
The Link between SARS-CoV-2 Infection, Inflammation and Hypercoagulability-Impact of Hemorheologic Alterations on Cardiovascular Mortality

Abstract
:1. Introduction
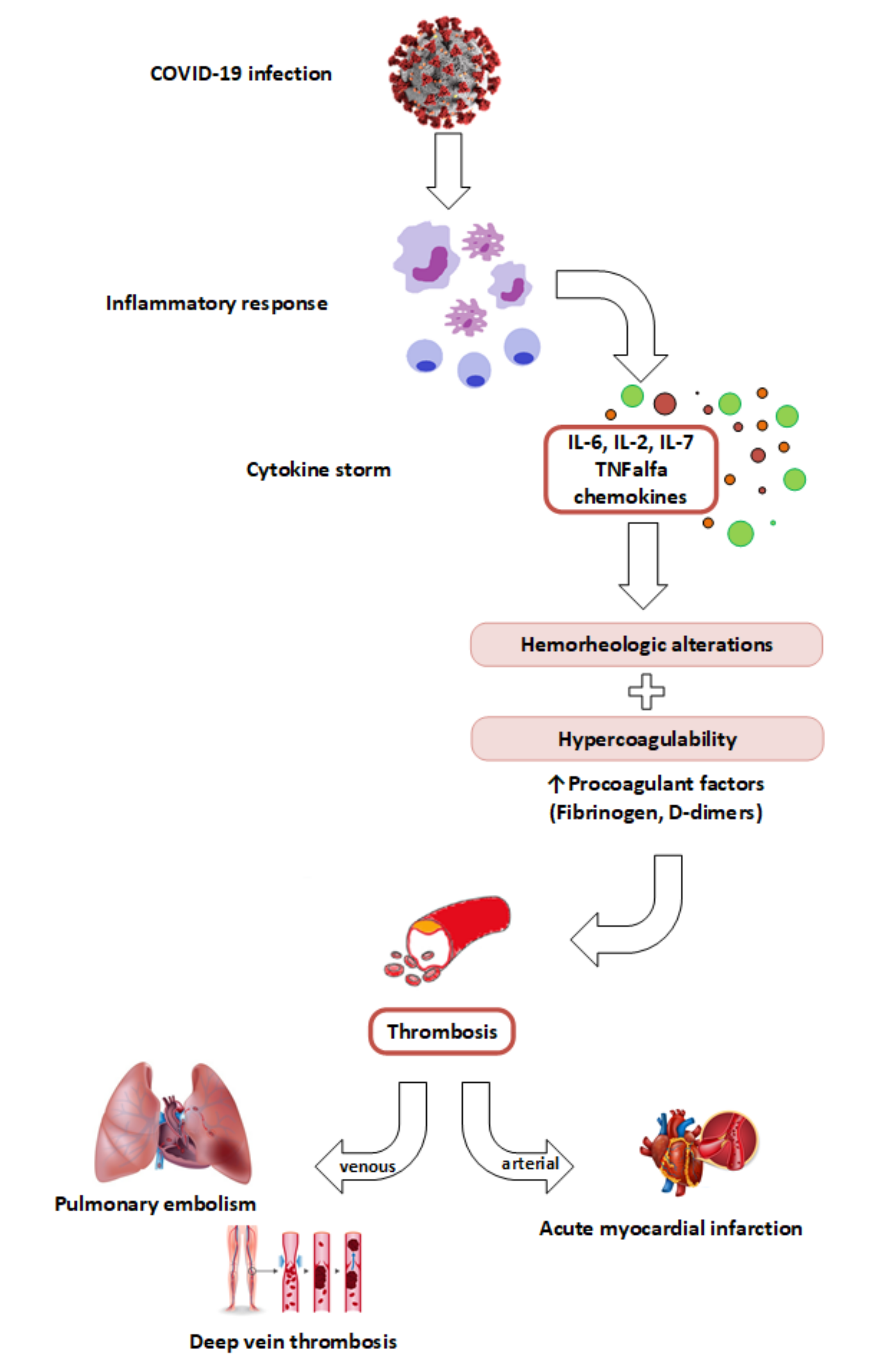
2. COVID-19, a Thromboinflammatory Disease
3. Platelet Activation and Acute Coronary Syndromes in COVID-19
4. The Link between SARS-CoV-2 Infection, Inflammatory Storm and Hypercoagulation
4.1. Cytokine Storm and Inflammation in COVID-19 Versus Sepsis
4.2. Cytokine Storm and Hypercoagulability
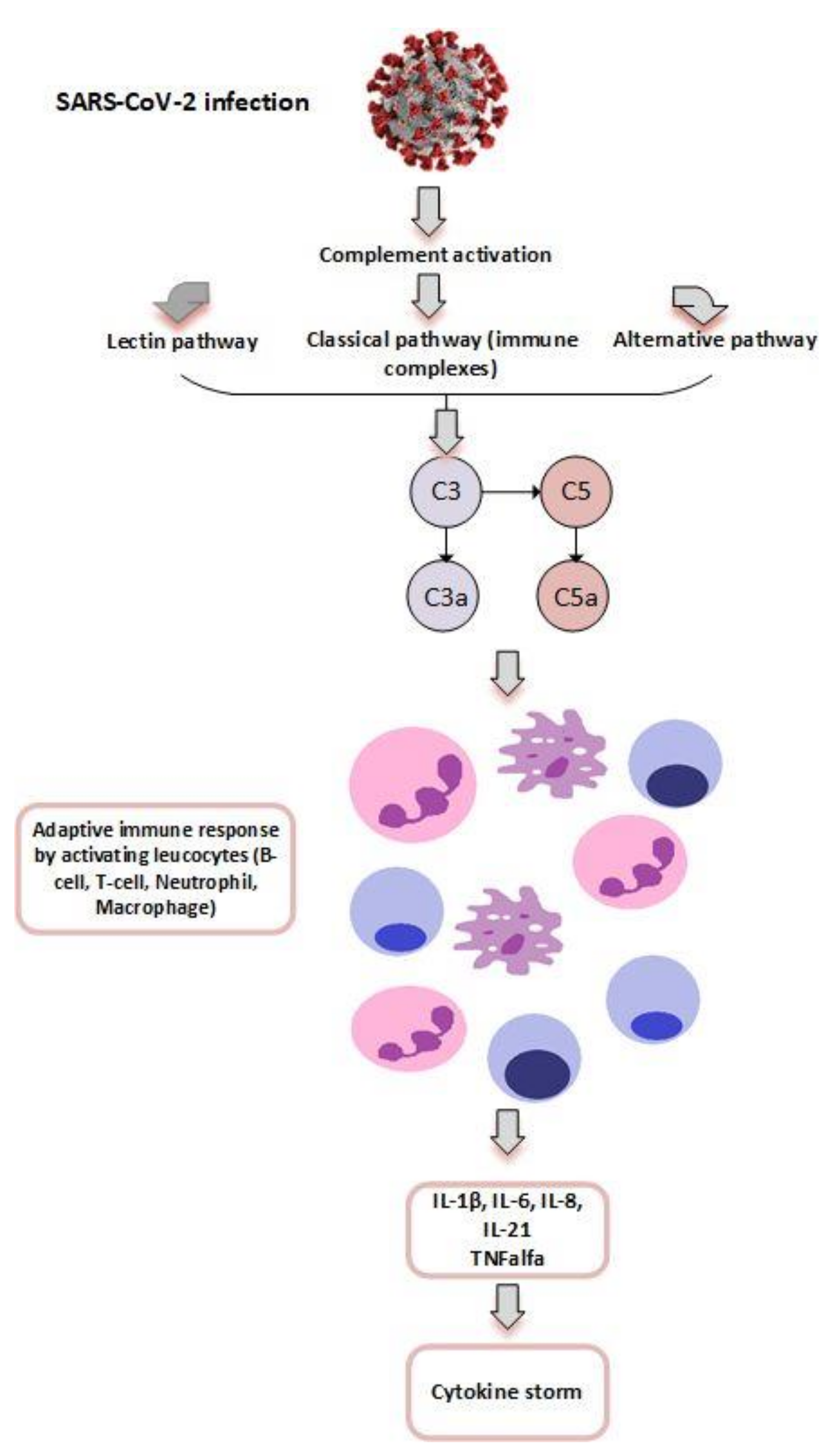
4.3. Complement, Coagulation and Inflammation in COVID-19
4.4. Hypercoagulability and Venous Thrombosis in COVID-19
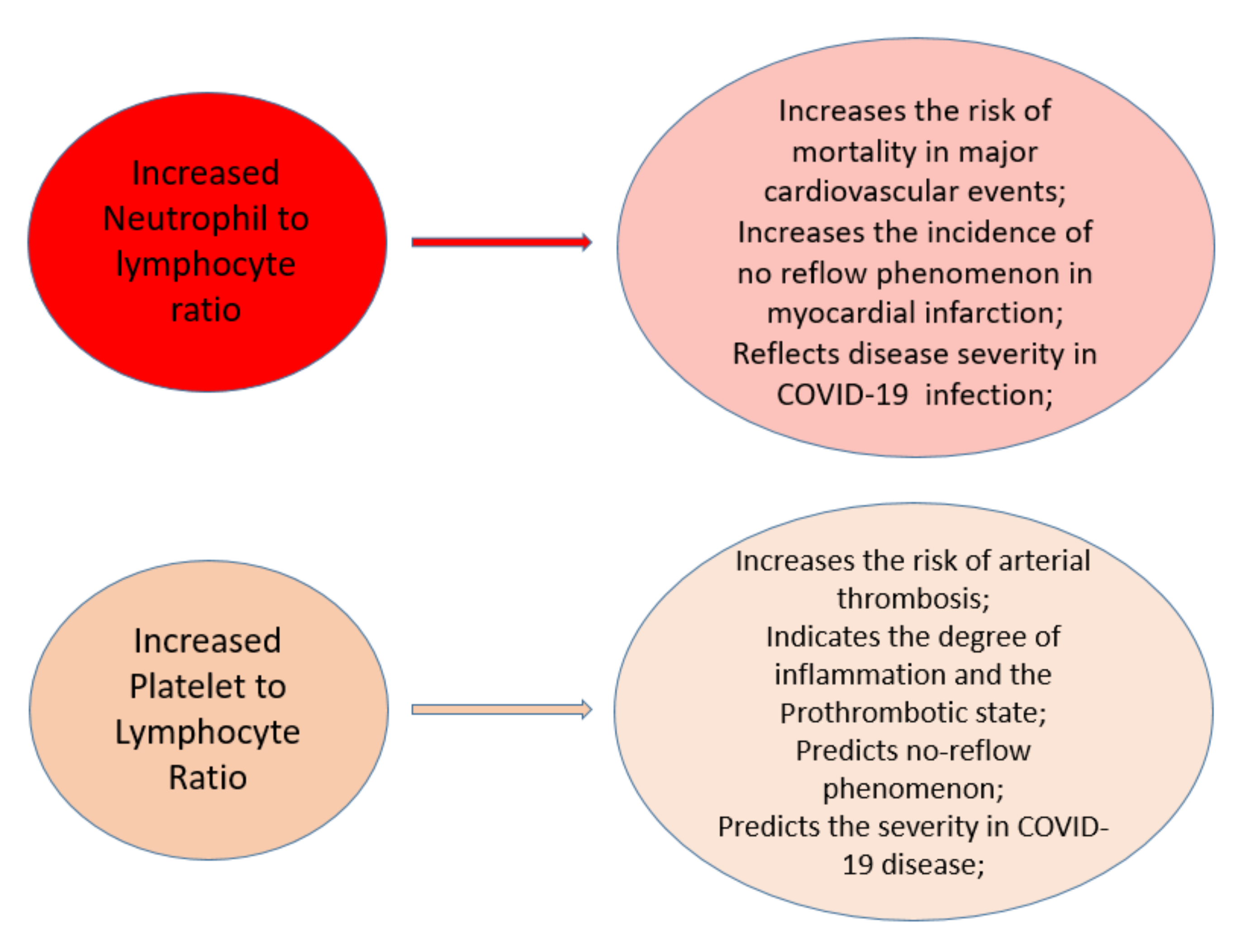
5. Biomarkers Associated with Worse Prognosis in COVID-19 Patients
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Matsushita, K.; Hess, S.; Marchandot, B.; Sato, C.; Truong, D.P.; Kim, N.T.; Weiss, A.; Jesel, L.; Ohlmann, P.; Morel, O. Clinical features of patients with acute coronary syndrome during the COVID-19 pandemic. J. Thromb. Thrombolysis 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Fried, J.A.; Ramasubbu, K.; Bhatt, R.; Topkara, V.K.; Clerkin, K.J.; Horn, E.; Rabbani, L.; Brodie, D.; Jain, S.S.; Kirtane, A.J.; et al. The Variety of Cardiovascular Presentations of COVID-19. Circulation 2020, 141, 1930–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmud, E.; Dauerman, H.L.; Welt, F.G.; Messenger, J.C.; Rao, S.V.; Grines, C.; Mattu, A.; Kirtane, A.J.; Jauhar, R.; Meraj, P.; et al. Management of Acute Myocardial Infarction During the COVID-19 Pandemic. J. Am. Coll. Cardiol. 2020, 76, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Harari, R.; Bangalore, S.; Chang, E.; Shah, B. COVID-19 complicated by acute myocardial infarction with extensive thrombus burden and cardiogenic shock. Catheter. Cardiovasc. Interv. 2021, 97. [Google Scholar] [CrossRef]
- Hendren, N.S.; Drazner, M.H.; Bozkurt, B.; Cooper, L.T., Jr. Description and Proposed Management of the Acute COVID-19 Cardiovascular Syndrome. Circulation 2020, 141, 1903–1914. [Google Scholar] [CrossRef] [PubMed]
- E Colling, M.; Kanthi, Y. COVID–19-associated coagulopathy: An exploration of mechanisms. Vasc. Med. 2020, 25, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.B.; Pasalic, L.; Hvas, A.-M. Platelets in Coronavirus Disease 2019. Semin. Thromb. Hemost. 2020, 46, 823–825. [Google Scholar] [CrossRef]
- Violi, F.; Pastori, D.; Cangemi, R.; Pignatelli, P.; Loffredo, L. Hypercoagulation and Antithrombotic Treatment in Coronavirus 2019: A New Challenge. Thromb. Haemost. 2020, 120, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Gąsecka, A.; Borovac, J.A.; Guerreiro, R.A.; Giustozzi, M.; Parker, W.; Caldeira, D.; Chiva-Blanch, G. Thrombotic Complications in Patients with COVID-19: Pathophysiological Mechanisms, Diagnosis, and Treatment. Cardiovasc. Drugs Ther. 2021, 35, 215–229. [Google Scholar] [CrossRef]
