
The Right Ventricle in COVID-19


Abstract
:1. Introduction
2. Methods
3. Right Ventricular Dysfunction in COVID-19: What Is the Evidence?

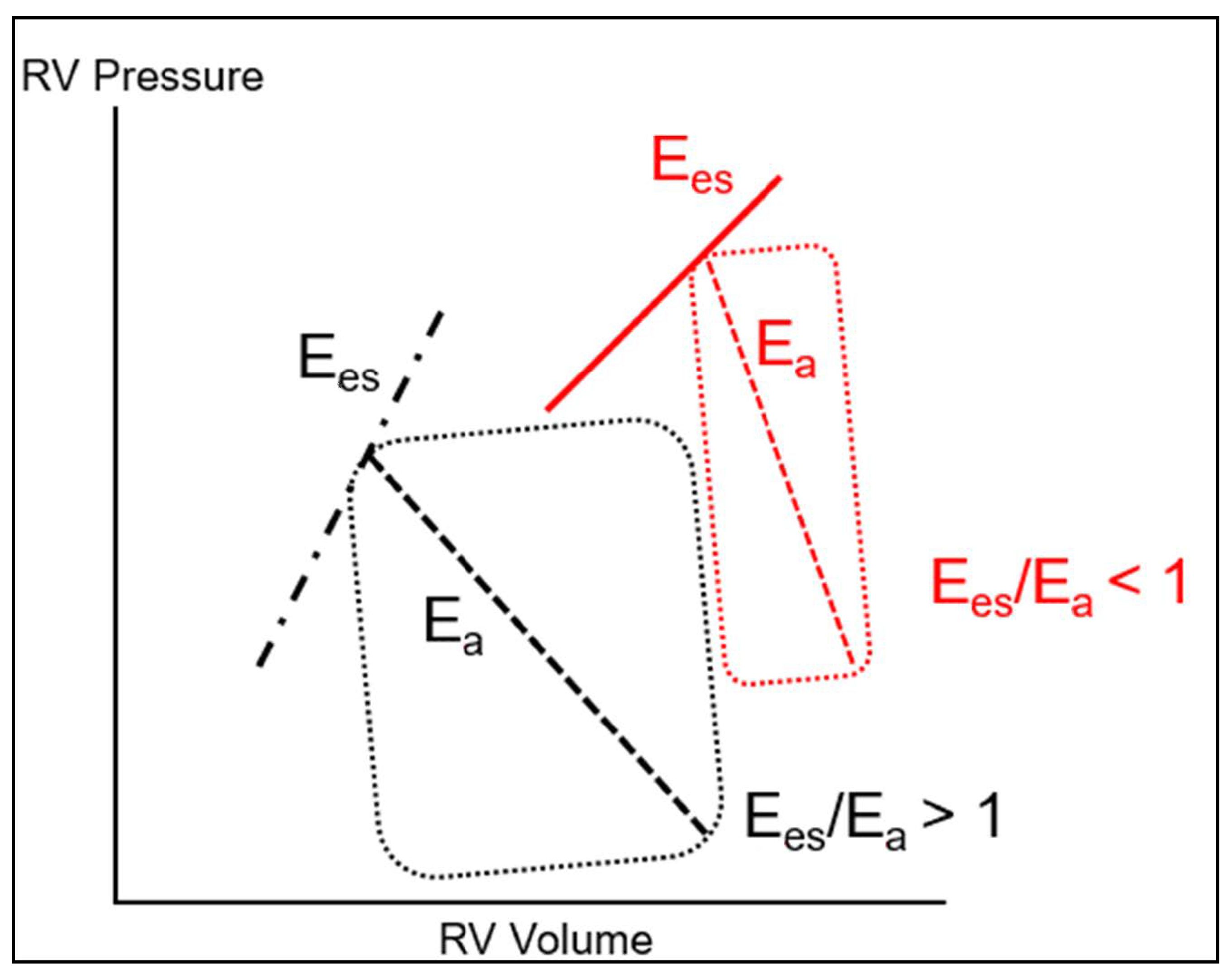{kind=link}
{kind=link}
{kind=link}
| N, Age, % Male | Patients | Echo Data | Pulmonary Circulation | Main Prognostic Findings | Ref |
|---|---|---|---|---|---|
| 29 (64) 70% | ICU |
|
|
| Beyls et al. [44] |
| 30 (61) 65% | ICU |
|
|
| Gonzalez et al. [45] |
| 35 (72) 79% | ICU |
|
|
| Stockenhuber et al. [46] |
| 12 (57) 42% | ICU |
|
|
| Krishnamoorthy et al. [47] |
| 32 (56) 66% | ICU |
|
|
| Gibson et al. [48] |
| 100 (59) 40% | ICU 22% |
|
|
| Baycan et al. [49] |
| 128 (61) 48% | ICU 15% |
|
|
| Zhang et al. [36] |
| 120 (61) 57% | ICU 21% |
|
|
| Li et al. [34] |
| 49 (66) 63% | ICU |
|
|
| Bursi et al. [50] |
| 214 (69) 55% | Non ICU |
|
|
| Lassen et al. [35] |
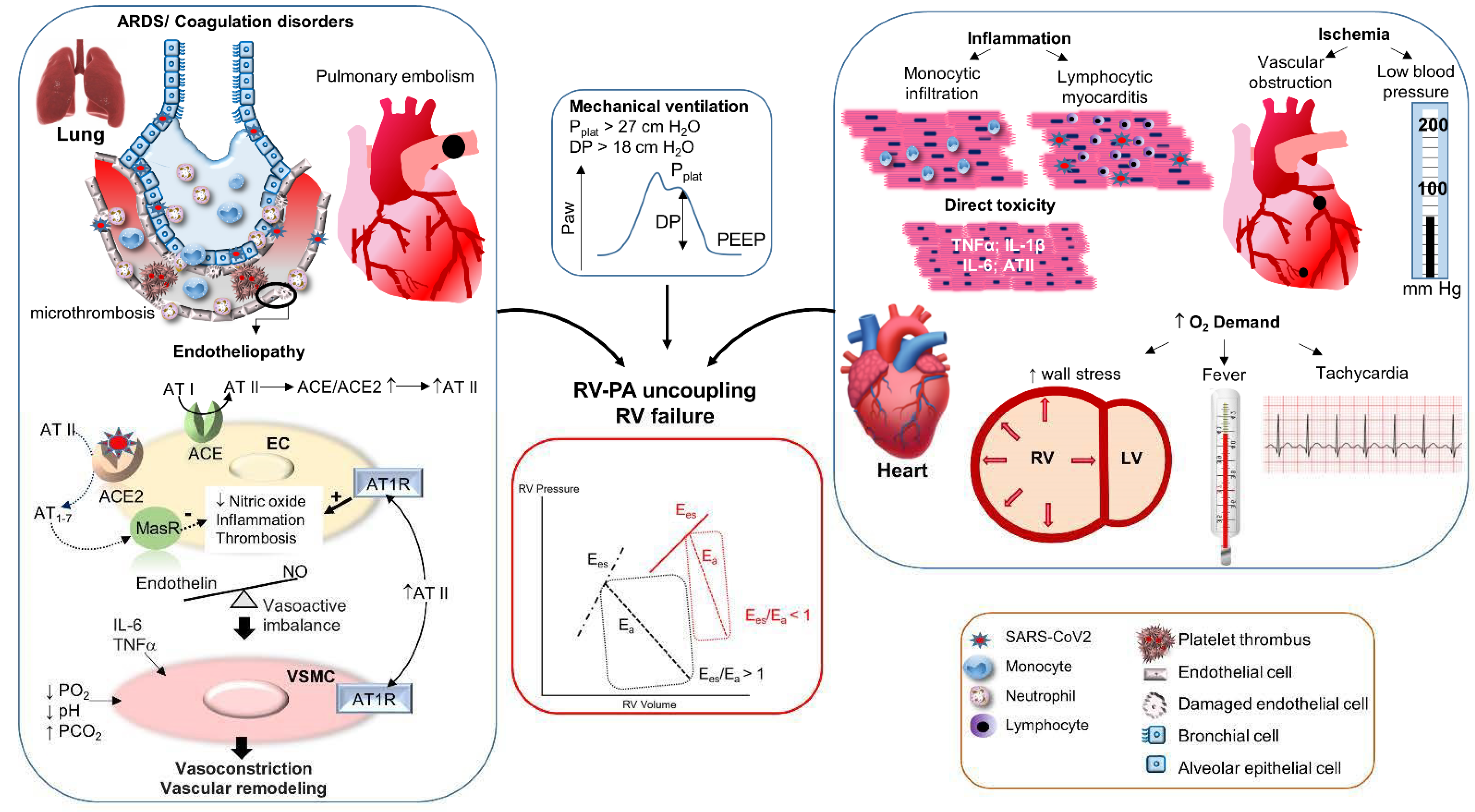
4. Pathophysiology of RV Dysfunction in COVID-19
4.1. Physiology and Pathophysiology of the Right Ventricle
4.2. Mechanisms of RV-PA Uncoupling in COVID-19
4.2.1. Increase of the Pulmonary Hydraulic Load
Pulmonary Vascular Obstruction
Disturbances of Pulmonary Vasomotor Tone
- Hypoxic vasoconstriction
- 2.
