
Neurogranin and Neuronal Pentraxin Receptor as Synaptic Dysfunction Biomarkers in Alzheimer’s Disease

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. The CSF Concentrations of Ng and NPTXR as Synaptic Biomarkers
2.2. Associations between CSF Levels of Ng, NPTXR and Neurochemical Biomarkers (Aβ42/40 Ratio, Tau, pTau181)
3. Discussion
4. Materials and Methods
4.1. Study Population and Diagnostic Criteria
4.2. Biochemical Evaluation
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor |
| Aβ | amyloid β |
| Aβo | amyloid β oligomers |
| CaM | calmodulin |
| CJD | Creutzfeldt–Jakob disease |
| CSF | Cerebrospinal Fluid |
| CT | computer tomography |
| CTRL | controls |
| ELISA | enzyme-linked immunosorbent assay |
| GAP-43 | Growth Associated Protein 43 |
| GluA4 | glutamate ionotropic receptor AMPA type subunit 4 |
| LTD | long-term depression |
| LTP | long-term potentiation |
| MCI | Mild cognitive impairment |
| MRI | magnetic resonance image |
| NDD | neurochemical dementia biomarkers |
| Ng | Neurogranin |
| NMDAR | N-methyl-D-aspartate receptor |
| NPTXR | Neuronal pentraxin receptor |
| PET | Positron Emission Tomography |
| pTau181 | phosphorylation Tau protein (Threonine 181) |
| SNAP-25 | Synaptosomal-Associated Protein 25 |
References
- Dhiman, K.; Blennow, K.; Zetterberg, H.; Martins, R.N.; Gupta, V.B. Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer’s disease pathogenesis. Cell. Mol. Life Sci. 2019, 76, 1833–1863. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Camporesi, E.; Nilsson, J.; Brinkmalm, A.; Becker, B.; Ashton, N.J.; Blennow, K.; Zetterberg, H. Fluid Biomarkers for Synaptic Dysfunction and Loss. Biomark. Insights 2020, 15, 117727192095031. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Abraham, W.C.; Jones, O.D.; Glanzman, D.L. Is plasticity of synapses the mechanism of long-term memory storage? NPJ Sci. Learn. 2019, 4, 9. [Google Scholar] [CrossRef]
- Blennow, K. A Review of Fluid Biomarkers for Alzheimer’s Disease: Moving from CSF to Blood. Neurol. Ther. 2017, 6, 15–24. [Google Scholar] [CrossRef]
- Jackson, J.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasinelli, P.; Ramakers, G.M.J.; Urban, I.J.A.; Hens, J.J.H.; Oestreicher, A.B.; de Graan, P.N.E.; Gispen, W.H. Long-term potentiation and synaptic protein phosphorylation. Behav. Brain Res. 1995, 66, 53–59. [Google Scholar] [CrossRef] [Green Version]
- LYNCH, M.A. Long-Term Potentiation and Memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H. Calmodulin Binding Proteins and Alzheimer’s Disease: Biomarkers, Regulatory Enzymes and Receptors That Are Regulated by Calmodulin. Int. J. Mol. Sci. 2020, 21, 7344. [Google Scholar] [CrossRef]
- Wang, J.H.; Kelly, P.T. Postsynaptic injection of Ca2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron 1995, 15, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Gengler, S.; Hamilton, A.; Hölscher, C. Synaptic Plasticity in the Hippocampus of a APP/PS1 Mouse Model of Alzheimer’s Disease Is Impaired in Old but Not Young Mice. PLoS ONE 2010, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mango, D.; Saidi, A.; Cisale, G.Y.; Feligioni, M.; Corbo, M.; Nisticò, R. Targeting Synaptic Plasticity in Experimental Models of Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hopf, C.; Reddy, R.; Cho, R.W.; Guo, L.; Lanahan, A.; Petralia, R.S.; Wenthold, R.J.; O’Brien, R.J.; Worley, P. Narp