
Breast Cancer Cell Re-Dissemination from Lung Metastases—A Mechanism for Enhancing Metastatic Burden


{kind=link}
{kind=link}
Abstract
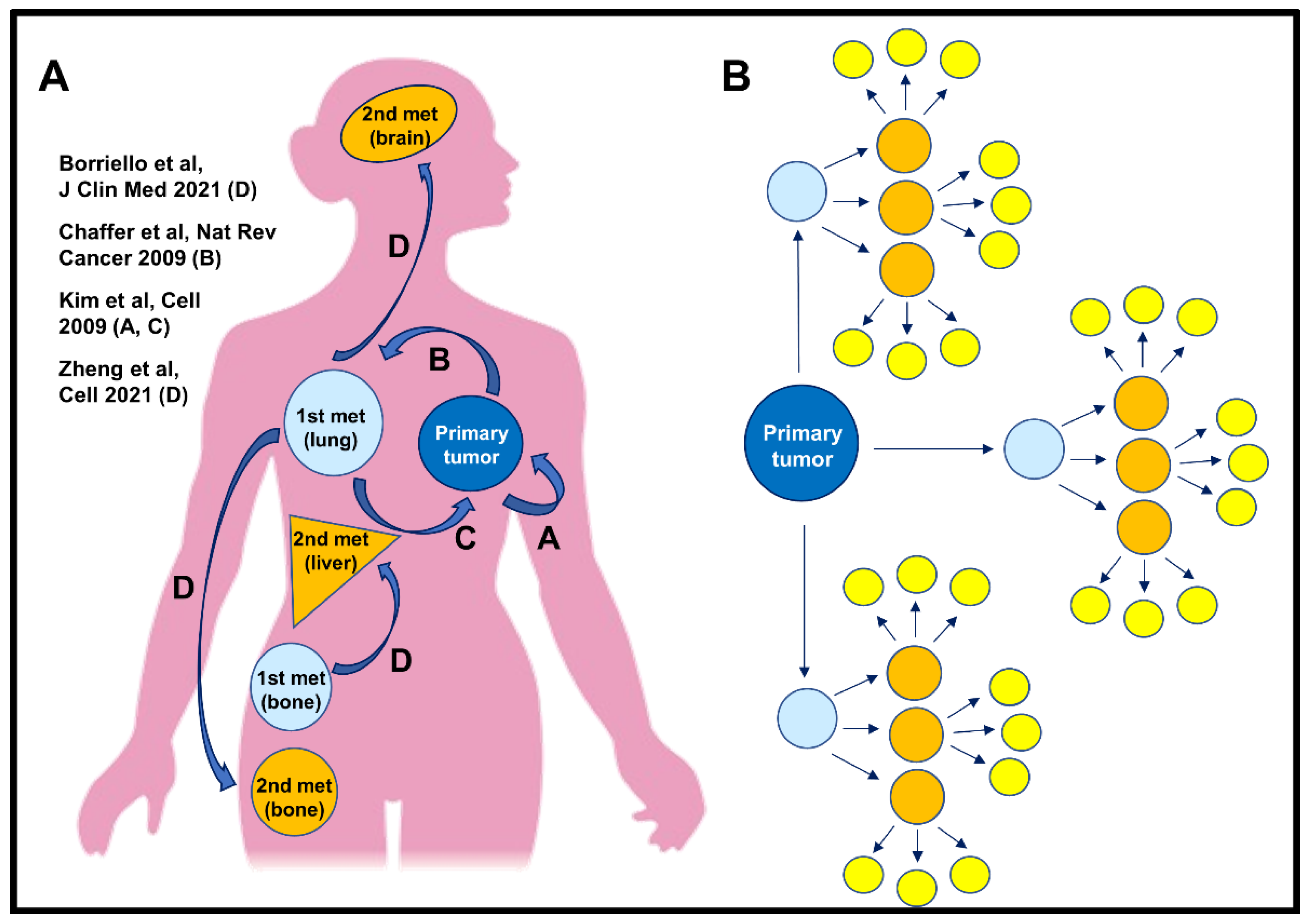
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Animals
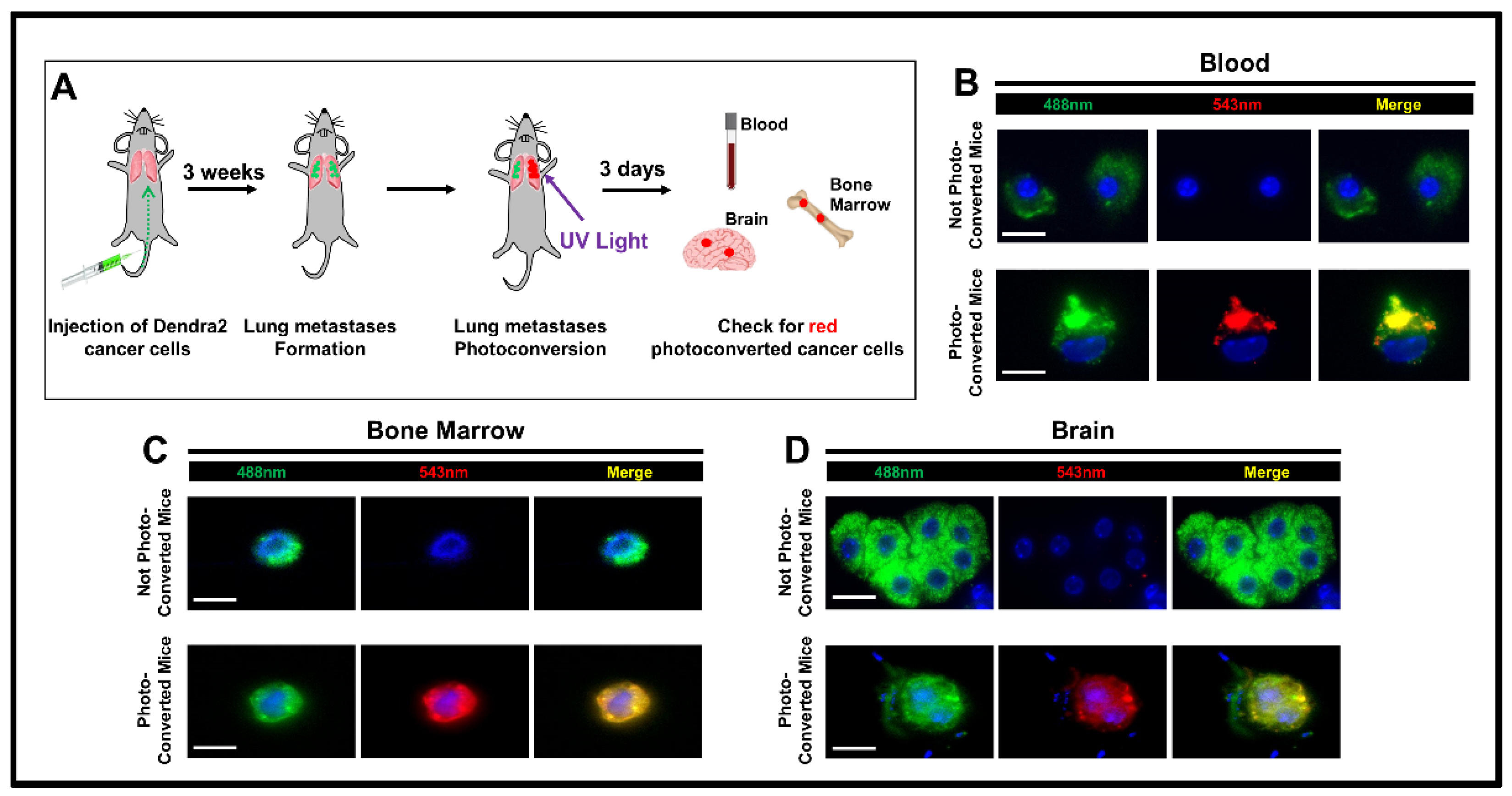
2.3. Experimental Metastasis
2.4. Photoconversion
2.5. Processing of Samples and Detection of Photoconverted Cancer Cells
3. Results
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Mph, K.D.M.; Sauer, A.G.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 438–451. [Google Scholar] [CrossRef]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Gupta, G.P.; Massagué, J. Cancer Metastasis: Building a Framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, N.G.R.J.; et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature 2016, 540, 589–612. [Google Scholar] [CrossRef]
- Risson, E.; Nobre, A.R.; Maguer-Satta, V.; Aguirre-Ghiso, J.A. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Rev. Cancer 2020, 1, 672–680. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. How dormant cancer persists and reawakens. Science 2018, 361, 1314–1315. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.-F.; Norton, L.; Massagué, J. Tumor Self-Seeding by Circulating Cancer Cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Norton, L.; Massagué, J. Is cancer a disease of self-seeding? Nat. Med. 2006, 12, 875–878. [Google Scholar] [CrossRef]
- Coste, A.; Karagiannis, G.S.; Wang, Y.; Xue, E.A.; Lin, Y.; Skobe, M.; Jones, J.G.; Oktay, M.H.; Condeelis, J.S.; Entenberg, D. Hematogenous Dissemination of Breast Cancer Cells from Lymph Nodes is Mediated by Tumor MicroEnvironment of Metastasis Doorways. Front. Oncol. 2020, 10, 2187. [Google Scholar] [CrossRef]
- Pereira, E.R.; Kedrin, D.; Seano, G.; Gautier, O.; Meijer, E.F.J.; Jones, D.; Chin, S.-M.; Kitahara, S.; Bouta, E.M.; Chang, J.; et al. Lymph node metastases can invade local blood vessels, exit the node, and colonize distant organs in mice. Science 2018, 359, 1403–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.; Assen, F.P.; Leithner, A.; Abe, J.; Schachner, H.; Asfour, G.; Bago-Horvath, Z.; Stein, J.V.; Uhrin, P.; Sixt, M.; et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 2018, 359, 1408–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoover, H.C.; Ketcham, A.S. Metastasis of metastases. Am. J. Surg. 1975, 130, 405–411. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. On the theory of tumor self-seeding: Implications for metastasis progression in humans. Breast Cancer Res. 2010, 12, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Comen, E.; Norton, L.; Massagué, J. Clinical implications of cancer self-seeding. Nat. Rev. Clin. Oncol. 2011, 8, 369–377. [Google Scholar] [CrossRef]
- Turajlic, S.; Swanton, C. Metastasis as an evolutionary process. Science 2016, 352, 169–175. [Google Scholar] [CrossRef]
- De Groot, A.E.; Roy, S.; Brown, J.S.; Pienta, K.J.; Amend, S.R. Revisiting Seed and Soil: Examining the Primary Tumor and Cancer Cell Foraging in Metastasis. Mol. Cancer Res. 2017, 15, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.-Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage–Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Robinson, B.D.; Sica, G.L.; Liu, Y.-F.; Rohan, T.E.; Gertler, F.B.; Condeelis, J.S.; Jones, J.G. Tumor Microenvironment of Metastasis in Human Breast Carcinoma: A Potential Prognostic Marker Linked to Hematogenous Dissemination. Clin. Cancer Res. 2009, 15, 2433–2441. [Google Scholar] [CrossRef] [Green Version]
- Rohan, T.E.; Xue, X.; Lin, H.-M.; D’Alfonso, T.M.; Ginter, P.S.; Oktay, M.H.; Robinson, B.D.; Ginsberg, M.; Gertler, F.B.; Glass, A.G.; et al. Tumor Microenvironment of Metastasis and Risk of Distant Metastasis of Breast Cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [Green Version]
- Oktay, M.H.; Jones, J.G. TMEM: A novel breast cancer dissemination marker for the assessment of metastatic risk. Biomarkers Med. 2015, 9, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Borriello, L.; Karagiannis, G.S.; Duran, C.L.; Coste, A.; Oktay, M.H.; Entenberg, D.; Condeelis, J.S. The role of the tumor microenvironment in tumor cell intravasation and dissemination. Eur. J. Cell Biol. 2020, 99, 151098. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Pastoriza, J.M.; Borriello, L.; Jafari, R.; Coste, A.; Condeelis, J.S.; Oktay, M.H.; Entenberg, D. Assessing TMEM Doorway-Mediated Vascular Permeability Associated with Cancer Cell Dissemination, using Intravital Imaging and Fixed Tissue Analysis. Jove 2019. [Google Scholar] [CrossRef] [PubMed]