- Hypercapnic acidosis
- 3.
- Angiotensin II-mediated vasoconstriction
- 4.
- Vasoactive mediator imbalance
- 5.
- Additional mechanisms of increased pulmonary vascular tone
Mechanical Ventilation
4.2.2. Reduction of Right Ventricular Contractility
Evidence of Myocardial Injury in COVID-19
Potential Mechanisms of Cardiac Injury and Dysfunction in COVID-19
- Myocardial inflammation
- 2.
- Myocardial ischemic injury
- 3.
- Dysregulated RAS and inflammatory cytokines
5. Treatment of COVID-19-Associated Right Ventricular Dysfunction
5.1. Anticoagulation
5.2. Anti-Inflammatory Therapies
5.3. Specific Management of RV Failure
- Perfusion pressure
- 2.
- Volume management
- 3.
- Afterload reduction
- 4.
- Inotropic support
- 5.
- Extracorporeal membrane oxygenation (ECMO)
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Creel-Bulos, C.; Hockstein, M.; Amin, N.; Melhem, S.; Truong, A.; Sharifpour, M. Acute Cor Pulmonale in Critically Ill Patients with Covid-19. N. Engl. J. Med. 2020, 382, e70. [Google Scholar] [CrossRef]
- Rauch, S.; Regli, I.B.; Clara, A.; Seraglio, P.M.; Bock, M.; Poschenrieder, F.; Resch, M. Right ventricular myopericarditis in COVID-19: A call for regular echocardiography. Minerva Anestesiol. 2020, 86, 1253–1254. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, G.; Fazzari, F.; Cozzi, O.; Maurina, M.; Bragato, R.; D’Orazio, F.; Torrisi, C.; Lanza, E.; Indolfi, E.; Donghi, V.; et al. Risk factors for myocardial injury and death in patients with COVID-19: Insights from a cohort study with chest computed tomography. Cardiovasc. Res. 2020, 116, 2239–2246. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kiernan, M.S.; Bernstein, D.; Bozkurt, B.; Jacob, M.; Kapur, N.K.; Kociol, R.D.; Lewis, E.F.; Mehra, M.R.; Pagani, F.D.; et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement from the American Heart Association. Circulation 2018, 137, e578–e622. [Google Scholar] [CrossRef] [PubMed]
- García-Cruz, E.; Manzur-Sandoval, D.; Baeza-Herrera, L.A.; Díaz-Méndez, A.; López-Zamora, A.; González-Ruiz, F.; Ángel, R.E.; Melano-Carranza, E.; Rojas-Velasco, G.; Álvarez-Álvarez, R.J.; et al. Acute right ventricular failure in COVID-19 infection: A case series. J. Cardiol. Cases 2021. [Google Scholar] [CrossRef] [PubMed]
- Schott, J.P.; Mertens, A.N.; Bloomingdale, R.; O’Connell, T.F.; Gallagher, M.J.; Dixon, S.; Abbas, A.E. Transthoracic echocardiographic findings in patients admitted with SARS-CoV-2 infection. Echocardiography 2020, 37, 1551–1556. [Google Scholar] [CrossRef]
- Argulian, E.; Sud, K.; Vogel, B.; Bohra, C.; Garg, V.P.; Talebi, S.; Lerakis, S.; Narula, J. Right Ventricular Dilation in Hospitalized Patients with COVID-19 Infection. JACC Cardiovasc. Imaging 2020, 13, 2459–2461. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-L.; Zheng, J.-B.; Jin, Y.; Tang, R.; Li, M.; Xiu, C.-H.; Dai, Q.-Q.; Zuo, S.; Wang, H.-Q.; Wang, H.-L.; et al. Acute right ventricular dysfunction in severe COVID-19 pneumonia. Rev. Cardiovasc. Med. 2020, 21, 635–641. [Google Scholar] [CrossRef]
- Szekely, Y.; Lichter, Y.; Taieb, P.; Banai, A.; Hochstadt, A.; Merdler, I.; Oz, A.G.; Rothschild, E.; Baruch, G.; Peri, Y.; et al. Spectrum of Cardiac Manifestations in COVID-19: A Systematic Echocardiographic Study. Circulation 2020, 142, 342–353. [Google Scholar] [CrossRef]
- Zeng, J.-H.; Wu, W.-B.; Qu, J.-X.; Wang, Y.; Dong, C.-F.; Luo, Y.-F.; Zhou, D.; Feng, W.-X.; Feng, C. Cardiac manifestations of COVID-19 in Shenzhen, China. Infection 2020, 48, 861–870. [Google Scholar] [CrossRef]
- Van den Heuvel, F.M.A.; Vos, J.L.; Koop, Y.; Van Dijk, A.P.J.; Duijnhouwer, A.L.; De Mast, Q.; Van De Veerdonk, F.L.; Bosch, F.; Kok, B.; Netea, M.G.; et al. Cardiac function in relation to myocardial injury in hospitalised patients with COVID-19. Neth. Hear. J. 2020, 28, 410–417. [Google Scholar] [CrossRef]
- Vasudev, R.; Guragai, N.; Habib, H.; Hosein, K.; Virk, H.; Goldfarb, I.; Bikkina, M.; Shamoon, F.; Pullatt, R. The utility of bedside echocardiography in critically ill COVID-19 patients: Early observational findings from three Northern New Jersey hospitals. Echocardiography 2020, 37, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.; Scarafile, R.; Riegler, L.; Liccardo, B.; Crescibene, F.; Cocchia, R.; Bossone, E. Right Ventricular Function and Pulmonary Pressures as Independent Predictors of Survival in Patients with COVID-19 Pneumonia. JACC Cardiovasc. Imaging 2020, 13, 2467–2468. [Google Scholar] [CrossRef] [PubMed]
- Pagnesi, M.; Baldetti, L.; Beneduce, A.; Calvo, F.; Gramegna, M.; Pazzanese, V.; Ingallina, G.; Napolano, A.; Finazzi, R.; Ruggeri, A.; et al. Pulmonary hypertension and right ventricular involvement in hospitalised patients with COVID-19. Heart 2020, 106, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Rath, D.; Petersen-Uribe, A.; Avdiu, A.; Witzel, K.; Jaeger, P.; Zdanyte, M.; Heinzmann, D.; Tavlaki, E.; Müller, K.; Gawaz, M.P. Impaired cardiac function is associated with mortality in patients with acute COVID-19 infection. Clin. Res. Cardiol. 2020, 109, 1491–1499. [Google Scholar] [CrossRef]
- Jain, S.S.; Liu, Q.; Raikhelkar, J.; Fried, J.; Elias, P.; Poterucha, T.J.; DeFilippis, E.M.; Rosenblum, H.; Wang, E.Y.; Redfors, B.; et al. Indications for and Findings on Transthoracic Echocardiography in COVID-19. J. Am. Soc. Echocardiogr. 2020, 33, 1278–1284. [Google Scholar] [CrossRef]
- Moody, W.E.; Mahmoud-Elsayed, H.M.; Senior, J.; Gul, U.; Khan-Kheil, A.M.; Horne, S.; Banerjee, A.; Bradlow, W.M.; Huggett, R.; Hothi, S.S.; et al. Impact of Right Ventricular Dysfunction on Mortality in Patients Hospitalized With COVID-19, According to Race. CJC Open 2021, 3, 91–100. [Google Scholar] [CrossRef]
- Dweck, M.R.; Bularga, A.; Hahn, R.T.; Bing, R.; Lee, K.K.; Chapman, A.R.; White, A.; Di Salvo, G.; Sade, L.E.; Pearce, K.; et al. Global evaluation of echocardiography in patients with COVID-19. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 949–958. [Google Scholar] [CrossRef]
- Mahmoud-Elsayed, H.M.; Moody, W.E.; Bradlow, W.M.; Khan-Kheil, A.M.; Senior, J.; Hudsmith, L.E.; Steeds, R.P. Echocardiographic Findings in Patients With COVID-19 Pneumonia. Can. J. Cardiol. 2020, 36, 1203–1207. [Google Scholar] [CrossRef]
- Kim, J.; Volodarskiy, A.; Sultana, R.; Pollie, M.P.; Yum, B.; Nambiar, L.; Tafreshi, R.; Mitlak, H.W.; RoyChoudhury, A.; Horn, E.M.; et al. Prognostic Utility of Right Ventricular Remodeling Over Conventional Risk Stratification in Patients with COVID-19. J. Am. Coll. Cardiol. 2020, 76, 1965–1977. [Google Scholar] [CrossRef]
- Bleakley, C.; Singh, S.; Garfield, B.; Morosin, M.; Surkova, E.; Mandalia, M.S.; Dias, B.; Androulakis, E.; Price, L.C.; McCabe, C.; et al. Right ventricular dysfunction in critically ill COVID-19 ARDS. Int. J. Cardiol. 2021, 327, 251–258. [Google Scholar] [CrossRef]