and NP1 Form Heterocomplexes that Function in Developmental and Activity-Dependent Synaptic Plasticity. Neuron 2003, 39, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y. Long-term potentiation: Two pathways meet at neurogranin. EMBO J. 2009, 28, 2859–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauterborn, J.C.; Scaduto, P.; Cox, C.D.; Schulmann, A.; Lynch, G.; Gall, C.M.; Keene, C.D.; Limon, A. Increased excitatory to inhibitory synaptic ratio in parietal cortex samples from individuals with Alzheimer’s disease. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Radde, R.; Bolmont, T.; Kaeser, S.A.; Coomaraswamy, J.; Lindau, D.; Stoltze, L.; Calhoun, M.E.; Jäggi, F.; Wolburg, H.; Gengler, S.; et al. Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006, 7, 940–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, N.J.; Wegenast-Braun, B.M.; Radde, R.; Calhoun, M.E.; Jucker, M. Early onset amyloid lesions lead to severe neuritic abnormalities and local, but not global neuron loss in APPPS1 transgenic mice. Neurobiol. Aging 2011, 32, 2324.e1–2324.e6. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. The Past and the Future of Alzheimer’s Disease Fluid Biomarkers. J. Alzheimers Dis. 2018, 62, 1125–1140. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.F.; White, G.L.; Jones, M.W.; Cooper-Blacketer, D.; Marshall, V.J.; Irizarry, M.; Younkin, L.; Good, M.A.; Bliss, T.V.P.; Hyman, B.T.; et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat. Neurosci. 1999, 2, 271–276. [Google Scholar] [CrossRef]
- Parihar, M.S.; Brewer, G.J. Amyloid-β as a Modulator of Synaptic Plasticity. J. Alzheimers Dis. 2010, 22, 741–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The role of NMDA receptors in Alzheimer’s disease. Front. Neurosci. 2019, 13, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Freitas, A.E.; Gorodetski, L.; Wang, J.; Tian, R.; Lee, Y.R.; Grewal, A.S.; Zou, Y. Planar cell polarity signaling components are a direct target of β-amyloid–associated degeneration of glutamatergic synapses. Sci. Adv. 2021, 7, eabh2307. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Luscher, C.; Malenka, R.C. NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [Green Version]
- Citri, A.; Malenka, R.C. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [Green Version]
- Neveu, D.; Zucker, R.S. Postsynaptic Levels of [Ca2+]i Needed to Trigger LTD and LTP. Neuron 1996, 16, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Hanley, J.G. The Regulation of AMPA Receptor Endocytosis by Dynamic Protein-Protein Interactions. Front. Cell. Neurosci. 2018, 12, 1–10. [Google Scholar] [CrossRef]
- Pelkey, K.A.; Barksdale, E.; Craig, M.T.; Yuan, X.; Sukumaran, M.; Vargish, G.A.; Mitchell, R.M.; Wyeth, M.S.; Petralia, R.S.; Chittajallu, R.; et al. Pentraxins Coordinate Excitatory Synapse Maturation and Circuit Integration of Parvalbumin Interneurons. Neuron 2015, 85, 1257–1272. [Google Scholar] [CrossRef] [Green Version]
- Öhrfelt, A.; Brinkmalm, A.; Dumurgier, J.; Zetterberg, H.; Bouaziz-Amar, E.; Hugon, J.; Paquet, C.; Blennow, K. A Novel ELISA for the Measurement of Cerebrospinal Fluid SNAP-25 in Patients with Alzheimer’s Disease. Neuroscience 2019, 420, 136–144. [Google Scholar] [CrossRef]