- Khandait, H.; Gandotra, G.; Sachdeva, S.; Kramer, C.A.; Nye, D.; Golamari, R.; Jain, R. COVID-19 and Hematology—What Do We Know So Far? SN Compr. Clin. Med. 2020, 2, 2631–2636. [Google Scholar] [CrossRef] [PubMed]
- Vernuccio, F.; Lombardo, F.; Cannella, R.; Panzuto, F.; Giambelluca, D.; Arzanauskaite, M.; Midiri, M.; Cabassa, P. Thromboembolic complications of COVID-19: The combined effect of a pro-coagulant pattern and an endothelial thrombo-inflammatory syndrome. Clin. Radiol. 2020, 75, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.-D.; Sacco, C.; Bertuzzi, A.; et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, G.G.; Montorfano, M.; Trabattoni, D.; Andreini, D.; Ferrante, G.; Ancona, M.B.M.; Metra, M.; Curello, S.; Maffeo, D.; Pero, G.; et al. ST-Elevation Myocardial Infarction in Patients With COVID-19. Circulation 2020, 141, 2113–2116. [Google Scholar] [CrossRef]
- Bouleti, C.; Mewton, N.; Germain, S. The no-reflow phenomenon: State of the art. Arch. Cardiovasc. Dis. 2015, 108, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Kurtul, A.; Yarlioglues, M.; Murat, S.N.; Ergun, G.; Duran, M.; Kasapkara, H.A.; Demircelik, M.B.; Cetin, M.; Ocek, A.H. Usefulness of the Platelet-to-Lymphocyte Ratio in Predicting Angiographic Reflow after Primary Percutaneous Coronary Intervention in Patients with Acute ST-Segment Elevation Myocardial Infarction. Am. J. Cardiol. 2014, 114, 342–347. [Google Scholar] [CrossRef]
- Toprak, C.; Tabakci, M.M.; Simsek, Z.; Arslantas, U.; Durmus, H.I.; Ocal, L.; Demirel, M.; Ozturkeri, B.; Ozal, E.; Kargin, R. Platelet/lymphocyte ratio was associated with impaired myocardial perfusion and both in-hospital and long-term adverse outcome in patients with ST-segment elevation acute myocardial infarction undergoing primary coronary intervention. Adv. Interv. Cardiol. 2015, 4, 288–297. [Google Scholar] [CrossRef]
- Dong, G.; Huang, A.; Liu, L. Platelet-to-lymphocyte ratio and prognosis in STEMI: A meta-analysis. Eur. J. Clin. Investig. 2021, 51. [Google Scholar] [CrossRef]
- Kounis, N.G.; Koniari, I.; Plotas, P.; Soufras, G.D.; Tsigkas, G.; Davlouros, P.; Hahalis, G. Inflammation, Thrombosis, and Platelet-to-Lymphocyte Ratio in Acute Coronary Syndromes. Angiology 2021, 72, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Vakili, H.; Shirazi, M.; Charkhkar, M.; Khaheshi, I.; Memaryan, M.; Naderian, M. Correlation of platelet-to-lymphocyte ratio and neutrophil-to-lymphocyte ratio with thrombolysis in myocardial infarction frame count in ST-segment elevation myocardial infarction. Eur. J. Clin. Investig. 