- Sud, K.; Vogel, B.; Bohra, C.; Garg, V.; Talebi, S.; Lerakis, S.; Narula, J.; Argulian, E. Echocardiographic Findings in Patients with COVID-19 with Significant Myocardial Injury. J. Am. Soc. Echocardiogr. 2020, 33, 1054–1055. [Google Scholar] [CrossRef] [PubMed]
- Badkoubeh, R.S.; Khoshavi, M.; Far, V.L.; Mehrakizadeh, A.; Eslami, M.; Salahshour, F.; Sardari, A.; Safari, S.; Larti, F.; Nissen, S. Imaging data in COVID-19 patients: Focused on echocardiographic findings. Int. J. Cardiovasc. Imaging 2021, 37, 1629–1636. [Google Scholar] [CrossRef]
- Scudiero, F.; Silverio, A.; Di Maio, M.; Russo, V.; Citro, R.; Personeni, D.; Cafro, A.; D’Andrea, A.; Attena, E.; Pezzullo, S.; et al. Pulmonary embolism in COVID-19 patients: Prevalence, predictors and clinical outcome. Thromb. Res. 2021, 198, 34–39. [Google Scholar] [CrossRef]
- D’Alto, M.; Marra, A.M.; Severino, S.; Salzano, A.; Romeo, E.; De Rosa, R.; Stagnaro, F.M.; Pagnano, G.; Verde, R.; Murino, P.; et al. Right ventricular-arterial uncoupling independently predicts survival in COVID-19 ARDS. Crit. Care 2020, 24, 670. [Google Scholar] [CrossRef]
- Giustino, G.; Croft, L.B.; Stefanini, G.G.; Bragato, R.; Silbiger, J.J.; Vicenzi, M.; Danilov, T.; Kukar, N.; Shaban, N.; Kini, A.; et al. Characterization of Myocardial Injury in Patients with COVID-19. J. Am. Coll. Cardiol. 2020, 76, 2043–2055. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, C.; Bonizzoli, M.; Batacchi, S.; Cianchi, G.; Franci, A.; Fulceri, G.E.; Peris, A. Cardiac Involvment in COVID-19–Related Acute Respiratory Distress Syndrome. Am. J. Cardiol. 2020, 132, 147–149. [Google Scholar] [CrossRef]
- Bagate, F.; Masi, P.; D’Humières, T.; Al-Assaad, L.; Chakra, L.A.; Razazi, K.; de Prost, N.; Carteaux, G.; Derumeaux, G.; Dessap, A.M. Advanced echocardiographic phenotyping of critically ill patients with coronavirus-19 sepsis: A prospective cohort study. J. Intensiv. Care 2021, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Rudski, L.G.; Lai, W.W.; Afilalo, J.; Hua, L.; Handschumacher, M.D.; Chandrasekaran, K.; Solomon, S.D.; Louie, E.K.; Schiller, N.B. Guidelines for the Echocardiographic Assessment of the Right Heart in Adults: A Report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J. Am. Soc. Echocardiogr. 2010, 23, 685–713. [Google Scholar] [CrossRef]
- Wibowo, A.; Pranata, R.; Astuti, A.; Tiksnadi, B.B.; Martanto, E.; Martha, J.W.; Purnomowati, A.; Akbar, M.R. Left and right ventricular longitudinal strains are associated with poor outcome in COVID-19: A systematic review and meta-analysis. J. Intensiv. Care 2021, 9, 9. [Google Scholar] [CrossRef]
- Carluccio, E.; Biagioli, P.; Alunni, G.; Murrone, A.; Zuchi, C.; Coiro, S.; Riccini, C.; Mengoni, A.; D’Antonio, A.; Ambrosio, G. Prognostic Value of Right Ventricular Dysfunction in Heart Failure With Reduced Ejection Fraction: Superiority of Longitudinal Strain Over Tricuspid Annular Plane Systolic Excursion. Circ. Cardiovasc. Imaging 2018, 11, e006894. [Google Scholar] [CrossRef] [Green Version]
- Bonizzoli, M.; Cipani, S.; Lazzeri, C.; Chiostri, M.; Ballo, P.; Sarti, A.; Peris, A. Speckle tracking echocardiography and right ventricle dysfunction in acute respiratory distress syndrome: A pilot study. Echocardiography 2018, 35, 1982–1987. [Google Scholar] [CrossRef]
- Lemarié, J.; Maigrat, C.-H.; Kimmoun, A.; Dumont, N.; Bollaert, P.-E.; Selton-Suty, C.; Gibot, S.; Huttin, O. Feasibility, reproducibility and diagnostic usefulness of right ventricular strain by 2-dimensional speckle-tracking echocardiography in ARDS patients: The ARD strain study. Ann. Intensiv. Care 2020, 10, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, H.; Zhu, S.; Xie, Y.; Wang, B.; He, L.; Zhang, D.; Zhang, Y.; Yuan, H.; Wu, C.; et al. Prognostic Value of Right Ventricular Longitudinal Strain in Patients with COVID-19. JACC Cardiovasc. Imaging 2020, 13, 2287–2299. [Google Scholar] [CrossRef] [PubMed]
- Lassen, M.C.H.; Skaarup, K.G.; Lind, J.N.; Alhakak, A.S.; Sengeløv, M.; Nielsen, A.B.; Espersen, C.; Ravnkilde, K.; Hauser, R.; Schöps, L.B.; et al. Echocardiographic abnormalities and predictors of mortality in hospitalized COVID-19 patients: The ECHOVID-19 study. ESC Hear. Fail. 2020, 7, 4189–4197. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, W.; Wu, C.; Zhang, Y.; Cui, L.; Xie, Y.; Wang, B.; He, L.; Yuan, H.; Zhang, Y.; et al. Prognostic Value of Right Ventricular Ejection Fraction Assessed by 3D Echocardiography in COVID-19 Patients. Front. Cardiovasc. Med. 2021, 8, 641088. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.; Hu, B.; Zhang, Y.; Wang, H.; Zhou, X.; Hu, W.; Cheng, Y.; Yan, J.; Ping, H.; Zhou, Q. Suspected myocardial injury in patients with COVID-19: Evidence from front-line clinical observation in Wuhan, China. Int. J. Cardiol. 2020, 311, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Kroboth, S.; Ignatowski, D.; Khandheria, B.K. Seroprevalence of SARS-CoV-2 Antibody in Echocardiography and Stress Laboratory. J. Patient. Cent. Res. Rev. 2021, 8, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Orde, S.; Slama, M.; Hilton, A.; Yastrebov, K.; McLean, A. Pearls and pitfalls in comprehensive critical care echocardiography. Crit. Care 2017, 21, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negishi, T.; Negishi, K.; Thavendiranathan, P.; Cho, G.-Y.; Popescu, B.A.; Vinereanu, D.; Kurosawa, K.; Penicka, M.; Marwick, T.H.; Aakhus, S.; et al. Effect of Experience and Training on the Concordance and Precision of Strain Measurements. JACC Cardiovasc. Imaging 2017, 10, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Willder, J.M.; McCall, P.; Messow, C.-M.; Gillies, M.; Berry, C.; Shelley, B. Study protocol for COVID-RV: A multicentre prospective observational cohort study of right ventricular dysfunction in ventilated patients with COVID-19. BMJ Open 2021, 11, e042098. [Google Scholar] [CrossRef] [PubMed]
- Beyls, C.; Bohbot, Y.; Huette, P.; Booz, T.; Daumin, C.; Abou-Arab, O.; Mahjoub, Y. Usefulness of Right Ventricular Longitudinal Shortening Fraction to Detect Right Ventricular Dysfunction in Acute Cor Pulmonale Related to COVID-19. J. Cardiothorac. Vasc. Anesth. 2021. [Google Scholar] [CrossRef]
- Gonzalez, F.; Gomes, R.; Bacariza, J.; Michard, F. Could strain echocardiography help to assess systolic function in critically ill COVID-19 patients? J. Clin. Monit. Comput. 2021, 1–6. [Google Scholar] [CrossRef]
- Stockenhuber, A.; Vrettos, A.; Androschuck, V.; George, M.; Robertson, C.; Bowers, N.; Clifford, P.; Firoozan, S. A pilot study on right ventricular longitudinal strain as a predictor of outcome in COVID-19 patients with evidence of cardiac involvement. Echocardiography 2021, 38, 222–229. [Google Scholar] [CrossRef]
- Krishnamoorthy, P.; Croft, L.B.; Ro, R.; Anastasius, M.; Zhao, W.; Giustino, G.; Argulian, E.; Goldman, M.E.; Sharma, S.K.; Kini, A.; et al. Biventricular strain by speckle tracking echocardiography in COVID-19: Findings and possible prognostic implications. Future Cardiol. 2020. [Google Scholar] [CrossRef]