- Clarke, M.T.M.; Brinkmalm, A.; Foiani, M.S.; Woollacott, I.O.C.; Heller, C.; Heslegrave, A.; Keshavan, A.; Fox, N.C.; Schott, J.M.; Warren, J.D.; et al. CSF synaptic protein concentrations are raised in those with atypical Alzheimer’s disease but not frontotemporal dementia. Alzheimers Res. Ther. 2019, 11, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerendasy, D. Homeostatic tuning of Ca2+ signal transduction by members of the calpacitin protein family. J. Neurosci. Res. 1999, 58, 107–119. [Google Scholar] [CrossRef]
- Merluzzi, A.P.; Vogt, N.M.; Norton, D.; Jonaitis, E.; Clark, L.R.; Carlsson, C.M.; Johnson, S.C.; Asthana, S.; Blennow, K.; Zetterberg, H.; et al. Differential effects of neurodegeneration biomarkers on subclinical cognitive decline. Alzheimers Dement. Transl. Res. Clin. Interv. 2019, 5, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Kvartsberg, H.; Duits, F.H.; Ingelsson, M.; Andreasen, N.; Öhrfelt, A.; Andersson, K.; Brinkmalm, G.; Lannfelt, L.; Minthon, L.; Hansson, O.; et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimers Dement. 2015, 11, 1180–1190. [Google Scholar] [CrossRef]
- Tarawneh, R.; Head, D.; Allison, S.; Buckles, V.; Fagan, A.M.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. Cerebrospinal Fluid Markers of Neurodegeneration and Rates of Brain Atrophy in Early Alzheimer Disease. JAMA Neurol. 2015, 72, 656. [Google Scholar] [CrossRef] [Green Version]
- Lim, B.; Sando, S.B.; Grøntvedt, G.R.; Bråthen, G.; Diamandis, E.P. Cerebrospinal fluid neuronal pentraxin receptor as a biomarker of long-term progression of Alzheimer’s disease: A 24-month follow-up study. Neurobiol. Aging 2020, 93. [Google Scholar] [CrossRef]
- Cummings, D.M.; Benway, T.A.; Ho, H.; Tedoldi, A.; Fernandes Freitas, M.M.; Shahab, L.; Murray, C.E.; Richard-Loendt, A.; Brandner, S.; Lashley, T.; et al. Neuronal and Peripheral Pentraxins Modify Glutamate Release and may Interact in Blood–Brain Barrier Failure. Cereb. Cortex 2017, 27, 3437–3448. [Google Scholar] [CrossRef] [PubMed]
- Lleó, A.; Núñez-Llaves, R.; Alcolea, D.; Chiva, C.; Balateu-Paños, D.; Colom-Cadena, M.; Gomez-Giro, G.; Muñoz, L.; Querol-Vilaseca, M.; Pegueroles, J.; et al. Changes in Synaptic Proteins Precede Neurodegeneration Markers in Preclinical Alzheimer’s Disease Cerebrospinal Fluid. Mol. Cell. Proteom. 2019, 18, 546–560. [Google Scholar] [CrossRef] [Green Version]
- Mecca, A.P.; Chen, M.; O’Dell, R.S.; Naganawa, M.; Toyonaga, T.; Godek, T.A.; Harris, J.E.; Bartlett, H.H.; Zhao, W.; Nabulsi, N.B.; et al. In vivo measurement of widespread synaptic loss in Alzheimer’s disease with SV2A PET. Alzheimers Dement. 2020, 16, 974–982. [Google Scholar] [CrossRef]
- Park, S.A.; Han, S.M.; Kim, C.E. New fluid biomarkers tracking non-amyloid-β and non-tau pathology in Alzheimer’s disease. Exp. Mol. Med. 2020, 52, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Gerges, N.Z. Neurogranin and synaptic plasticity balance. Commun. Integr. Biol. 2010, 3, 340–342. [Google Scholar] [CrossRef]
- Petersen, A.; Gerges, N.Z. Neurogranin regulates CaM dynamics at dendritic spines. Sci. Rep. 2015, 5, 11135. [Google Scholar] [CrossRef] [Green Version]
