
GSH-Independent Induction of ER Stress during Hypoglycaemia in the Retinal Cells of Mice



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Lines
2.2. Cell Culture Conditions
2.3. RT-PCR and qPCR
2.4. Western Blotting Analysis
2.5. Endoplasmic Reticulum (ER) Isolation
2.6. GSH Analysis
2.7. Tunicamycin Injections
2.8. Terminal dUTP Nick End-Labelling (TUNEL) of Fragmented DNA
2.9. Immunostaining
2.10. Hyperinsulinemic Clamp
2.11. Statistical Analysis
3. Results
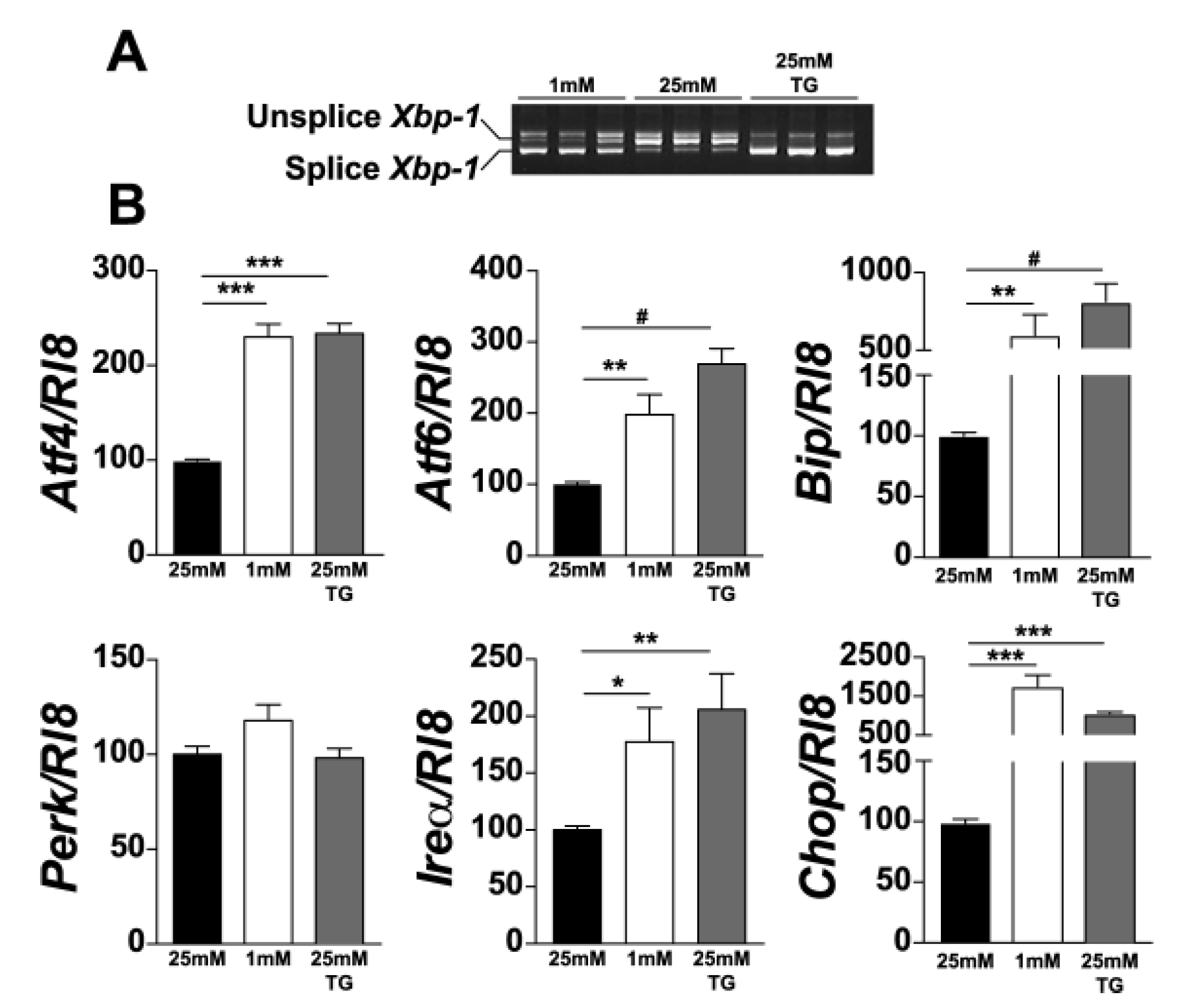
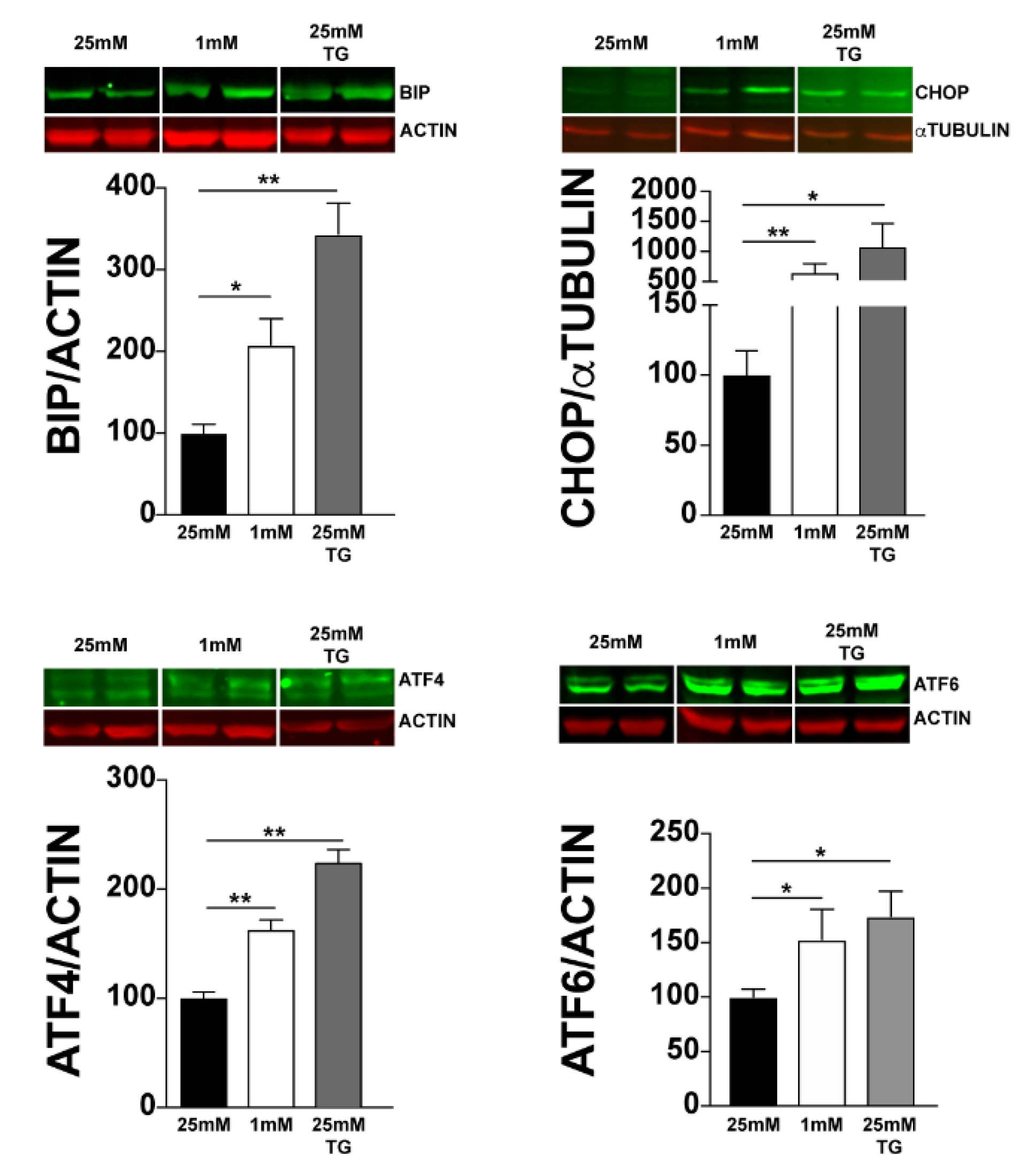
3.1. Expression of UPR-Related Genes Is Increased in 661W Cells under Hypoglycaemic Stress
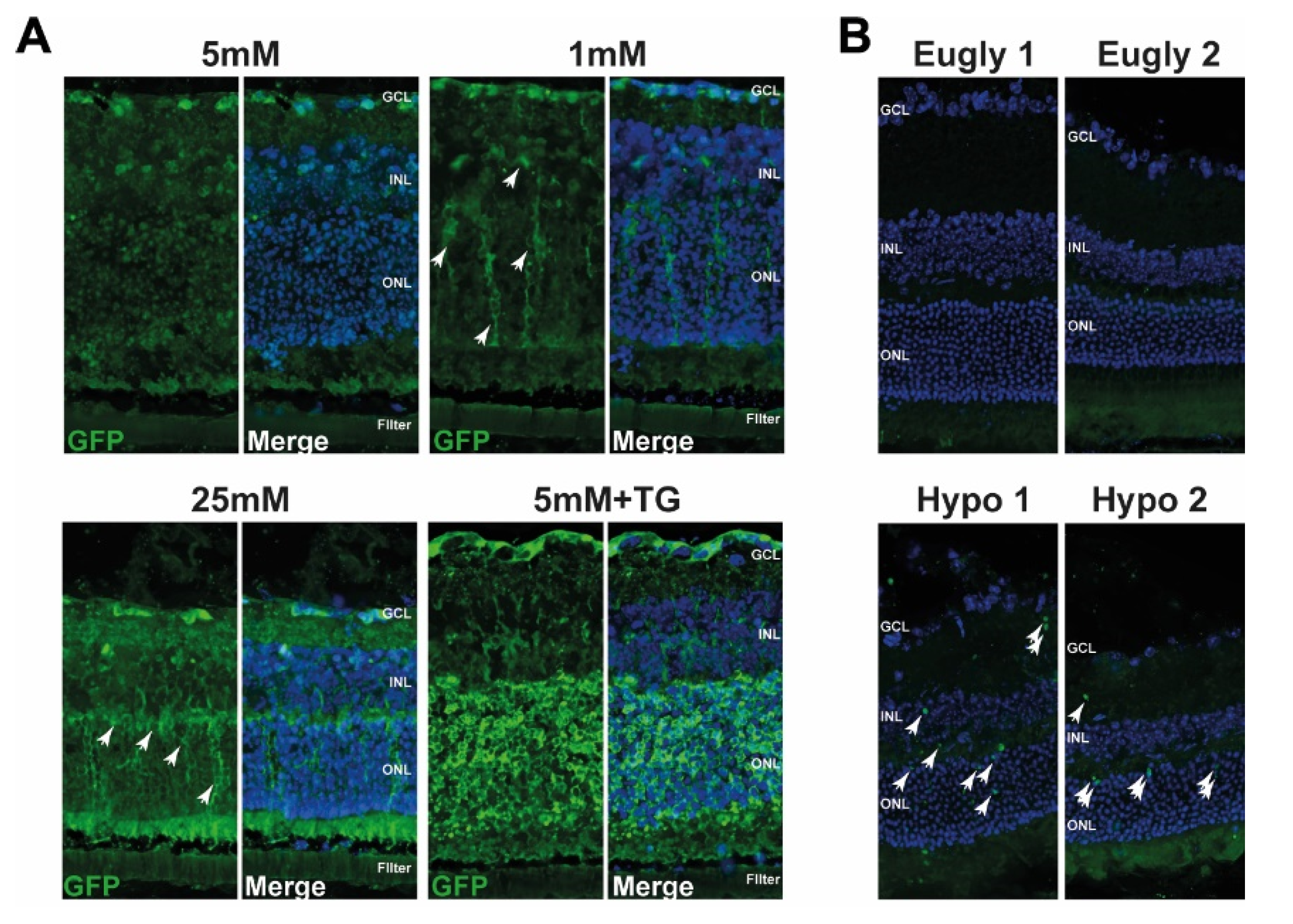
3.2. UPR Is Also Triggered Ex Vivo and In Vivo under Low Glucose Conditions
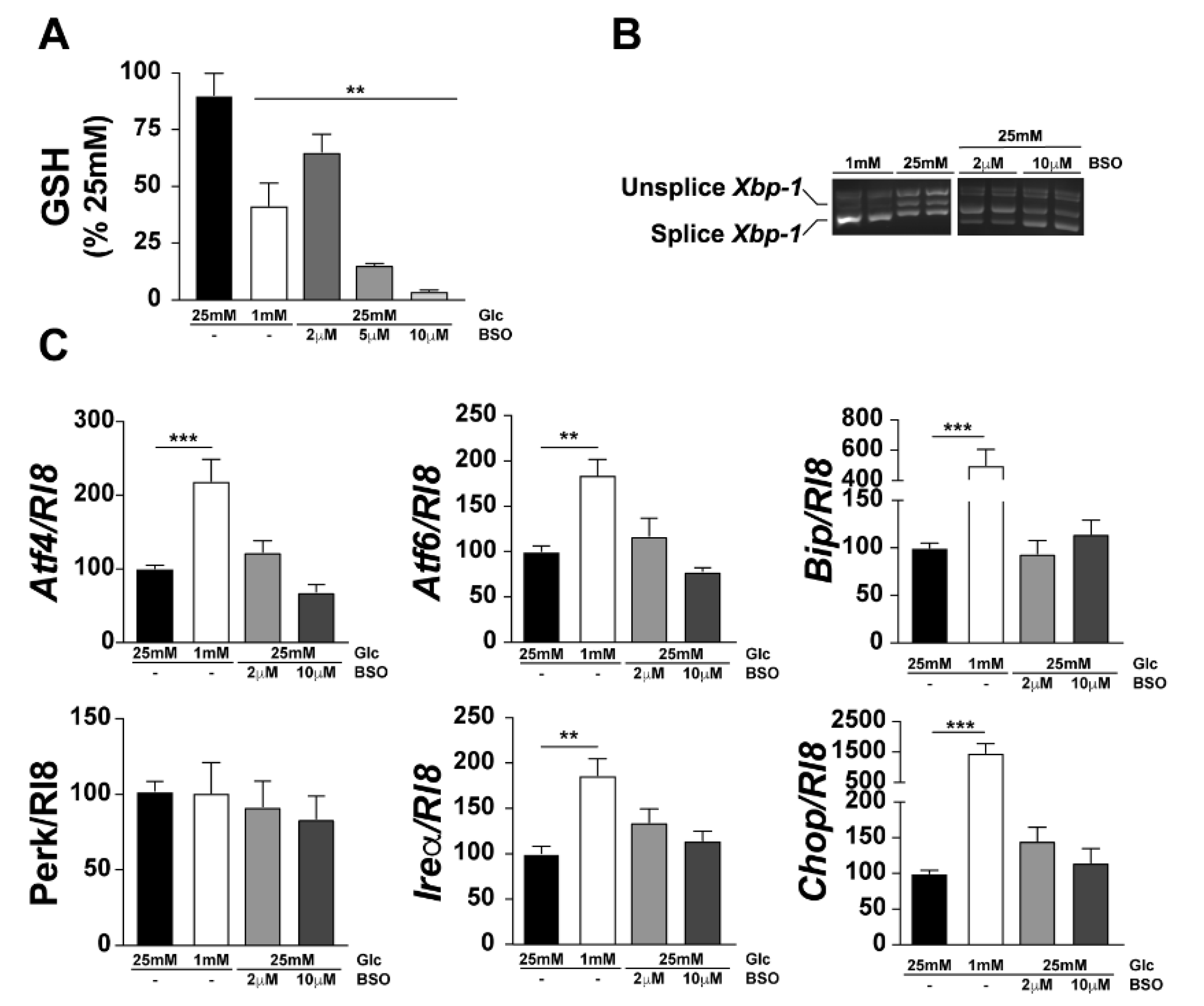
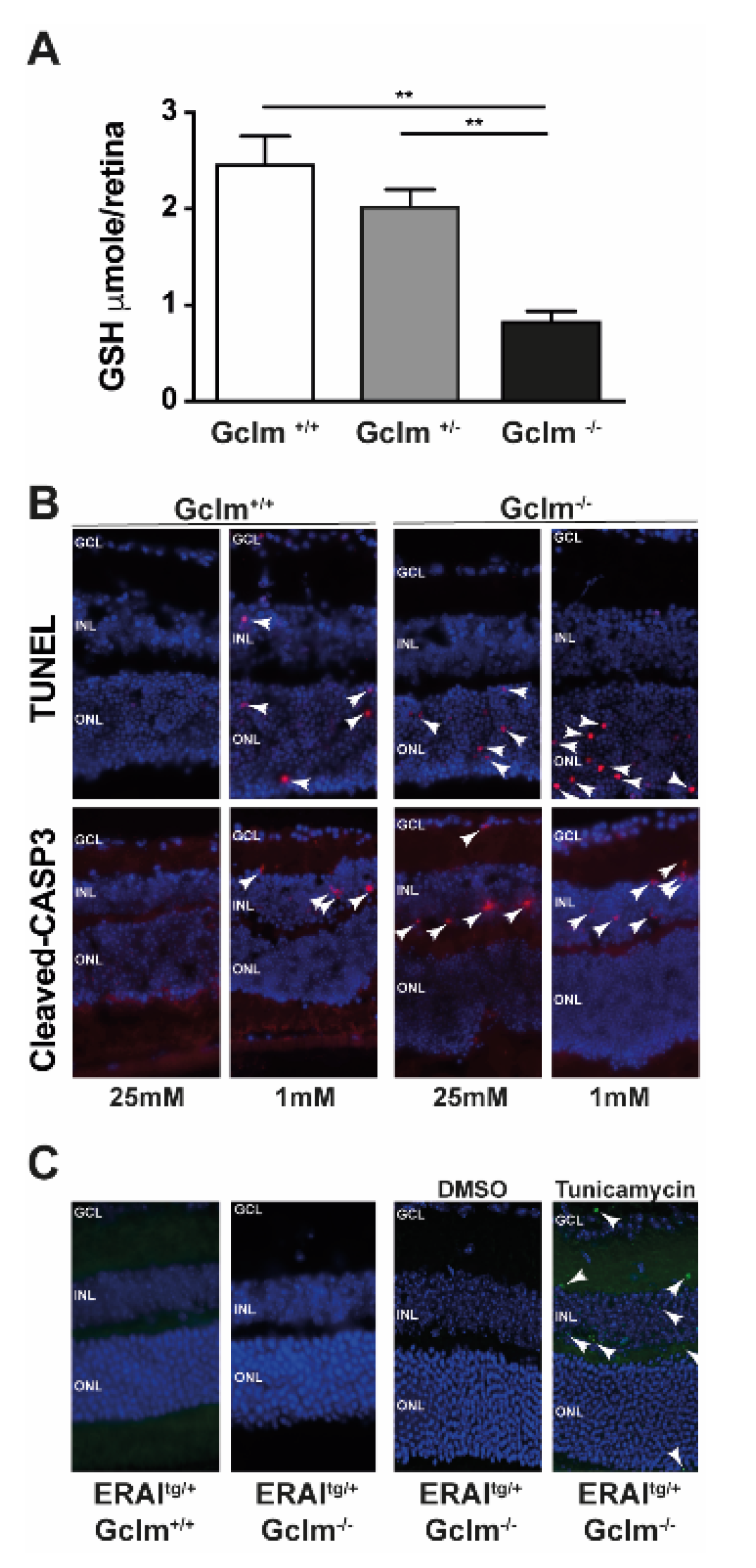
3.3. GSH Decrease Is Not Involved in UPR Activation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Yau, J.W.Y.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.-J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global Prevalence and Major Risk Factors of Diabetic Retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, C.; Simó, R. Neuroprotection in Diabetic Retinopathy. Curr. Diabetes Rep. 2012, 12, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Xu, X.; Bi, H.; Zhu, Q.; Wu, J.; Xia, X.; Ren, Q.; Ho, P.C.P. Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: The role of reactive oxygen species in diabetic retinopathy. Exp. Eye Res. 2006, 83, 807–816. [Google Scholar] [CrossRef]
- Sohn, E.H.; van Dijk, H.W.; Jiao, C.; Kok, P.H.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; van Velthoven, M.E.; et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef] [Green Version]
- Simo, R.; Hernandez, C. Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog. Retin. Eye Res. 2015, 48, 160–180. [Google Scholar] [CrossRef]