- Sparano, J.A.; Gray, R.; Oktay, M.H.; Entenberg, D.; Rohan, T.; Xue, X.; Donovan, M.; Peterson, M.; Shuber, A.; Hamilton, D.A.; et al. A metastasis biomarker (MetaSite Breast™ Score) is associated with distant recurrence in hormone receptor-positive, HER2-negative early-stage breast cancer. npj Breast Cancer 2017, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Entenberg, D.; Voiculescu, S.; Guo, P.; Borriello, L.; Wang, Y.; Karagiannis, G.S.; Jones, J.; Baccay, F.; Oktay, M.; Condeelis, J. A permanent window for the murine lung enables high-resolution imaging of cancer metastasis. Nat. Methods 2018, 15, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Tripathy, D.; Frenkel, E.P.; Shete, S.; Naftalis, E.Z.; Huth, J.F.; Beitsch, P.D.; Leitch, M.; Hoover, S.; Euhus, D.; et al. Circulating Tumor Cells in Patients with Breast Cancer Dormancy. Clin. Cancer Res. 2004, 10, 8152–8162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, V.; Stahmann, N.; Riethdorf, S.; Rau, T.; Zabel, T.; Goetz, A.; Jänicke, F.; Pantel, K. Circulating Tumor Cells in Breast Cancer: Correlation to Bone Marrow Micrometastases, Heterogeneous Response to Systemic Therapy and Low Proliferative Activity. Clin. Cancer Res. 2005, 11, 3678–3685. [Google Scholar] [CrossRef] [Green Version]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Rack, B.K.; Schindlbeck, C.; Andergassen, U.; Schneeweiss, A.; Zwingers, T.; Lichtenegger, W.; Beckmann, M.; Sommer, H.L.; Pantel, K.; Janni, W.; et al. Use of circulating tumor cells (CTC) in peripheral blood of breast cancer patients before and after adjuvant chemotherapy to predict risk for relapse: The SUCCESS trial. J. Clin. Oncol. 2010, 28, 1003. [Google Scholar] [CrossRef]
- Kimura, H.; Hayashi, K.; Yamauchi, K.; Yamamoto, N.; Tsuchiya, H.; Tomita, K.; Kishimoto, H.; Bouvet, M.; Hoffman, R.M. Real-time imaging of single cancer-cell dynamics of lung metastasis. J. Cell. Biochem. 2009, 109, 58–64. [Google Scholar] [CrossRef]
- Entenberg, D.; Wyckoff, J.; Gligorijevic, B.; Roussos, E.T.; Verkhusha, V.; Pollard, J.W.; Condeelis, J. Setup and use of a two-laser multiphoton microscope for multichannel intravital fluorescence imaging. Nat. Protoc. 2011, 6, 1500–1520. [Google Scholar] [CrossRef] [Green Version]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D.; et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borriello, L.; Nakata, R.; Sheard, M.A.; Fernandez, G.E.; Sposto, R.; Malvar, J.; Blavier, L.; Shimada, H.; Asgharzadeh, S.; Seeger, R.C.; et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017, 77, 5142–5157. [Google Scholar] [CrossRef] [Green Version]