- Zhabotinsky, A.M.; Camp, R.N.; Epstein, I.R.; Lisman, J.E. Role of the Neurogranin Concentrated in Spines in the Induction of Long-Term Potentiation Synaptic plasticity in CA1 hippocampal neurons depends on Ca 2 elevation and the resulting activation of calmodulin-dependent enzymes. Induction of long-term depression (LTD) depends on calcineurin, whereas long-term potentiation (LTP) depends on Ca 2/calmodulin-dependent protein kinase II (CaMKII). J. Neurosci. 2006, 26, 7337–7347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Gerges, N.Z. Neurogranin Regulates Metaplasticity. Front. Mol. Neurosci. 2020, 12, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tarawneh, R.; D’Angelo, G.; Crimmins, D.; Herries, E.; Griest, T.; Fagan, A.M.; Zipfel, G.J.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M.; et al. Diagnostic and Prognostic Utility of the Synaptic Marker Neurogranin in Alzheimer Disease. JAMA Neurol. 2016, 73, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Zetterberg, H.; Skillbäck, T.; Törnqvist, U.; Andreasson, U.; Trojanowski, J.Q.; Weiner, M.W.; Shaw, L.M.; Mattsson, N.; Blennow, K. Cerebrospinal fluid neurogranin: Relation to cognition and neurodegeneration in Alzheimer’s disease. Brain 2015, 138, 3373–3385. [Google Scholar] [CrossRef] [Green Version]
- Kvartsberg, H.; Lashley, T.; Murray, C.E.; Brinkmalm, G.; Cullen, N.C.; Höglund, K.; Zetterberg, H.; Blennow, K.; Portelius, E. The intact postsynaptic protein neurogranin is reduced in brain tissue from patients with familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2019, 137, 89–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, J.B.; Westman, E.; Hansson, O. Association between cerebrospinal fluid and plasma neurodegeneration biomarkers with brain atrophy in Alzheimer’s disease. Neurobiol. Aging 2017, 58, 14–29. [Google Scholar] [CrossRef]
- Mattsson, N.; Insel, P.S.; Palmqvist, S.; Portelius, E.; Zetterberg, H.; Weiner, M.; Blennow, K.; Hansson, O. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer’s disease. EMBO Mol. Med. 2016, 8, 1184–1196. [Google Scholar] [CrossRef]
- Dulewicz, M.; Kulczyńska-Przybik, A.; Mroczko, B. Neurogranin and VILIP-1 as Molecular Indicators of Neurodegeneration in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 8335. [Google Scholar] [CrossRef]
- Dodds, D.C.; Omeis, I.A.; Cushman, S.J.; Helms, J.A.; Perin, M.S. Neuronal Pentraxin Receptor, a Novel Putative Integral Membrane Pentraxin That Interacts with Neuronal Pentraxin 1 and 2 and Taipoxin-associated Calcium-binding Protein 49. J. Biol. Chem. 1997, 272, 21488–21494. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, L.L.; Matzuk, M.M.; Dodds, D.C.; Perin, M.S. Biochemical Interactions of the Neuronal Pentraxins. J. Biol. Chem. 2000, 275, 17786–17792. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Wei, M.; Zhang, C.; Maxeiner, S.; Pak, C.H.; Botelho, S.C.; Trotter, J.; Sterky, F.H.; Südhof, T.C. Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses. J. Neurosci. 2017, 37, 1062–1080. [Google Scholar] [CrossRef]
- O’Brien, R.; Xu, D.; Mi, R.; Tang, X.; Hopf, C.; Worley, P. Synaptically Targeted Narp Plays an Essential Role in the Aggregation of AMPA Receptors at Excitatory Synapses in Cultured Spinal Neurons. J. Neurosci. 2002, 22, 4487–4498. [Google Scholar] [CrossRef] [PubMed]
- Begcevic, I.; Tsolaki, M.; Brinc, D.; Brown, M.; Martinez-Morillo, E.; Lazarou, I.; Kozori, M.; Tagaraki, F.; Nenopoulou, S.; Gkioka, M.; et al. Neuronal pentraxin receptor-1 is a new cerebrospinal fluid biomarker of Alzheimer’s disease progression. F1000Research 2018, 7, 1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.; Fowler, C.; Li, Q.-X.; Rowe, C.; Dhiman, K.; Gupta, V.B.; Masters, C.L.; Doecke, J.D.; Martins, R.N.; Collins, S.; et al. Decreased cerebrospinal fluid neuronal pentraxin receptor is associated with PET-Aβ load and cerebrospinal fluid Aβ in a pilot study of Alzheimer’s disease. Neurosci. Lett. 2020, 731, 135078. [Google Scholar] [CrossRef]