- Gibson, L.E.; Di Fenza, R.; Lang, M.; Capriles, M.I.; Li, M.D.; Kalpathy-Cramer, J.; Little, B.P.; Arora, P.; Mueller, A.L.; Ichinose, F.; et al. Right Ventricular Strain Is Common in Intubated COVID-19 Patients and Does Not Reflect Severity of Respiratory Illness. J. Intensiv. Care Med. 2021. [Google Scholar] [CrossRef]
- Baycan, O.F.; Barman, H.A.; Atici, A.; Tatlisu, A.; Bolen, F.; Ergen, P.; Icten, S.; Gungor, B.; Caliskan, M. Evaluation of biventricular function in patients with COVID-19 using speckle tracking echocardiography. Int. J. Cardiovasc. Imaging 2021, 37, 135–144. [Google Scholar] [CrossRef]
- Bursi, F.; Santangelo, G.; Sansalone, D.; Valli, F.; Vella, A.M.; Toriello, F.; Barbieri, A.; Carugo, S. Prognostic utility of quantitative offline 2D-echocardiography in hospitalized patients with COVID-19 disease. Echocardiography 2020, 37, 2029–2039. [Google Scholar] [CrossRef]
- Dandel, M. Heart–lung interactions in COVID-19: Prognostic impact and usefulness of bedside echocardiography for monitoring of the right ventricle involvement. Heart Fail. Rev. 2021, 1–15. [Google Scholar] [CrossRef]
- Hassani, N.S.; Shojaee, A.; Khodaprast, Z.; Sepahvandi, R.; Shahrestanaki, E.; Rastad, H. Echocardiographic Features of Cardiac Injury Related to COVID-19 and Their Prognostic Value: A Systematic Review. J. Intensiv. Care Med. 2021, 36, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Martha, J.W.; Pranata, R.; Wibowo, A.; Lim, M.A. Tricuspid annular plane systolic excursion (TAPSE) measured by echocardiography and mortality in COVID-19: A systematic review and meta-analysis. Int. J. Infect. Dis. 2021, 105, 351–356. [Google Scholar] [CrossRef]
- Vieillard-Baron, A.; Naeije, R.; Haddad, F.; Bogaard, H.J.; Bull, T.M.; Fletcher, N.; Lahm, T.; Magder, S.; Orde, S.; Schmidt, G.; et al. Diagnostic workup, etiologies and management of acute right ventricle failure: A state-of-the-art paper. Intensive Care Med. 2018, 44, 774–790. [Google Scholar] [CrossRef]
- Dandel, M. Cardiac manifestations of COVID-19 infection: The role of echocardiography in patient management. Infection 2021, 49, 187–189. [Google Scholar] [CrossRef]
- Noordegraaf, A.V.; Chin, K.M.; Haddad, F.; Hassoun, P.M.; Hemnes, A.R.; Hopkins, S.R.; Kawut, S.M.; Langleben, D.; Lumens, J.; Naeije, R. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: An update. Eur. Respir. J. 2019, 53, 1801900. [Google Scholar] [CrossRef] [PubMed]
- Noordegraaf, A.V.; Westerhof, B.E.; Westerhof, N. The Relationship between the Right Ventricle and its Load in Pulmonary Hypertension. J. Am. Coll. Cardiol. 2017, 69, 236–243. [Google Scholar] [CrossRef]
- Naeije, R. Physiology of the Pulmonary Circulation and the Right Heart. Curr. Hypertens. Rep. 2013, 15, 623–631. [Google Scholar] [CrossRef]
- Chemla, D.; Castelain, V.; Zhu, K.; Papelier, Y.; Creuzé, N.; Hoette, S.; Parent, F.; Simonneau, G.; Humbert, M.; Hervé, P. Estimating Right Ventricular Stroke Work and the Pulsatile Work Fraction in Pulmonary Hypertension. Chest 2013, 143, 1343–1350. [Google Scholar] [CrossRef]
- Hoette, S.; Creuzé, N.; Günther, S.; Montani, D.; Savale, L.; Jaïs, X.; Parent, F.; Sitbon, O.; Rochitte, C.E.; Simonneau, G.; et al. RV Fractional Area Change and TAPSE as Predictors of Severe Right Ventricular Dysfunction in Pulmonary Hypertension: A CMR Study. Lung 2018, 196, 157–164. [Google Scholar] [CrossRef]
- Saouti, N.; Westerhof, N.; Helderman, F.; Marcus, J.T.; Boonstra, A.; Postmus, P.E.; Vonk-Noordegraaf, A. Right Ventricular Oscillatory Power Is a Constant Fraction of Total Power Irrespective of Pulmonary Artery Pressure. Am. J. Respir. Crit. Care Med. 2010, 182, 1315–1320. [Google Scholar] [CrossRef] [Green Version]
- Zorzi, M.-F.; Cancelli, E.; Rusca, M.; Kirsch, M.; Yerly, P.; Liaudet, L.; Foetisch, E. The prognostic value of pulmonary artery compliance in cardiogenic shock. Pulm. Circ. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jenner, W.J.; Kanji, R.; Mirsadraee, S.; Gue, Y.X.; Price, S.; Prasad, S.; Gorog, D.A. Thrombotic complications in 2928 patients with COVID-19 treated in intensive care: A systematic review. J. Thromb. Thrombolysis 2021, 51, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Kaptein, F.; Stals, M.; Grootenboers, M.; Braken, S.; Burggraaf, J.; van Bussel, B.; Cannegieter, S.; Cate, H.T.; Endeman, H.; Gommers, D.; et al. Incidence of thrombotic complications and overall survival in hospitalized patients with COVID-19 in the second and first wave. Thromb. Res. 2021, 199, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Meziani, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.V.; Arachchillage, D.J.; Ridge, C.A.; Bianchi, P.; Doyle, J.F.; Garfield, B.; Ledot, S.; Morgan, C.; Passariello, M.; Price, S.; et al. Pulmonary Angiopathy in Severe COVID-19: Physiologic, Imaging, and Hematologic Observations. Am. J. Respir. Crit. Care Med. 2020, 202, 690–699. [Google Scholar] [CrossRef]
- García-Ortega, A.; Oscullo, G.; Calvillo, P.; López-Reyes, R.; Méndez, R.; Gómez-Olivas, J.D.; Bekki, A.; Fonfría, C.; Trilles-Olaso, L.; Zaldívar, E.; et al. Incidence, risk factors, and thrombotic load of pulmonary embolism in patients hospitalized for COVID-19 infection. J. Infect. 2021, 82, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Van Dam, L.; Kroft, L.; van der Wal, L.; Cannegieter, S.; Eikenboom, J.; de Jonge, E.; Huisman, M.; Klok, F. Clinical and computed tomography characteristics of COVID-19 associated acute pulmonary embolism: A different phenotype of thrombotic disease? Thromb. Res. 2020, 193, 86–89. [Google Scholar] [CrossRef]
- McGonagle, D.; Bridgewood, C.; Ramanan, A.V.; Meaney, J.F.M.; Watad, A. COVID-19 vasculitis and novel vasculitis mimics. Lancet. Rheumatol. 2021, 3, e224–e233. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Bösmüller, H.; Matter, M.; Fend, F.; Tzankov, A. The pulmonary pathology of COVID-19. Virchows Archiv 2021, 478, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2021, 18, 194–209. [Google Scholar] [CrossRef]
- García-Ortega, A.; de la Rosa, D.; Oscullo, G.; Castillo-Villegas, D.; López-Reyes, R.; Martínez-García, M.A. Coagulation disorders and thromboembolic disease in COVID-19: Review of current evidence in search of a better approach. J. Thorac. Dis. 2021, 13, 1239–1255. [Google Scholar] [CrossRef]
- Ciceri, F.; Beretta, L.; Scandroglio, A.M.; Colombo, S.; Landoni, G.; Ruggeri, A.; Zangrillo, A.; Peccatori, J.; D’Angelo, A.; de Cobelli, S. Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): An atypical acute respiratory distress syndrome working hypothesis. Crit. Care Resusc. 2020, 22, 95–97. [Google Scholar]
- McGonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar] [CrossRef]
- Iba, T.; Warkentin, T.E.; Thachil, J.; Levi, M.; Levy, J.H. Proposal of the Definition for COVID-19-Associated Coagulopathy. J. Clin. Med. 2021, 10, 191. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Paz, L.; Capodanno, D.; Montalescot, G.; Angiolillo, D.J. Coronavirus Disease 2019–Associated Thrombosis and Coagulopathy: Review of the Pathophysiological Characteristics and Implications for Antithrombotic Management. J. Am. Hear. Assoc. 2021, 10, e019650. [Google Scholar] [CrossRef]