2017, 47, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Seaoud, E.; Mohamed, A.A.H.A.; Elkot, M.A. The Role of the Platelet/Lymphocyte Ratio and Neutrophil/Lymphocyte Ratio in Predicting High-Risk Heart Score in Patients Admitted with Non-ST Elevation Acute Coronary Syndrome. Pulse 2020, 8, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Oylumlu, M.; Oylumlu, M.; Arslan, B.; Polat, N.; Özbek, M.; Demir, M.; Yildiz, A.; Toprak, N. Platelet-to-lymphocyte ratio is a predictor of long-term mortality in patients with acute coronary syndrome. Postępy Kardiol. Interwencyjnej Adv. Interv. Cardiol 2020, 16, 170–176. [Google Scholar] [CrossRef]
- Li, H.; Zhou, Y.; Ma, Y.; Han, S.; Zhou, L. The prognostic value of the platelet-to-lymphocyte ratio in acute coronary syndrome: A systematic review and meta-analysis. Kardiol. Polska 2017, 75, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Liu, Q.; Tang, Y. Platelet to lymphocyte ratio in the prediction of adverse outcomes after acute coronary syndrome: A meta-analysis. Sci. Rep. 2017, 7, 40426. [Google Scholar] [CrossRef]
- Li, L.; Ma, Y.; Geng, X.; Tan, Z.; Wang, J.; Cui, C.; Wang, H.-L.; Shang, X. Platelet-to-lymphocyte ratio relates to poor prognosis in elderly patients with acute myocardial infarction. Aging Clin. Exp. Res. 2021, 33, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, A.; Yuksel, M.; Oylumlu, M.; Polat, N.; Akyuz, A.; Acet, H.; Aydin, M.; Ülgen, M.S. The Utility of the Platelet–Lymphocyte Ratio for Predicting No Reflow in Patients With ST-Segment Elevation Myocardial Infarction. Clin. Appl. Thromb. 2014, 21, 223–228. [Google Scholar] [CrossRef]
- Ayça, B.; Akın, F.; Celik, O.; Sahin, I.; Yildiz, S.S.; Avci, I.I.; Gulsen, K.; Okuyan, E.; Dinckal, M.H. Neutrophil to Lymphocyte Ratio is Related to Stent Thrombosis and High Mortality in Patients With Acute Myocardial Infarction. Angiology 2014, 66, 545–552. [Google Scholar] [CrossRef]
- Anwar, I.W.; Wijaya, I.P.; Sukrisman, L.; A Nasution, S.; Rumende, C.M. Diagnostic Accuracy of Platelet/Lymphocyte Ratio for Screening Complex Coronary Lesion in Different Age Group of Patients with Acute Coronary Syndrome. Acta Med. Indones. 2018, 50, 185–192. [Google Scholar] [PubMed]
- Mansiroglu, A.K.; Sincer, I.; Gunes, Y. Assessment of neutrophil and neutrophil/lymphocyte ratio in coronary collateral developed patients with acute coronary syndrome. Revista da Associação Médica Brasileira 2020, 66, 954–959. [Google Scholar] [CrossRef]
- Tamhane, U.U.; Aneja, S.; Montgomery, D.; Rogers, E.-K.; Eagle, K.A.; Gurm, H.S. Association Between Admission Neutrophil to Lymphocyte Ratio and Outcomes in Patients With Acute Coronary Syndrome. Am. J. Cardiol. 2008, 102, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Tenekecioglu, E.; Arslan, B.; Bekler, A.; Ozluk, O.A.; Karaagac, K.; Agca, F.V.; Peker, T.; Akgumus, A. White Blood Cell Subtypes and Neutrophil–Lymphocyte Ratio in Prediction of Coronary Thrombus Formation in Non-ST-Segment Elevated Acute Coronary Syndrome. Clin. Appl. Thromb. 2013, 21, 446–452. [Google Scholar] [CrossRef]