- Garcia-Ramírez, M.; Hernández, C.; Villarroel, M.; Canals, F.; Alonso, M.A.; Fortuny, R.; Masmiquel, L.; Navarro, A.; García-Arumí, J.; Simó, R. Interphotoreceptor retinoid-binding protein (IRBP) is downregulated at early stages of diabetic retinopathy. Diabetologia 2009, 52, 2633–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco, E.; Hernandez, C.; Miralles, A.; Huguet, P.; Farres, J.; Simo, R. Lower somatostatin expression is an early event in diabetic retinopathy and is associated with retinal neurodegeneration. Diabetes Care 2007, 30, 2902–2908. [Google Scholar] [CrossRef] [Green Version]
- Hammes, H.-P.P. Diabetic retinopathy: Hyperglycaemia, oxidative stress and beyond. Diabetologia 2018, 61, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Rodríguez, M.A.; Mendoza-Núñez, V.M. Oxidative Stress Indexes for Diagnosis of Health or Disease in Humans. Oxid Med. Cell Longev. 2019, 2019, 1–32. [Google Scholar] [CrossRef]
- McLaughlin, T.; Siddiqi, M.; Wang, J.J.; Zhang, S.X. Loss of XBP1 Leads to Early-Onset Retinal Neurodegeneration in a Mouse Model of Type I Diabetes. J. Clin. Med. 2019, 8, 906. [Google Scholar] [CrossRef] [Green Version]
- Roybal, C.N.; Yang, S.; Sun, C.-W.; Hurtado, D.; Jagt, D.L.V.; Townes, T.M.; Abcouwer, S.F. Homocysteine Increases the Expression of Vascular Endothelial Growth Factor by a Mechanism Involving Endoplasmic Reticulum Stress and Transcription Factor ATF4. J. Biol. Chem. 2004, 279, 14844–14852. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Chen, C.; McLaughlin, T.; Wang, Y.; Le, Y.Z.; Wang, J.J.; Zhang, S.X. Loss of X-box binding protein 1 in Muller cells augments retinal inflammation in a mouse model of diabetes. Diabetologia 2019, 62, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B. Transport from the ER through the Golgi Apparatus in Molecular Biology of the Cell, 5th ed.; Garland Science: New