- Thachil, J.; Cushman, M.; Srivastava, A. A proposal for staging COVID-19 coagulopathy. Res. Pr. Thromb. Haemost. 2020, 4, 731–736. [Google Scholar] [CrossRef]
- Rodríguez, C.; Luque, N.; Blanco, I.; Sebastian, L.; Barberà, J.A.; Peinado, V.I.; Tura-Ceide, O. Pulmonary Endothelial Dysfunction and Thrombotic Complications in Patients with COVID-19. Am. J. Respir. Cell Mol. Biol. 2020, 64, 407–415. [Google Scholar] [CrossRef]
- FitzGerald, E.S.; Chen, Y.; Fitzgerald, K.A.; Jamieson, A.M. Lung Epithelial Cell Transcriptional Regulation as a Factor in COVID-19 Associated Coagulopathies. Am. J. Respir. Cell Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Roberts, K.A.; Colley, L.; Agbaedeng, T.A.; Ellison-Hughes, G.M.; Ross, M.D. Vascular Manifestations of COVID-19—Thromboembolism and Microvascular Dysfunction. Front. Cardiovasc. Med. 2020, 7, 598400. [Google Scholar] [CrossRef]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021, 17, 46–64. [Google Scholar] [CrossRef]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Joppich, M.; Hoffknecht, M.; Gold, C.; Engel, A.; Polewka, V.; Muenchhoff, M.; et al. Vascular neutrophilic inflammation and immunothrombosis distinguish severe COVID-19 from influenza pneumonia. J. Thromb. Haemost. 2021, 19, 574–581. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Krishnamachary, B.; Cook, C.; Spikes, L.; Chalise, P.; Dhillon, N.K. The Potential Role of Extracellular Vesicles in COVID-19 Asso-ciated Endothelial injury and Pro-inflammation. medRxiv 2020. [Google Scholar] [CrossRef]
- Mancuso, P.; Gidaro, A.; Gregato, G.; Raveane, A.; Cremonesi, P.; Quarna, J.; Caccia, S.; Gusso, L.; Rusconi, S.; Giacomelli, A.; et al. Circulating endothelial progenitors are increased in COVID-19 patients and correlate with SARS-CoV-2 RNA in severe cases. J. Thromb. Haemost. 2020, 18, 2744–2750. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.; Mehra, M.R.; Schuepbach, R.A.; Ruzchitzka, F.; Moch, H. Faculty Opinions recommendation of Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, in press. [Google Scholar] [CrossRef]
- Liaudet, L.; Szabo, C. Blocking mineralocorticoid receptor with spironolactone may have a wide range of therapeutic actions in severe COVID-19 disease. Crit. Care 2020, 24, 318. [Google Scholar] [CrossRef]
- McLoughlin, P. Hypoxic pulmonary vasoconstriction: Building a solid base. Exp. Physiol. 2018, 103, 1181–1182. [Google Scholar] [CrossRef]
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P.T. Hypoxic Pulmonary Vasoconstriction. Physiol. Rev. 2012, 92, 367–520. [Google Scholar] [CrossRef] [PubMed]
- Repessé, X.; Vieillard-Baron, A. Right heart function during acute respiratory distress syndrome. Ann. Transl. Med. 2017, 5, 295. [Google Scholar] [CrossRef] [Green Version]
- Swenson, E.R.; Bärtsch, P. High-Altitude Pulmonary Edema. Compr. Physiol. 2012, 2, 2753–2773. [Google Scholar] [CrossRef] [PubMed]
- Habashi, N.M.; Camporota, L.; Gatto, L.A.; Nieman, G.F. Functional pathophysiology of SARS-CoV-2-induced acute lung injury and clinical implications. J. Appl. Physiol. 2021, 130, 877–891. [Google Scholar] [CrossRef]
- Busana, M.; Gasperetti, A.; Giosa, L.; Forleo, G.B.; Schiavone, M.; Mitacchione, G.; Bonino, C.; Villa, P.; Galli, M.; Tondo, C.; et al. Prevalence and outcome of silent hypoxemia in COVID-19. Minerva Anestesiol. 2021, 87, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Caplan, M.; Goutay, J.; Bignon, A.; Jaillette, E.; Favory, R.; Mathieu, D.; Parmentier-Decrucq, E.; Poissy, J.; Duburcq, T. Almitrine Infusion in Severe Acute Respiratory Syndrome Coronavirus 2-Induced Acute Respiratory Distress Syndrome: A Single-Center Observational Study. Crit. Care Med. 2021, 49, e191–e198. [Google Scholar] [CrossRef]
- Caravita, S.; Baratto, C.; Di Marco, F.; Calabrese, A.; Balestrieri, G.; Russo, F.; Faini, A.; Soranna, D.; Perego, G.B.; Badano, L.P.; et al. Haemodynamic characteristics ofCOVID-19 patients with acute respiratory distress syndrome requiring mechanical ventilation. An invasive assessment using right heart catheterization. Eur. J. Hear. Fail. 2020, 22, 2228–2237. [Google Scholar] [CrossRef] [PubMed]
- Dessap, A.M.; Boissier, F.; Charron, C.; Bégot, E.; Repessé, X.; Legras, A.; Brun-Buisson, C.; Vignon, P.; Vieillard-Baron, A. Acute cor pulmonale during protective ventilation for acute respiratory distress syndrome: Prevalence, predictors, and clinical impact. Intensive Care Med. 2016, 42, 862–870. [Google Scholar] [CrossRef]
- Zochios, V.; Parhar, K.K.S.; Tunnicliffe, W.; Roscoe, A.; Gao, F. The Right Ventricle in ARDS. Chest 2017, 152, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Revercomb, L.; Hanmandlu, A.; Wareing, N.; Akkanti, B.; Karmouty-Quintana, H. Mechanisms of Pulmonary Hypertension in Acute Respiratory Distress Syndrome (ARDS). Front. Mol. Biosci. 2020, 7, 624093. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1702638. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nat. Cell Biol. 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Eleuteri, D.; Montini, L.; Cutuli, S.L.; Rossi, C.; Alcaro, F.; Antonelli, M. Renin–angiotensin system dysregulation in critically ill patients with acute respiratory distress syndrome due to COVID-19: A preliminary report. Crit. Care 2021, 25, 91. [Google Scholar] [CrossRef]
- Ozkan, S.; Cakmak, F.; Konukoglu, D.; Biberoglu, S.; Ipekci, A.; Akdeniz, Y.S.; Bolayirli, I.M.; Balkan, I.I.; Dumanli, G.Y.; Ikizceli, I. Efficacy of Serum Angiotensin II Levels in Prognosis of Patients With Coronavirus Disease 2019. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- Houweling, B.; Merkus, D.; Dekker, M.M.D.; Duncker, D.J. Nitric oxide blunts the endothelin-mediated pulmonary vasoconstriction in exercising swine. J. Physiol. 2005, 568, 629–638. [Google Scholar] [CrossRef]
- Albertine, K.H.; Wang, Z.M.; Michael, J.R. Expression of endothelial nitric oxide synthase, inducible nitric oxide synthase, and endothelin-1 in lungs of subjects who died with ARDS. Chest 1999, 116, 101S–102S. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Jiang, J.; Su, L.; Shu, T.; Liu, H.; Lai, S.; Ghiladi, R.A.; Wang, J. The role of NO in COVID-19 and potential therapeutic strategies. Free Radic. Biol. Med. 2021, 163, 153–162. [Google Scholar] [CrossRef]
- Karmouty-Quintana, H.; Thandavarayan, R.A.; Keller, S.P.; Sahay, S.; Pandit, L.M.; Akkanti, B. Emerging Mechanisms of Pulmonary Vasoconstriction in SARS-CoV-2-Induced Acute Respiratory Distress Syndrome (ARDS) and Potential Therapeutic Targets. Int. J. Mol. Sci. 2020, 21, 8081. [Google Scholar] [CrossRef] [PubMed]
- Hellman, U.; Karlsson, M.G.; Engström-Laurent, A.; Cajander, S.; Dorofte, L.; Ahlm, C.; Laurent, C.; Blomberg, A. Presence of hyaluronan in lung alveoli in severe Covid-19: An opening for new treatment options? J. Biol. Chem. 2020, 295, 15418–15422. [Google Scholar] [CrossRef] [PubMed]