- Oylumlu, M.; Yildiz, A.; Oylumlu, M.; Yuksel, M.; Polat, N.; Bilik, M.Z.; Akyuz, A.; Aydin, M.; Acet, H.; Soydinc, S. Platelet-to-lymphocyte ratio is a predictor of in-hospital mortality patients with acute coronary syndrome. Anatol. J. Cardiol. 2015, 15, 277–283. [Google Scholar] [CrossRef]
- Wagdy, S.; Sobhy, M.; Loutfi, M. Neutrophil/Lymphocyte Ratio as a Predictor of In-Hospital Major Adverse Cardiac Events, New-Onset Atrial Fibrillation, and No-Reflow Phenomenon in Patients with ST Elevation Myocardial Infarction. Clin. Med. Insights Cardiol. 2016, 10, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Badran, H.M.; Fatah, A.A.; Soltan, G. Platelet/ lymphocyte ratio for prediction of no reflow phenomenon in ST elevation myocardial infarction managed with primary percutanous coronary intervention. J. Clin. Transl. Res. 2020, 6, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xie, Z.; Liu, X.; Huang, X.; Lin, J.; Huang, D.; Yu, B.; Hou, J. Association of Platelet to lymphocyte ratio with non-culprit atherosclerotic plaque vulnerability in patients with acute coronary syndrome: An optical coherence tomography study. BMC Cardiovasc. Disord. 2017, 17, 175. [Google Scholar] [CrossRef] [Green Version]
- Seyit, M.; Avci, E.; Nar, R.; Senol, H.; Yilmaz, A.; Ozen, M.; Oskay, A.; Aybek, H. Neutrophil to lymphocyte ratio, lymphocyte to monocyte ratio and platelet to lymphocyte ratio to predict the severity of COVID-19. Am. J. Emerg. Med. 2021, 40, 110–114. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Shang, Y.; Wang, J.; Zhang, X.; Su, D.; Zhao, S.; Wang, Q.; Liu, L.; Li, Y.; et al. Ratios of neutrophil-to-lymphocyte and platelet-to-lymphocyte predict all-cause mortality in inpatients with coronavirus disease 2019 (COVID-19): A retrospective cohort study in a single medical centre. Epidemiol. Infect. 2020, 148, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.S.; Rout, A. Use of Neutrophil-to-Lymphocyte and Platelet-to-Lymphocyte Ratios in COVID-19. J. Clin. Med. Res. 2020, 12, 448–453. [Google Scholar] [CrossRef]
- Wang, L.; He, W.; Yu, X.; Hu, D.; Bao, M.; Liu, H.; Zhou, J.; Jiang, H. Coronavirus disease 2019 in elderly patients: Characteristics and prognostic factors based on 4-week follow-up. J. Infect. 2020, 80, 639–645. [Google Scholar] [CrossRef]
- Gao, S.; Jiang, F.; Jin, W.; Shi, Y.; Yang, L.; Xia, Y.; Jia, L.; Wang, B.; Lin, H.; Cai, Y.; et al. Risk factors influencing the prognosis of elderly patients infected with COVID-19: A clinical retrospective study in Wuhan, China. Aging 2020, 12, 12504–12516. [Google Scholar] [CrossRef]