York, NY, USA; Taylor & Francis, Distributor: London, UK, 2008. [Google Scholar]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; García-Poyatos, C.; Martín-García, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2α Axis. Mol. Cell 2019, 74, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, M.S.; Knox, T.; LaVail, M.M.; Gorbatyuk, O.S.; Noorwez, S.M.; Hauswirth, W.W.; Lin, J.H.; Muzyczka, N.; Lewin, A.S. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. USA 2010, 107, 5961–5966. [Google Scholar] [CrossRef] [Green Version]
- Ghaderi, S.; Ahmadian, S.; Soheili, Z.-S.; Ahmadieh, H.; Samiei, S.; Kheitan, S.; Pirmardan, E.R. AAV delivery of GRP78/BiP promotes adaptation of human RPE cell to ER stress. J. Cell. Biochem. 2018, 119, 1355–1367. [Google Scholar] [CrossRef]
- Ikesugi, K.; Mulhern, M.L.; Madson, C.J.; Hosoya, K.; Terasaki, T.; Kador, P.F.; Shinohara, T. Induction of endoplasmic reticulum stress in retinal pericytes by glucose deprivation. Curr. Eye Res. 2006, 31, 947–953. [Google Scholar] [CrossRef]
- Deneke, S.M.; Fanburg, B.L. Regulation of cellular glutathione. Am. J. Physiol. 1989, 257, 163–173. [Google Scholar] [CrossRef]
- Ganea, E.; Harding, J.J. Glutathione-Related Enzymes and the Eye. Curr. Eye Res. 2006, 31, 1–11. [Google Scholar] [CrossRef]
- Delaunay-Moisan, A.; Ponsero, A.; Toledano, M.B. Reexamining the Function of Glutathione in Oxidative Protein Folding and Secretion. Antioxid. Redox Sign. 2017, 27, 1178–1199. [Google Scholar] [CrossRef]
- Dixon, B.M.; Heath, S.-H.D.; Kim, R.; Suh, J.H.; Hagen, T.M. Assessment of endoplasmic reticulum glutathione redox status is confounded by extensive ex vivo oxidation. Antioxid. Redox Sign. 2008, 10, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Veenstra, A.; Palczewski, K.; Kern, T.S. Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc. Natl. Acad. Sci. USA 2013, 110, 16586–16591. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Park, J.W.; Park, S.J.; Kim, K.Y.; Chung, J.W.; Chun, M.H.; Oh, S.J. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia 2003, 46, 1260–1268. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kern, T.S.; Engerman, R.L. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. Iv. Antioxid. Def. Syst. Free Radic Biol. Med. 1997, 22, 587–592. [Google Scholar] [CrossRef]
- Kern, T.S. Do photoreceptor cells cause the development of retinal vascular disease? Vis. Res. 2017, 139, 65–71. [Google Scholar] [CrossRef]
- Coffe, V.; Carbajal, R.C.; Salceda, R. Glycogen metabolism in the rat retina. J. Neurochem. 2004, 88, 885–890. [Google Scholar] [CrossRef]
- Chertov, A.O.; Holzhausen, L.; Kuok, I.T.; Couron, D.; Parker, E.; Linton, J.D.; Sadilek, M.; Sweet, I.R.; Hurley, J.B. Roles of glucose in photoreceptor survival. J. Biol. Chem. 2011, 286, 34700–34711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.I.; Barlow, R.B.; Weinstock, R.S. Acute hypoglycemia decreases central retinal function in the human eye. Vis. Res. 2011, 51, 1623–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryer, P.E. Hypoglycaemia: The limiting factor in the glycaemic management of Type I and Type II diabetes. Diabetologia 2002, 45, 937–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryer, P.E. The barrier of hypoglycemia in diabetes. Diabetes 2008, 57, 3169–3176. [Google Scholar] [CrossRef] [Green Version]
- Emery, M.; Schorderet, D.F.; Roduit, R. Acute hypoglycemia induces retinal cell death in mouse. PLoS ONE 2011, 6, e21586. [Google Scholar] [CrossRef]
- Emery, M.; Nanchen, N.; Preitner, F.; Ibberson, M.; Roduit, R. Biological Characterization of Gene Response to Insulin-Induced Hypoglycemia in Mouse Retina. PLoS ONE 2016, 11, e0150266. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Kohno, K.; Miura, M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med. 2004, 10, 98–102. [Google Scholar] [CrossRef]