- Hellwig, K.; Kvartsberg, H.; Portelius, E.; Andreasson, U.; Oberstein, T.J.; Lewczuk, P.; Blennow, K.; Kornhuber, J.; Maler, J.M.; Zetterberg, H.; et al. Neurogranin and YKL-40: Independent markers of synaptic degeneration and neuroinflammation in Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanfilippo, C.; Forlenza, O.; Zetterberg, H.; Blennow, K. Increased neurogranin concentrations in cerebrospinal fluid of Alzheimer’s disease and in mild cognitive impairment due to AD. J. Neural Transm. 2016, 123, 1443–1447. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Diaz-Lucena, D.; Zetterberg, H.; Villar-Pique, A.; Karch, A.; Vidal, E.; Hermann, P.; Schmitz, M.; Ferrer Abizanda, I.; Zerr, I.; et al. CSF neurogranin as a neuronal damage marker in CJD: A comparative study with AD. J. Neurol. Neurosurg. Psychiatry 2019, 90, 846–853. [Google Scholar] [CrossRef]
- Wellington, H.; Paterson, R.W.; Portelius, E.; Törnqvist, U.; Magdalinou, N.; Fox, N.C.; Blennow, K.; Schott, J.M.; Zetterberg, H. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology 2016, 86, 829–835. [Google Scholar] [CrossRef] [Green Version]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Boros, B.D.; Greathouse, K.M.; Gentry, E.G.; Curtis, K.A.; Birchall, E.L.; Gearing, M.; Herskowitz, J.H. Dendritic spines provide cognitive resilience against Alzheimer’s disease. Ann. Neurol. 2017, 82, 602–614. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Kaleka, K.S.; Gerges, N.Z. Neurogranin restores amyloid β-mediated synaptic transmission and long-term potentiation deficits. Exp. Neurol. 2016, 277, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Gerges, N.Z. Neurogranin Targets Calmodulin and Lowers the Threshold for the Induction of Long-Term Potentiation. PLoS ONE 2012, 7, e41275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.W.; Schumacher, E.; Coulter, P.M.; Vinters, H.V.; Watson, J.B. Dendritic Translocation of RC3/Neurogranin mRNA in Normal Aging, Alzheimer Disease and Fronto-Temporal Dementia. J. Neuropathol. Exp. Neurol. 1997, 56, 1105–1118. [Google Scholar] [CrossRef] [PubMed]
- Merluzzi, A.P.; Carlsson, C.M.; Johnson, S.C.; Schindler, S.E.; Asthana, S.; Blennow, K.; Zetterberg, H.; Bendlin, B.B. Neurodegeneration, synaptic dysfunction, and gliosis are phenotypic of Alzheimer dementia. Neurology 2018, 91, E436–E443. [Google Scholar] [CrossRef]
- De Vos, A.; Jacobs, D.; Struyfs, H.; Fransen, E.; Andersson, K.; Portelius, E.; Andreasson, U.; De Surgeloose, D.; Hernalsteen, D.; Sleegers, K.; et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimers Dement. 2015, 11, 1461–1469. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.G.; Kang, M.; Kim, Y.-S.; Kim, D.-H.; Nam, D.W.; Song, E.J.; Mook-Jung, I.; Moon, M. Intrahippocampal injection of a lentiviral vector expressing neurogranin enhances cognitive function in 5XFAD mice. Exp. Mol. Med. 2018, 50, e461. [Google Scholar] [CrossRef] [Green Version]