- Gao, Y.M.; Xu, G.; Wang, B.; Liu, B.C. Cytokine storm syndrome in coronavirus disease 2019: A narrative review. J. Intern. Med. 2021, 289, 147–161. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sinha, A.; Banach, M.; Mittoo, S.; Weissert, R.; Kass, J.S. Cytokine storm in COVID-19—Immunopathological mechanisms, clinical considerations, and therapeutic approaches: The REPROGRAM consortium position paper. Front. Immunol. 2020, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine storm in COVID-19: The current evidence and treatment strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef]
- Bastug, A.; Bodur, H.; Erdogan, S.; Gokcinar, D.; Kazancioglu, S.; Kosovali, B.D. Clinical and laboratory features of COVID-19: Predictors of severe prognosis. Int. Immunopharmacol. 2020, 88, 106950. [Google Scholar] [CrossRef]
- Olwal, C.O.; Nganyewo, N.N.; Tapela, K.; Djomkam Zune, A.L.; Owoicho, O.; Bediako, Y.; Duodu, S. Parallels in Sepsis and COVID-19 Conditions: Implications for Managing Severe COVID-19. Front. Immunol. 2021, 12, 602848. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Connors, J.M.; Warkentin, T.E.; Thachil, J.; Levi, M. The unique characteristics of COVID-19 coagulopathy. Crit. Care 2020, 24, 360. [Google Scholar] [CrossRef]
- Sims, J.T.; Krishnan, V.; Chang, C.Y.; Engle, S.M.; Casalini, G.; Rodgers, G.H. Characterization of the cytokine storm reflects hyperinflammatory endothelial dysfunction in COVID-19. J. Allergy Clin. Immunol. 2021, 147, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Englisch, C.N.; Tschernig, T.; Flockerzi, F.; Meier, C.; Bohle, R.M. Lesions in the lungs of fatal corona virus disease Covid-19. Ann. Anat. Anat. Anz. 2021, 234, 151657. [Google Scholar] [CrossRef]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef] [PubMed]
- Ulhaq, Z.S.; Soraya, G.V. Interleukin-6 as a potential biomarker of COVID-19 progression. Med. Mal. Infect. 2020, 50, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; McGuire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. In Vivo 2013, 27, 669–684. [Google Scholar] [PubMed]
- Webb, B.J.; Peltan, I.D.; Jensen, P.; Hoda, D.; Hunter, B.; Silver, A. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: A cohort study. Lancet Rheumatol. 2020, 2, e754–e763. [Google Scholar] [CrossRef]
- Ponti, G.; Maccaferri, M.; Ruini, C.; Tomasi, A.; Ozben, T. Biomarkers associated with COVID-19 disease progression. Crit. Rev. Clin. Lab. Sci. 2020, 57, 389–399. [Google Scholar] [CrossRef]
- Katneni, U.K.; Alexaki, A.; Hunt, R.C.; Schiller, T.; DiCuccio, M.; Buehler, P.W. Coagulopathy and thrombosis as a result of severe COVID-19 infection: A microvascular focus. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Benedek, T. Pulmonary Embolism—A Thrombo-Inflammatory Complication of a Viral Infection. The Paradigm Shift in the COVID Era. J. Cardiovasc. Emergencies 2021, 7, 1–2. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, X.; Fan, Q.; Liu, H.; Liu, X.; Liu, Z.; Zhang, Z. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J. Thromb. Haemost. 2020, 18, 1324–1329. [Google Scholar] [CrossRef]