- Yang, Y.; Dieter, M.Z.; Chen, Y.; Shertzer, H.G.; Nebert, D.W.; Dalton, T.P. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(-/-) knockout mouse. Novel model system for a severely compromised oxidative stress response. J. Biol. Chem. 2002, 277, 49446–49452. [Google Scholar] [CrossRef] [Green Version]
- Romijn, H.J.; Jong, B.M.d.; Ruijter, J.M. A procedure for culturing rat neocortex explants in a serum-free nutrient medium. J. Neurosci. Meth. 1988, 23, 75–83. [Google Scholar] [CrossRef]
- Tan, E.; Ding, X.Q.; Saadi, A.; Agarwal, N.; Naash, M.I.; Al-Ubaidi, M.R. Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Invest. Ophthalmol. Vis. Sci. 2004, 45, 764–768. [Google Scholar] [CrossRef] [Green Version]
- Cook, S.; Hugli, O.; Egli, M.; Menard, B.; Thalmann, S.; Sartori, C.; Perrin, C.; Nicod, P.; Thorens, B.; Vollenweider, P.; et al. Partial gene deletion of endothelial nitric oxide synthase predisposes to exaggerated high-fat diet-induced insulin resistance and arterial hypertension. Diabetes 2004, 53, 2067–2072. [Google Scholar] [CrossRef] [Green Version]
- Roh, Y.-J.; Moon, C.; Kim, S.; Park, M.; Bae, Y.; Chun, M.-H.; Moon, J.-I. Glutathione depletion induces differential apoptosis in cells of mouse retina, in vivo. Neurosci. Lett. 2007, 417, 266–270. [Google Scholar] [CrossRef]
- Balmer, D.; Emery, M.; Andreux, P.; Auwerx, J.; Ginet, V.; Puyal, J.; Schorderet, D.F.; Roduit, R. Autophagy defect is associated with low glucose-induced apoptosis in 661W photoreceptor cells. PLoS ONE 2013, 8, e74162. [Google Scholar] [CrossRef]
- Jing, G.; Wang, J.J.; Zhang, S.X. ER stress and apoptosis: A new mechanism for retinal cell death. Exp. Diabetes Res. 2012, 2012, 589589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C. Glutathione-and non-glutathione-based oxidant control in the endoplasmic reticulum. J. Cell Sci. 2011, 124, 847–855. [Google Scholar] [CrossRef] [Green Version]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef] [Green Version]
