-Invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosel, D.; Fernandes, M.; Sanz-Moreno, V.; Brábek, J. Migrastatics: Redirecting R&D in Solid Cancer Towards Metastasis? Trends Cancer 2019, 5, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Kishino, A.; Konishi, O.; Kumagai, K.; Hosotani, N.; Saji, I.; Nakayama, C.; Kimura, T. In Vitro and in Vivo Characterization of a Novel Semaphorin 3A Inhibitor, SM-216289 or Xanthofulvin. J. Biol. Chem. 2003, 278, 42985–42991. [Google Scholar] [CrossRef] [Green Version]
- Martínez-García, D.; Manero-Rupérez, N.; Quesada, R.; Korrodi-Gregório, L.; Soto-Cerrato, V. Therapeutic Strategies Involving Survivin Inhibition in Cancer. Med. Res. Rev. 2019, 39, 887–909. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Doubling Time (h) | Slope of the Exponential Growth Phase |
|---|---|---|
| JUN-1 | 9.5 | 0.032 |
| JUN-2 | 10.4 | 0.029 |
| JUN-2fos-3 | 30.9 | 0.010 |
| JUN-3 | 7.4 | 0.041 |
| Motility and invasiveness-related genes | JUN3↑ JUN2f3↑ JUN2↓ and JUN3↓ JUN2f3↓ JUN2↑ (activators of motility) (suppressors of motility) |
| Proliferation-related genes | JUN3↑ JUN2f3↓ JUN2↓ and JUN3↓ JUN2f3↑ JUN2↑ (activators of proliferation) (suppressors of proliferation) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kripnerová, M.; Parmar, H.S.; Šána, J.; Kopková, A.; Radová, L.; Sopper, S.; Biernacki, K.; Jedlička, J.; Kohoutová, M.; Kuncová, J.; et al. Complex Interplay of Genes Underlies Invasiveness in Fibrosarcoma Progression Model. J. Clin. Med. 2021, 10, 2297. https://doi.org/10.3390/jcm10112297
Kripnerová M, Parmar HS, Šána J, Kopková A, Radová L, Sopper S, Biernacki K, Jedlička J, Kohoutová M, Kuncová J, et al. Complex Interplay of Genes Underlies Invasiveness in Fibrosarcoma Progression Model. Journal of Clinical Medicine. 2021; 10(11):2297. https://doi.org/10.3390/jcm10112297
Chicago/Turabian StyleKripnerová, Michaela, Hamendra Singh Parmar, Jiří Šána, Alena Kopková, Lenka Radová, Sieghart Sopper, Krzysztof Biernacki, Jan Jedlička, Michaela Kohoutová, Jitka Kuncová, and et al. 2021. "Complex Interplay of Genes Underlies Invasiveness in Fibrosarcoma Progression Model" Journal of Clinical Medicine 10, no. 11: 2297. https://doi.org/10.3390/jcm10112297
APA StyleKripnerová, M., Parmar, H. S., Šána, J., Kopková, A., Radová, L., Sopper, S., Biernacki, K., Jedlička, J., Kohoutová, M., Kuncová, J., Peychl, J., Rudolf, E., Červinka, M., Houdek, Z., Dvořák, P., Houfková, K., Pešta, M., Tůma, Z., Dolejšová, M., ... Hatina, J. (2021). Complex Interplay of Genes Underlies Invasiveness in Fibrosarcoma Progression Model. Journal of Clinical Medicine, 10(11), 2297. https://doi.org/10.3390/jcm10112297