- Chauhan, A.J.; Wiffen, L.J.; Brown, T.P. COVID-19: A collision of complement, coagulation and inflammatory pathways. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2020, 17, 46–64. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. Am. Soc. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertano, D.M.; Sutanto, H.; Wungu, C.D.K. Immunomodulation as a potent COVID pharmacotherapy:past, present and future. Preprints 2021. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef]
- Wichmann, D.; Sperhake, J.P.; Lütgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A. Autopsy findings and venous thromboembolism in patients with COVID-19: A prospective cohort study. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef]
- Hippensteel, J.A.; Burnham, E.L.; Jolley, S.E. Prevalence of venous thromboembolism in critically ill patients with COVID-19. Br. J. Haematol. 2020, 190, e134–e137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Feng, X.; Zhang, D.; Jiang, C.; Mei, H.; Wang, J. Deep vein thrombosis in hospitalized patients with COVID-19 in Wuhan, China: Prevalence, risk factors, and outcome. Circulation 2020, 142, 114–128. [Google Scholar] [CrossRef]
- Correale, M.; Tricarico, L.; Fortunato, M.; Dattilo, G.; Iacoviello, M.; Brunetti, N.D. Infection, atherothrombosis and thromboembolism beyond the COVID-19 disease: What similar in physiopathology and researches. Aging Clin. Exp. Res. 2021, 1–6. [Google Scholar] [CrossRef]
- Li, X.; Guan, B.; Su, T.; Liu, W.; Chen, M.; Bin Waleed, K.; Guan, X.; Gary, T.; Zhu, Z. Impact of cardiovascular disease and cardiac injury on in-hospital mortality in patients with COVID-19: A systematic review and meta-analysis. Heart 2020, 106, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Ssentongo, P.; Ssentongo, A.E.; Heilbrunn, E.S.; Ba, D.M.; Chinchilli, V.M. Association of cardiovascular disease and 10 other pre-existing comorbidities with COVID-19 mortality: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0238215. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Gheibi Hayat, S.M.; Taghizadeh, H.; Akbari, A.; Inabadi, M.; Savardashtaki, A. COVID-19 and cardiac injury: Clinical manifestations, biomarkers, mechanisms, diagnosis, treatment, and follow up. Expert Rev. Anti-Infect. Ther. 2020, 1–13. [Google Scholar] [CrossRef]
- Myhre, P.L.; Prebensen, C.; Strand, H.; Røysland, R.; Jonassen, C.M.; Rangberg, A. Growth Differentiation Factor 15 Provides Prognostic Information Superior to Established Cardiovascular and Inflammatory Biomarkers in Unselected Patients Hospitalized With COVID-19. Circulation 2020, 142, 2128–2137. [Google Scholar] [CrossRef]