- Collum, S.D.; Chen, N.; Hernandez, A.M.; Hanmandlu, A.; Sweeney, H.; Mertens, T.C.J.; Weng, T.; Luo, F.; Molina, J.G.; Davies, J.; et al. Inhibition of hyaluronan synthesis attenuates pulmonary hypertension associated with lung fibrosis. Br. J. Pharmacol. 2017, 174, 3284–3301. [Google Scholar] [CrossRef] [Green Version]
- Pinsky, M.R. Cardiopulmonary Interactions: Physiologic Basis and Clinical Applications. Ann. Am. Thorac. Soc. 2018, 15, S45–S48. [Google Scholar] [CrossRef]
- Marik, P.E.; Monnet, X.; Teboul, J.-L. Hemodynamic parameters to guide fluid therapy. Ann. Intensiv. Care 2011, 1, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, J.J.; Rocco, P.R.M.; Gattinoni, L. Static and Dynamic Contributors to Ventilator-induced Lung Injury in Clinical Practice. Pressure, Energy, and Power. Am. J. Respir. Crit. Care Med. 2020, 201, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Menk, M.; Estenssoro, E.; Sahetya, S.K.; Neto, A.S.; Sinha, P.; Slutsky, A.S.; Summers, C.; Yoshida, T.; Bein, T.; Ferguson, N.D. Current and evolving standards of care for patients with ARDS. Intensive Care Med. 2020, 46, 2157–2167. [Google Scholar] [CrossRef]
- Jardin, F.; Vieillard-Baron, A. Is there a safe plateau pressure in ARDS? The right heart only knows. Intensive Care Med. 2007, 33, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Lavie, C.J.; Sanchis-Gomar, F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): Evidence from a meta-analysis. Prog. Cardiovasc. Dis. 2020, 63, 390–391. [Google Scholar] [CrossRef]
- Lala, A.; Johnson, K.; Januzzi, J.L.; Russak, A.J.; Paranjpe, I.; Richter, F.; Zhao, S.; Somani, S.; Van Vleck, T.; Vaid, A.; et al. Prevalence and Impact of Myocardial Injury in Patients Hospitalized With COVID-19 Infection. J. Am. Coll. Cardiol. 2020, 76, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Giannitsis, E.; Jaffe, A.S.; Huber, K.; Mair, J.; Cullen, L.; Hammarsten, O.; Mills, N.L.; Möckel, M.; Krychtiuk, K.; et al. Cardiovascular biomarkers in patients with COVID-19. Eur. Heart J. Acute Cardiovasc. Care 2021, 10, 310–319. [Google Scholar] [CrossRef]
- Gordon, J.S.; Drazner, M.H. Biomarkers of Cardiac Stress and Cytokine Release Syndrome in COVID-19: A Review. Curr. Heart Fail. Rep. 2021, 1–6. [Google Scholar] [CrossRef]
- Cheng, M.P.; Cau, A.; Lee, T.C.; Brodie, D.; Slutsky, A.; Marshall, J.; Murthy, S.; Lee, T.; Singer, J.; Demir, K.K.; et al. Acute Cardiac Injury in Coronavirus Disease 2019 and Other Viral Infections—A Systematic Review and Meta-Analysis. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- Sheth, A.; Modi, M.; Dawson, D.; Dominic, P. Prognostic value of cardiac biomarkers in COVID-19 infection. Sci. Rep. 2021, 11, 4930. [Google Scholar] [CrossRef]
- Chung, M.K.; Zidar, D.A.; Bristow, M.R.; Cameron, S.J.; Chan, T.; Iii, C.V.H.; Kwon, D.H.; Singh, T.; Tilton, J.C.; Tsai, E.J.; et al. COVID-19 and Cardiovascular Disease: From Bench to Bedside. Circ. Res. 2021, 128, 1214–1236. [Google Scholar] [CrossRef] [PubMed]
- Ojha, V.; Verma, M.; Pandey, N.N.; Mani, A.; Malhi, A.S.; Kumar, S.; Jagia, P.; Roy, A.; Sharma, S. Cardiac Magnetic Resonance Imaging in Coronavirus Disease 2019 (COVID-19): A System-atic Review of Cardiac Magnetic Resonance Imaging Findings in 199 Patients. J. Thorac. Imaging 2021, 36, 73–83. [Google Scholar] [CrossRef]
- Kawakami, R.; Sakamoto, A.; Kawai, K.; Gianatti, A.; Pellegrini, D.; Nasr, A.; Kutys, B.; Guo, L.; Cornelissen, A.; Mori, M.; et al. Pathological Evidence for SARS-CoV-2 as a Cause of Myocarditis JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Haslbauer, J.D.; Tzankov, A.; Mertz, K.D.; Schwab, N.; Nienhold, R.; Twerenbold, R.; Leibundgut, G.; Stalder, A.K.; Matter, M.; Glatz, K. Characterisation of cardiac pathology in 23 autopsies of lethal COVID-19. J. Pathol. Clin. Res. 2021. [Google Scholar] [CrossRef]
- Bailey, A.L.; Dmytrenko, O.; Greenberg, L.; Bredemeyer, A.L.; Ma, P.; Liu, J.; Penna, V.; Winkler, E.S.; Sviben, S.; Brooks, E.; et al. SARS-CoV-2 Infects Human Engineered Heart Tissues and Models COVID-19 Myocarditis. JACC Basic Transl. Sci. 2021, 6, 331–345. [Google Scholar] [CrossRef]
- Basso, C.; Leone, O.; Rizzo, S.; De Gaspari, M.; Van Der Wal, A.C.; Aubry, M.-C.; Bois, M.C.; Lin, P.T.; Maleszewski, J.J.; Stone, J.R. Pathological features of COVID-19-associated myocardial injury: A multicentre cardiovascular pathology study. Eur. Hear. J. 2020, 41, 3827–3835. [Google Scholar] [CrossRef]
- Weckbach, L.T.; Curta, A.; Bieber, S.; Kraechan, A.; Brado, J.; Hellmuth, J.C.; Muenchhoff, M.; Scherer, C.; Schroeder, I.; Irlbeck, M.; et al. Myocardial Inflammation and Dysfunction in COVID-19–Associated Myocardial Injury. Circ. Cardiovasc. Imaging 2021, 14, e012220. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The significance of COVID-19-associated myocardial injury: How overinterpretation of scientific findings can fuel media sensationalism and spread misinformation. Eur. Hear. J. 2020, 41, 3836–3838. [Google Scholar] [CrossRef] [PubMed]
- Bangalore, S.; Sharma, A.; Slotwiner, A.; Yatskar, L.; Harari, R.; Shah, B.; Ibrahim, H.; Friedman, G.H.; Thompson, C.; Alviar, C.L.; et al. ST-Segment Elevation in Patients with Covid-19—A Case Series. N. Engl. J. Med. 2020, 382, 2478–2480. [Google Scholar] [CrossRef]
- Pellegrini, D.; Kawakami, R.; Guagliumi, G.; Sakamoto, A.; Kawai, K.; Gianatti, A.; Nasr, A.; Kutys, R.; Guo, L.; Cornelissen, A.; et al. Microthrombi as a Major Cause of Cardiac Injury in COVID-19: A Pathologic Study. Circulation 2021, 143, 1031–1042. [Google Scholar] [CrossRef]
- Bois, M.C.; Boire, N.A.; Layman, A.J.; Aubry, M.-C.; Alexander, M.P.; Roden, A.C.; Hagen, C.E.; Quinton, R.A.; Larsen, C.; Erben, Y.; et al. COVID-19–Associated Nonocclusive Fibrin Microthrombi in the Heart. Circulation 2021, 143, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Sala, S.; Caforio, A.L.P. Acute myocardial injury, MINOCA, or myocarditis? Improving characterization of coronavirus-associated myocardial involvement. Eur. Hear. J. 2020, 41, 2124–2125. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.E.; Yadid, M.; McCain, M.L.; Sheehy, S.P.; Pasqualini, F.; Park, S.-J.; Cho, A.; Campbell, P.; Parker, K.K. Angiotensin II Induced Cardiac Dysfunction on a Chip. PLoS ONE 2016, 11, e0146415. [Google Scholar] [CrossRef] [Green Version]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef]
- Casini, A.; Alberio, L.; Angelillo-Scherrer, A.; Fontana, P.; Gerber, B.; Graf, L.; Hegemann, I.; Korte, W.; Hovinga, J.K.; Lecompte, T.; et al. Thromboprophylaxis and laboratory monitoring for in-hospital patients with Covid-19—A Swiss consensus statement by the Working Party Hemostasis. Swiss Med. Wkly. 2020, 150, w20247. [Google Scholar] [CrossRef] [Green Version]