- Badiola, N.; Penas, C.; Miñano-Molina, A.; Barneda-Zahonero, B.; Fadó, R.; Sánchez-Opazo, G.; Comella, J.X.; Sabriá, J.; Zhu, C.; Blomgren, K.; et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011, 2, e149. [Google Scholar] [CrossRef] [Green Version]
- Iurlaro, R.; Püschel, F.; León-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramón, D.; Lucendo, E.; Muñoz-Pinedo, C. Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Mol. Cell. Biol. 2017, 37, e00479-16. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.-R.; Choi, J.A.; Koh, J.-Y.; Yoon, Y.H. Ursodeoxycholic Acid Attenuates Endoplasmic Reticulum Stress-Related Retinal Pericyte Loss in Streptozotocin-Induced Diabetic Mice. J. Diabetes Res. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.J.; Yu, Q.; Wang, M.; Zhang, S.X. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 2009, 583, 1521–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Fruttiger, M.; Zhu, L.; Chung, S.H.; Barnett, N.L.; Kirk, J.K.; Lee, S.; Coorey, N.J.; Killingsworth, M.; Sherman, L.S.; et al. Conditional Müller Cell Ablation Causes Independent Neuronal and Vascular Pathologies in a Novel Transgenic Model. J. Neurosci. 2012, 32, 15715–15727. [Google Scholar] [CrossRef] [PubMed]
- Banki, K.; Hutter, E.; Colombo, E.; Gonchoroff, N.J.; Perl, A. Glutathione Levels and Sensitivity to Apoptosis Are Regulated by Changes in Transaldolase Expression. J. Biol. Chem. 1996, 271, 32994–33001. [Google Scholar] [CrossRef] [Green Version]
- Chiba, T.; Takahashi, S.; Sato, N.; Ishii, S.; Kikuchi, K. Fas-mediated apoptosis is modulated by intracellular glutathione in human T cells. Eur. J. Immunol. 1996, 26, 1164–1169. [Google Scholar] [CrossRef]