- Schiavone, M.; Gobbi, C.; Biondi-Zoccai, G.; D’ascenzo, F.; Palazzuoli, A.; Gasperetti, A. Acute coronary syndromes and Covid-19: Exploring the uncertainties. J. Clin. Med. 2020, 9, 1683. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.C.; Zhang, R.T.; Guo, L.J. Hypoxia and inflammation are risk factors for acute myocardial injury in patients with coronavirus disease 2019. Peking Univ. Health Sci. 2020, 53, 159–166. [Google Scholar] [CrossRef]
- Moriarty, P.M.; Gorby, L.K.; Stroes, E.S.; Kastelein, J.P.; Davidson, M.; Tsimikas, S. Lipoprotein (a) and its potential association with thrombosis and inflammation in COVID-19: A testable hypothesis. Curr. Atheroscler. Rep. 2020, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study | Data Collection Period | Patients | Age | Hemorheologic Ratio | Conclusions |
|---|---|---|---|---|---|
| Li et al. (2020) [26] | 2012–2016 | 1001 patients with acute myocardial infarction (AMI) and primary percutaneous coronary intervention (PPCI) | 441—49.7 ± 7.2 560—67.3 ± 5.6 | PLR 165 ± 79 PLR 190 ± 107 p = 0.001 | PLR is az independent predictor for aparition of adverse events during the hospitalization |
| Ayça et al. (2014) [28] | 2010–2013 | 102 patients with stent thrombosis 450 patients with STEMI | 54.6 + 11.1 58.3 + 7.4 | NLR 7.00 + 5.77 4.60 + 3.87 p < 0.001 | Higher NLR was associated with higher mortality rate in each group. Increased NLR can anticipate stent thrombosis and is associated with higher mortality rates in patients with STEMI. |
| Wahjuni et al. (2018) [29] | 2012–2015 | 125 patients with acute coronary syndrome | n = 60, ≤45 years Gensini score > 53 (n = 23) Gensini score ≤ 53 (n = 37) n = 65, >45 years Gensini score > 53 (n = 36) Gensini score ≤ 53 (n = 29) | PLR 171.08 ± 83.54 88.51 ± 24.28 209.91 ± 164.45 133.01 ± 108.22 | Optimum cut-off point for PLR was 111.06 for patients aged ≤45 years and 104.78 for patients aged >45 years |
| Mansiroglu et al. (2020) [30] | 2015–2018 | 426 patients who undervent coronary angiography for acute coronary syndrome n = 102 unstable angina pectoris n = 223 non-STEMI n = 103 STEMI | 64 ± 12 67 ± 12 67 ± 13 | NLR < 0.001 2.92 ± 2.39 5.19 ± 4.80 7.93 ± 6.38 | Statistically significant difference in the number of neutrophil counts and NLR between the types of acute coronary syndromes |
| Tamhane et al. (2008) [31] | 1998–2004 | 2833 patients with ACS n = 564 STEMI n = 2269 non-STEMI | Low NLR n = 935 61 ± 13 years Medium NLR n = 948 65 ± 14 years High NLR n = 948 67 ± 13.8 years | NLR 1.82 (0.75) 3.56 (1.36) 9.10 (7.27) | NLR at admission can be successfully used for prediction of in-hospital and 6-month mortality |
| Yilmaz et al. (2015) [32] | No data available | 251 patients with non-STEMI n = 82 without coronary thrombus n = 169 with coronary thrombus | 60.68 ± 11.73 years 61.37 ± 12.34 years | NLR p < 0.001 3.17 + 1.52 4.12 + 1.89 | Leukocyte count and NLR can be used to predict the presence of absence of a coronary thrombus. |