- Gligorijevic, B.; Kedrin, D.; Segall, J.E.; Condeelis, J.; Van Rheenen, J. Dendra2 Photoswitching through the Mammary Imaging Window. J. Vis. Exp. 2009, 18, e1278. [Google Scholar] [CrossRef] [Green Version]
- Gurskaya, N.; Verkhusha, V.; Shcheglov, A.S.; Staroverov, D.B.; Chepurnykh, T.V.; Fradkov, A.F.; Lukyanov, S.; Lukyanov, K. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol. 2006, 24, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yang, M.; Wang, L.; Williamson, I.; Tian, F.; Qin, M.; Shah, P.K.; Sharifi, B.G. Autofluorescence contributes to false-positive intracellular Foxp3 staining in macrophages: A lesson learned from flow cytometry. J. Immunol. Methods 2012, 386, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Bado, I.L.; Hu, J.; Wan, Y.-W.; Wu, L.; Wang, H.; Gao, Y.; Jeong, H.-H.; Xu, Z.; Hao, X.; et al. The bone microenvironment invigorates metastatic seeds for further dissemination. Cell 2021, 184, 2471–2486. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Jalgaonkar, S.P.; Middleton, J.D.; Hai, T. Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, E7159–E7168. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D’Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9, eaan0026. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borriello, L.; Condeelis, J.; Entenberg, D.; Oktay, M.H. Breast Cancer Cell Re-Dissemination from Lung Metastases—A Mechanism for Enhancing Metastatic Burden. J. Clin. Med. 2021, 10, 2340. https://doi.org/10.3390/jcm10112340
Borriello L, Condeelis J, Entenberg D, Oktay MH. Breast Cancer Cell Re-Dissemination from Lung Metastases—A Mechanism for Enhancing Metastatic Burden. Journal of Clinical Medicine. 2021; 10(11):2340. https://doi.org/10.3390/jcm10112340
Chicago/Turabian StyleBorriello, Lucia, John Condeelis, David Entenberg, and Maja H. Oktay. 2021. "Breast Cancer Cell Re-Dissemination from Lung Metastases—A Mechanism for Enhancing Metastatic Burden" Journal of Clinical Medicine 10, no. 11: 2340. https://doi.org/10.3390/jcm10112340
APA StyleBorriello, L., Condeelis, J., Entenberg, D., & Oktay, M. H. (2021). Breast Cancer Cell Re-Dissemination from Lung Metastases—A Mechanism for Enhancing Metastatic Burden. Journal of Clinical Medicine, 10(11), 2340. https://doi.org/10.3390/jcm10112340