- Wang, M.; Lau, L.-I.; Sreekumar, P.G.; Spee, C.; Hinton, D.R.; Sadda, S.R.; Kannan, R. Characterization and Regulation of Carrier Proteins of Mitochondrial Glutathione Uptake in Human Retinal Pigment Epithelium Cells. Invest. Ophth. Vis. Sci. 2019, 60, 500–516. [Google Scholar] [CrossRef] [Green Version]
- García-Giménez, J.L.; Markovic, J.; Dasí, F.; Queval, G.; Schnaubelt, D.; Foyer, C.H.; Pallardó, F.V. Nuclear glutathione. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 3304–3316. [Google Scholar] [CrossRef]
- Ponsero, A.J.; Igbaria, A.; Darch, M.A.; Miled, S.; Outten, C.E.; Winther, J.R.; Palais, G.; D’Autréaux, B.; Delaunay-Moisan, A.; Toledano, M.B. Endoplasmic Reticulum Transport of Glutathione by Sec61 Is Regulated by Ero1 and Bip. Mol. Cell 2017, 67, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthi, S.; Bulleid, N.J. Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J. Biol. Chem. 2004, 279, 39872–39879. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, M.; Chan, P.-S.; Kern, T.S.; Kowluru, R.A. Oxidative Damage in the Retinal Mitochondria of Diabetic Mice: Possible Protection by Superoxide Dismutase. Invest. Ophth. Vis. Sci. 2007, 48, 3805–3811. [Google Scholar] [CrossRef] [Green Version]
- Kritsiligkou, P.; Rand, J.D.; Weids, A.J.; Wang, X.; Kershaw, C.J.; Grant, C.M. Endoplasmic reticulum (ER) stress–induced reactive oxygen species (ROS) are detrimental for the fitness of a thioredoxin reductase mutant. J. Biol. Chem. 2018, 293, 11984–11995. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, S.; Bhootada, Y.; Gorbatyuk, O.; Gorbatyuk, M. Caspase-7 ablation modulates UPR, reprograms TRAF2-JNK apoptosis and protects T17M rhodopsin mice from severe retinal degeneration. Cell Death Dis. 2013, 4, e528. [Google Scholar] [CrossRef] [Green Version]