| Yildiz et al. (2014) [27] | No data available | 287 patients with STEMI grouped by PLR n = 96 PLR: 88.2 (84.6–91.8) n = 96 PLR: 135.2 (132.0–138.4) n = 95 PLR: 231.7 (220.5–242.8) | 57.6 + 13.3 years 60.2 + 13.7 years 64.5 + 13.2 years | NLR p < 0.001 3.44 + 1.47 5.24 + 2.20 8.44 + 3.83 | Elevated PLR and NLR were indipendently and strongly associated with the no-reflow phenomenon in STEMI |
| Oylumlu et al. (2014) [33] | 2012–2013 | 587 patients with acute coronary syndrome grouped by PLR n = 195 PLR: 83.9 ± 15.4 n = 196 PLR: 127.0 ± 13.8 n = 196 PLR: 214.0 ± 71.8 | 59.0 ± 12.2 61.7 ± 12.7 64.7 ± 13.7 | NLR p < 0.001 2.50 (1.86–3.57) 4.11 (2.88–5.46) 7.04 (4.57–10.15) | An increased PLR can be an independent predictor of in-hospital mortality |
| Wagdy et al. (2016) [34] | 2013 | 200 patients with STEMI grouped by the final TIMI flow n = 165-normal flow after PPCI n = 35–no reflow | No data available | NLR 5.44 ± 3.53 8.19 ± 3.05 | NLR can predict the no-reflow phenomenon or in-hospital major advers cardiac event with 90.4% sensitivity and 51.5% specificity |
| Badran et al. (2020) [35] | 2017–2019 | 200 patients with STEMI grouped by TIMI flow n = 58–TIMI 0-II n = 142–TIMI III | 52.9 ± 11.1 years | PLR p = 0.001 199.4 ± 52 102 ± 53 | Elevated pre-procedural PLR was predictive of the no-reflow phenomenon in STEMI |
| Wang et al. (2017) [36] | No data available | 119 non-culprit plaques from 71 patients with ACS grouped by PLR assesed with optical coherence tomography n = 35 patients with 50 plaques (high) PLR > 109 n = 36 patients with 69 plaques (low) PLR < 109 | 59.77 ± 8.88 years 57.97 ± 10.51 years | Platelets, ×109/L 230.89 ± 45.47 202.75 ± 43.57 | Increased PLR can be linked with vulnerable plaque features of non-culprit lesions |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandor-Keri, J.; Benedek, I.; Polexa, S.; Benedek, I. The Link between SARS-CoV-2 Infection, Inflammation and Hypercoagulability-Impact of Hemorheologic Alterations on Cardiovascular Mortality. J. Clin. Med. 2021, 10, 3015. https://doi.org/10.3390/jcm10143015
Sandor-Keri J, Benedek I, Polexa S, Benedek I. The Link between SARS-CoV-2 Infection, Inflammation and Hypercoagulability-Impact of Hemorheologic Alterations on Cardiovascular Mortality. Journal of Clinical Medicine. 2021; 10(14):3015. https://doi.org/10.3390/jcm10143015
Chicago/Turabian StyleSandor-Keri, Johanna, Istvan Benedek, Stefania Polexa, and Imre Benedek. 2021. "The Link between SARS-CoV-2 Infection, Inflammation and Hypercoagulability-Impact of Hemorheologic Alterations on Cardiovascular Mortality" Journal of Clinical Medicine 10, no. 14: 3015. https://doi.org/10.3390/jcm10143015
APA StyleSandor-Keri, J., Benedek, I., Polexa, S., & Benedek, I. (2021). The Link between SARS-CoV-2 Infection, Inflammation and Hypercoagulability-Impact of Hemorheologic Alterations on Cardiovascular Mortality. Journal of Clinical Medicine, 10(14), 3015. https://doi.org/10.3390/jcm10143015