- Thachil, J. The versatile heparin in COVID-19. J. Thromb. Haemost. 2020, 18, 1020–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talasaz, A.H.; Sadeghipour, P.; Kakavand, H.; Aghakouchakzadeh, M.; Kordzadeh-Kermani, E.; Van Tassell, B.W.; Gheymati, A.; Ariannejad, H.; Hosseini, S.H.; Jamalkhani, S.; et al. Recent Randomized Trials of Antithrombotic Therapy for Patients with COVID-19: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 77, 1903–1921. [Google Scholar] [CrossRef] [PubMed]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- The REMAP-CAP Investigators. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Kerbaul, F.; Rondelet, B.; Motte, S.; Fesler, P.; Hubloue, I.; Ewalenko, P.; Naeije, R.; Brimioulle, S. Effects of norepinephrine and dobutamine on pressure load-induced right ventricular failure. Crit. Care Med. 2004, 32, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Grignola, J.C.; Domingo, E. Acute Right Ventricular Dysfunction in Intensive Care Unit. BioMed Res. Int. 2017, 2017, 8217105. [Google Scholar] [CrossRef] [Green Version]
- Sanz, J.; Sánchez-Quintana, D.; Bossone, E.; Bogaard, H.J.; Naeije, R. Anatomy, Function, and Dysfunction of the Right Ventricle: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 1463–1482. [Google Scholar] [CrossRef] [PubMed]
- Paternot, A.; Repessé, X.; Vieillard-Baron, A. Rationale and Description of Right Ventricle-Protective Ventilation in ARDS. Respir. Care 2016, 61, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Guérin, C.; Albert, R.K.; Beitler, J.; Gattinoni, L.; Jaber, S.; Marini, J.J.; Munshi, L.; Papazian, L.; Pesenti, A.; Vieillard-Baron, A.; et al. Prone position in ARDS patients: Why, when, how and for whom. Intensiv. Care Med. 2020, 46, 2385–2396. [Google Scholar] [CrossRef]
- Guérin, C.; Reignier, J.; Richard, J.-C.; Beuret, P.; Gacouin, A.; Boulain, T.; Mercier, E.; Badet, M.; Mercat, A.; Baudin, O.; et al. Prone Positioning in Severe Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2013, 368, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Nasa, P.; Azoulay, E.; Khanna, A.K.; Jain, R.; Gupta, S.; Javeri, Y.; Juneja, D.; Rangappa, P.; Sundararajan, K.; Alhazzani, W.; et al. Expert consensus statements for the management of COVID-19-related acute respiratory failure using a Delphi method. Crit. Care 2021, 25, 106. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [Green Version]
- Lotz, C.; Muellenbach, R.M.; Meybohm, P.; Mutlak, H.; Lepper, P.M.; Rolfes, C.; Peivandi, A.; Stumpner, J.; Kredel, M.; Kranke, P.; et al. Effects of inhaled nitric oxide in COVID-19–induced ARDS—Is it worthwhile? Acta Anaesthesiol. Scand. 2021, 65, 629–632. [Google Scholar] [CrossRef]
- Tavazzi, G.; Pozzi, M.; Mongodi, S.; Dammassa, V.; Romito, G.; Mojoli, F. Inhaled nitric oxide in patients admitted to intensive care unit with COVID-19 pneumonia. Crit. Care 2020, 24, 508. [Google Scholar] [CrossRef]
- Abou-Arab, O.; Huette, P.; Debouvries, F.; Dupont, H.; Jounieaux, V.; Mahjoub, Y. Inhaled nitric oxide for critically ill Covid-19 patients: A prospective study. Crit. Care 2020, 24, 645. [Google Scholar] [CrossRef]
- Longobardo, A.; Montanari, C.; Shulman, R.; Benhalim, S.; Singer, M.; Arulkumaran, N. Inhaled nitric oxide minimally improves oxygenation in COVID-19 related acute respiratory distress syndrome. Br. J. Anaesth. 2021, 126, e44–e46. [Google Scholar] [CrossRef]
- Fakhr, B.S.; Wiegand, S.B.; Pinciroli, R.; Gianni, S.; Morais, C.C.A.; Ikeda, T.; Miyazaki, Y.; Marutani, E.; Di Fenza, R.; Larson, G.M.; et al. High Concentrations of Nitric Oxide Inhalation Therapy in Pregnant Patients with Severe Coronavirus Disease 2019 (COVID-19). Obstet. Gynecol. 2020, 136, 1109–1113. [Google Scholar] [CrossRef]
- Ferrari, M.; Santini, A.; Protti, A.; Andreis, D.T.; Iapichino, G.; Castellani, G.; Rendiniello, V.; Costantini, E.; Cecconi, M. Inhaled nitric oxide in mechanically ventilated patients with COVID-19. J. Crit. Care 2020, 60, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Garfield, B.; McFadyen, C.; Briar, C.; Bleakley, C.; Vlachou, A.; Baldwin, M.; Lees, N.; Price, S.; Ledot, S.; McCabe, C.; et al. Potential for personalised application of inhaled nitric oxide in COVID-19 pneumonia. Br. J. Anaesth. 2021, 126, e72–e75. [Google Scholar] [CrossRef] [PubMed]
- Robba, C.; Ball, L.; Battaglini, D.; Cardim, D.; Moncalvo, E.; Brunetti, I.; Bassetti, M.; Giacobbe, D.R.; Vena, A.; Patroniti, N.; et al. Early effects of ventilatory rescue therapies on systemic and cerebral oxygenation in mechanically ventilated COVID-19 patients with acute respiratory distress syndrome: A prospective observational study. Crit. Care 2021, 25, 111. [Google Scholar] [CrossRef]
- Zochios, V.; Jones, N. Acute right heart syndrome in the critically ill patient. Heart Lung Vessel. 2014, 6, 157–170. [Google Scholar] [PubMed]
- Missant, C.; Rex, S.; Segers, P.; Wouters, P.F. Levosimendan improves right ventriculovascular coupling in a porcine model of right ventricular dysfunction. Crit. Care Med. 2007, 35, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Lebrin, M.; Vaccaro, A.; Senard, J.-M.; Despas, F. Pharmacology of levosimendan: Inotropic, vasodilatory and cardioprotective effects. J. Clin. Pharm. Ther. 2013, 38, 341–349. [Google Scholar] [CrossRef]
- Morelli, A.; Teboul, J.-L.; Maggiore, S.M.; Vieillard-Baron, A.; Rocco, M.; Conti, G.; De Gaetano, A.; Picchini, U.; Orecchioni, A.; Carbone, I.; et al. Effects of levosimendan on right ventricular afterload in patients with acute respiratory distress syndrome: A pilot study. Crit. Care Med. 2006, 34, 2287–2293. [Google Scholar] [CrossRef]
- Combes, A.; Hajage, D.; Capellier, G.; Demoule, A.; Lavoué, S.; Guervilly, C.; Da Silva, D.; Zafrani, L.; Tirot, P.; Veber, B.; et al. Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2018, 378, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.H.; Ogino, M.T.; Brodie, D.; McMullan, D.M.; Lorusso, R.; MacLaren, G.; Stead, C.M.; Rycus, P.; Fraser, J.F.; Belohlavek, J.; et al. Initial ELSO Guidance Document: ECMO for COVID-19 Patients with Severe Cardiopulmonary Failure. ASAIO J. 2020, 66, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, R.P.; MacLaren, G.; Boonstra, P.S.; Iwashyna, T.J.; Slutsky, A.S.; Fan, E.; Bartlett, R.H.; Tonna, J.E.; Hyslop, R.; Fanning, J.J.; et al. Extracorporeal membrane oxygenation support in COVID-19: An international cohort study of the Extracorporeal Life Support Organization registry. Lancet 2020, 396, 1071–1078. [Google Scholar] [CrossRef]
- Henry, B.M.; Lippi, G. Poor survival with extracorporeal membrane oxygenation in acute respiratory distress syndrome (ARDS) due to coronavirus disease 2019 (COVID-19): Pooled analysis of early reports. J. Crit. Care 2020, 58, 27–28. [Google Scholar] [CrossRef] [PubMed]
- Abrams, D.; Lorusso, R.; Vincent, J.-L.; Brodie, D. ECMO during the COVID-19 pandemic: When is it unjustified? Crit. Care 2020, 24, 507. [Google Scholar] [CrossRef] [PubMed]