- Jeng, Y.-Y.; Lin, N.-T.; Chang, P.-H.; Huang, Y.-P.; Pang, V.F.; Liu, C.-H.; Lin, C.-T. Retinal ischemic injury rescued by sodium 4-phenylbutyrate in a rat model. Exp. Eye Res. 2007, 84, 486–492. [Google Scholar] [CrossRef]
- Kammoun, H.L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferré, P.; Foufelle, F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Investig. 2009, 119, 1201–1215. [Google Scholar] [CrossRef]
- Kang, M.-J.; Ryoo, H.D. Suppression of retinal degeneration in Drosophila by stimulation of ER-associated degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 17043–17048. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Hyttinen, J.M.T.; Toropainen, E.; Kaarniranta, K. Endoplasmic Reticulum Stress in Age-Related Macular Degeneration: Trigger for Neovascularization. Mol. Med. 2010, 16, 535–542. [Google Scholar] [CrossRef]
- Simó, R.; Stitt, A.W.; Gardner, T.W. Neurodegeneration in diabetic retinopathy: Does it really matter? Diabetologia 2018, 61, 1902–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fresia, D.; Cannizzaro, E.; Borgo, A.; Roduit, R. GSH-Independent Induction of ER Stress during Hypoglycaemia in the Retinal Cells of Mice. J. Clin. Med. 2021, 10, 2529. https://doi.org/10.3390/jcm10112529
Fresia D, Cannizzaro E, Borgo A, Roduit R. GSH-Independent Induction of ER Stress during Hypoglycaemia in the Retinal Cells of Mice. Journal of Clinical Medicine. 2021; 10(11):2529. https://doi.org/10.3390/jcm10112529
Chicago/Turabian StyleFresia, Daria, Enrica Cannizzaro, Angelica Borgo, and Raphaël Roduit. 2021. "GSH-Independent Induction of ER Stress during Hypoglycaemia in the Retinal Cells of Mice" Journal of Clinical Medicine 10, no. 11: 2529. https://doi.org/10.3390/jcm10112529
APA StyleFresia, D., Cannizzaro, E., Borgo, A., & Roduit, R. (2021). GSH-Independent Induction of ER Stress during Hypoglycaemia in the Retinal Cells of Mice. Journal of Clinical Medicine, 10(11), 2529. https://doi.org/10.3390/jcm10112529