- Badulak, J.; Antonini, M.V.; Stead, C.M.; Shekerdemian, L.; Raman, L.; Paden, M.L.; Agerstrand, C.; Bartlett, R.H.; Barrett, N.; Combes, A.; et al. Extracorporeal Membrane Oxygenation for COVID-19: Updated 2021 Guidelines from the Extracorporeal Life Support Organization. ASAIO J. 2021, 67, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhao, S.; Luo, H.; Wu, Z.; Wu, J.; Xia, H.; Chen, X. The role of extracorporeal membrane oxygenation in critically ill patients with COVID-19: A narrative review. BMC Pulm. Med. 2021, 21, 116. [Google Scholar] [CrossRef]
- Schmidt, M.; Hajage, D.; Lebreton, G.; Monsel, A.; Voiriot, G.; Levy, D.; Baron, E.; Beurton, A.; Chommeloux, J.; Meng, P.; et al. Extracorporeal membrane oxygenation for severe acute respiratory distress syndrome associated with COVID-19: A retrospective cohort study. Lancet Respir. Med. 2020, 8, 1121–1131. [Google Scholar] [CrossRef]
- Lorusso, R.; Combes, A.; Coco, V.L.; De Piero, M.E.; Belohlavek, J. ECMO for COVID-19 patients in Europe and Israel. Intensiv. Care Med. 2021, 47, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, G.; Schmidt, M.; Ponnaiah, M.; Folliguet, T.; Para, M.; Guihaire, J.; Lansac, E.; Sage, E.; Cholley, B.; Mégarbane, B.; et al. Extracorporeal membrane oxygenation network organisation and clinical outcomes during the COVID-19 pandemic in Greater Paris, France: A multicentre cohort study. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Shaefi, S.; Brenner, S.K.; Gupta, S.; O’Gara, B.P.; Krajewski, M.L.; Charytan, D.M.; Chaudhry, S.; Mirza, S.H.; Peev, V.; Leaf, D.E.; et al. Extracorporeal membrane oxygenation in patients with severe respiratory failure from COVID-19. Intensive Care Med. 2021, 47, 208–221. [Google Scholar] [CrossRef]
- Miranda, D.R.; Van Thiel, R.; Brodie, D.; Bakker, J. Right Ventricular Unloading after Initiation of Venovenous Extracorporeal Membrane Oxygenation. Am. J. Respir. Crit. Care Med. 2015, 191, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Brodie, D. Reply: Protecting the right ventricle in COVID-19 acute respiratory distress syndrome—More data required. J. Thorac. Cardiovasc. Surg. 2021, 161, e215–e216. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Alexander, P.J.; Joshi, D.J.; Tabachnick, D.R.; Cross, C.A.; Pappas, P.S.; Tatooles, A.J. Extracorporeal Membrane Oxygenation for Patients With COVID-19 in Severe Respiratory Failure. JAMA Surg. 2020, 155, 990. [Google Scholar] [CrossRef] [PubMed]
- Joyce, D.L. Mechanical ventilation: A necessary evil? J. Thorac. Cardiovasc. Surg. 2021, 161, e213–e214. [Google Scholar] [CrossRef] [PubMed]



| N (Age) % Male | Patients | Echography/Biomarkers | Pulmonary Circulation | Main Prognostic Findings | Ref |
|---|---|---|---|---|---|
| 332 (66.9) 71.4% | ICU 22% |
|
|
| Ferrante et al. [3] |
| 4 (50–67 y) 75% | ICU |
|
|
| Garcia-Cruz et al. [5] |
| 5 (42–76 y) 60% | ICU |
|
|
| Creel-Bulos et al. [1] |
| 66 (60) 57.6% | ICU 58% |
|
|
| Schott et al. [6] |
| 29 (NA) | ICU |
|
|
| Rauch et al. [2] |
| 110 (66) 64% | ICU |
|
|
| Argulian et al. [7] |
| 49 (64.3) 54.3% | ICU |
|
|
| Li et al. [8] |
| 100 (66.1) 63% | ICU |
|
|
| Szekely et al. [9] |
| 416 (47) 48% | ICU 8% Echo in 57 pts |
|
|
| Zeng et al. [10] |
| 51 (63) 80% | Non-ICU |
|
|
| Van den Heuvel et al. [11] |
| 45 (61) 51% | NA |
|
|
| Vasudev et al. [12] |
| 115 (64) 60% | ICU |
|
|
| D’Andrea et al. [13] |
| 200 (62) 66% | non ICU |
|
|
| Pagnesi et al. [14] |
| 98 (68) 77% | ICU 57% |
|
|
| Rath et al. [15] |
| 72 (18–80 y) 72% | ICU 56% |
|
|
| Jain et al. [16] |
| 164 (61) 78% | ICU |
|
|
| Moody et al. [17] |
| 1216 (62) 70% | ICU 60% |
|
|
| Dweck et al. [18] |
| 74 (59) 78% | ICU |
|
|
| Mahmoud-Elsayed et al. [19] |
| 510 (64) 66% | ICU 68% |
|
|
| Kim et al. [20] |
| 90 (52) 74.4% | ICU (ECMO 42%) |
|
|
| Bleakley et al. [21] |
| 24 (64.5) 54& | NA |
|
|
| Sud et al. [22] |
| 86 (58.8) 60% | ICU 37% |
|
|
| Sattarzadeh Badkoubeh et al. [23] |
| 224 (69) 62% | ICU 33% |
|
|
| Scudiero et al. [24] |
| 94 (64) 74% | ICU |
|
|
| D’Alto et al. [25] |
| 305 (63) 67% | ICU 44% |
|
|
| Giustino et al. [26] |
| 28 (61.7) 79% | ICU (ECMO 14%) |
|
|
| Lazzeri et al. [27] |
| 67 (61) 82% | ICU |
|
|
| Bagate et al. [28] |
| Drug | SVR | PVR | PVR/SVR | Main Adverse Effects |
|---|---|---|---|---|
| Vasopressors | ||||
| Norepinephrine | ↑↑↑ | ↑ | →/↓ | ↑ PA pressure (at >0.5 mg/kg/min), tachycardia |
| Phenylephrine | ↑↑ | ↑↑ | → | ↑ PA pressure, ↑ RV afterload |
| Vasopressin | ↑↑↑ | →/↓ | ↓↓ | Digital and mesenteric ischemia (keep < 0.03 U/min) |
| Inotropes | ||||
| Dobutamine | →/↓ | →/↓ | →/↓ | Tachycardia, ↑ myocardial O2 demand, hypotension |
| Epinephrine | ↑↑↑ | ↑↑ | →/↓ | Tachycardia, ↑ myocardial O2 demand, ↑ RV afterload |
| Milrinone | ↓↓ | ↓↓↓ | ↓↓ | Hypotension, tachycardia, ↑ myocardial O2 demand |
| Levosimendan | ↓↓↓ | ↓↓ | →/↓ | Hypotension |
| Reference | Design | n | Population | [iNO] | iNO Duration | Effect on P/F O2 | Effect on RV | Effect on PVR |
|---|---|---|---|---|---|---|---|---|
| Abou-Arab, O. et al. [151] | Prospective | 34 | ICU | 10 ppm | 30 min | Significant (20% increase in 65% pts) | Similar incidence of ACP in responders and non-responders | NR |
| Tavazzi, G. et al. [150] | Retrospective | 16 | ICU | 25 ppm | 30 min | Not significant (20% increase in 25% pts) | Better improvement of P/F O2 in pts with RV dysfunction | NR |
| Longobardo, A. et al. [152] | Retrospective case–control | 27 | ICU | 10–20 ppm | NR | Not significant (10% increase in 40% pts) | NR | NR |
| Safaee Fakhr, B et al. [153] | Prospective observational | 6 | Obstetric/ICU | 200 ppm (SB); 40 ppm (MV) | 30 min 2×/day (SB) Continuous administration (MV) | Significant increase after each inhalation period | NR | NR |
| Ferrari, M. et al. [154] | Retrospective | 10 | ICU | 20 ppm | 30 min | Not significant | NR | NR |
| Garfield, B. et al. [155] | Observational | 36 | ICU | 20 ppm | 24 h (144 h median) | Significant (30% increase in 57% pts) | NR | NR |
| Lotz, C. et al. [149] | Retrospective observational | 19 | ICU | 20 ppm | NR | Significant (20% mean increase) | NR | Median decrease of 15.9% (not significant) |
| Roba, c. et al. [156] | Prospective | 9 | ICU | 20 ppm | 1 h | Significant increase of P/F O2 and of cerebral saturation | NR | NR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonnemain, J.; Ltaief, Z.; Liaudet, L. The Right Ventricle in COVID-19. J. Clin. Med. 2021, 10, 2535. https://doi.org/10.3390/jcm10122535
Bonnemain J, Ltaief Z, Liaudet L. The Right Ventricle in COVID-19. Journal of Clinical Medicine. 2021; 10(12):2535. https://doi.org/10.3390/jcm10122535
Chicago/Turabian StyleBonnemain, Jean, Zied Ltaief, and Lucas Liaudet. 2021. "The Right Ventricle in COVID-19" Journal of Clinical Medicine 10, no. 12: 2535. https://doi.org/10.3390/jcm10122535
APA StyleBonnemain, J., Ltaief, Z., & Liaudet, L. (2021). The Right Ventricle in COVID-19. Journal of Clinical Medicine, 10(12), 2535. https://doi.org/10.3390/jcm10122535