- Bereczki, E.; Francis, P.T.; Howlett, D.; Pereira, J.B.; Höglund, K.; Bogstedt, A.; Cedazo-Minguez, A.; Baek, J.H.; Hortobágyi, T.; Attems, J.; et al. Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimers Dement. 2016, 12, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Pak, J.H.; Huang, F.L.; Li, J.; Balschun, D.; Reymann, K.G.; Chiang, C.; Westphal, H.; Huang, K.-P. Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: A study with knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11232–11237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Pak, J.H.; Huang, F.L.; Huang, K.P. N-methyl-D-aspartate induces neurogranin/RC3 oxidation in rat brain slices. J. Biol. Chem. 1999, 274, 1294–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peineau, S.; Rabiant, K.; Pierrefiche, O.; Potier, B. Synaptic plasticity modulation by circulating peptides and metaplasticity: Involvement in Alzheimer’s disease. Pharmacol. Res. 2018, 130, 385–401. [Google Scholar] [CrossRef]
- Falgàs, N.; Ruiz-Peris, M.; Pérez-Millan, A.; Sala-Llonch, R.; Antonell, A.; Balasa, M.; Borrego-Écija, S.; Ramos-Campoy, O.; Augé, J.M.; Castellví, M.; et al. Contribution of CSF biomarkers to early-onset Alzheimer’s disease and frontotemporal dementia neuroimaging signatures. Hum. Brain Mapp. 2020, 41, 2004–2013. [Google Scholar] [CrossRef]
- Sydow, A.; Van der Jeugd, A.; Zheng, F.; Ahmed, T.; Balschun, D.; Petrova, O.; Drexler, D.; Zhou, L.; Rune, G.; Mandelkow, E.; et al. Tau-Induced Defects in Synaptic Plasticity, Learning, and Memory Are Reversible in Transgenic Mice after Switching Off the Toxic Tau Mutant. J. Neurosci. 2011, 31, 2511–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acquarone, E.; Argyrousi, E.K.; van den Berg, M.; Gulisano, W.; Fà, M.; Staniszewski, A.; Calcagno, E.; Zuccarello, E.; D’Adamio, L.; Deng, S.-X.; et al. Synaptic and memory dysfunction induced by tau oligomers is rescued by up-regulation of the nitric oxide cascade. Mol. Neurodegener. 2019, 14, 26. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.-L.; et al. Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [Green Version]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Sia, G.M.; Béïque, J.C.; Rumbaugh, G.; Cho, R.; Worley, P.F.; Huganir, R.L. Interaction of the N-Terminal Domain of the AMPA Receptor GluR4 Subunit with the Neuronal Pentraxin NP1 Mediates GluR4 Synaptic Recruitment. Neuron 2007, 55, 87–102. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewczuk, P.; Zimmermann, R.; Wiltfang, J.; Kornhuber, J. Neurochemical dementia diagnostics: A simple algorithm for interpretation of the CSF biomarkers. J. Neural Transm. 2009, 116, 1163–1167. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Median (Interquartile Range) | ||
|---|---|---|
| AD n = 28 | CTRL n = 19 | |
| Age (mean in years) | 75.5 (65.5–80.5) | 67 (64–73) |
| Gender (Female/Male) | 21/7 | 12/7 |
| MMSE score (range 0–30 p.) | 22 (18.8–23) | 28.5 (27–30) |
| Tested Variables in CSF | Median (Range of Interquartile) | p (U-Mann–Whitney) | |
|---|---|---|---|
| AD | CTRL | ||
| Aβ42/40 ratio | 0.032 (0.03–0.04) | 0.066 (0.06–0.08) | <0.001 |
| Aβ42 | 513 (460–655) | 926 (815–1004) | <0.001 |
| Tau (pg/mL) | 676 (591–1058) | 222 (191–273) | <0.001 |
| pTau181 (pg/mL) | 86.7 (73.2–122) | 37.5 (34–42.9) | <0.001 |
| Ng (ng/mL) | 920 (737–1202) | 487 (435–580) | <0.001 |
| NPTXR (pg/mL) | 13.2 (10.8–16.3) | 19 (16.9–21.6) | <0.001 |
| NPTXR/Ng ratio | 0.014 (0.009–0.019) | 0.395(0.039–0.044) | <0.001 |
| Spearman’s Rho | p | ||||
|---|---|---|---|---|---|
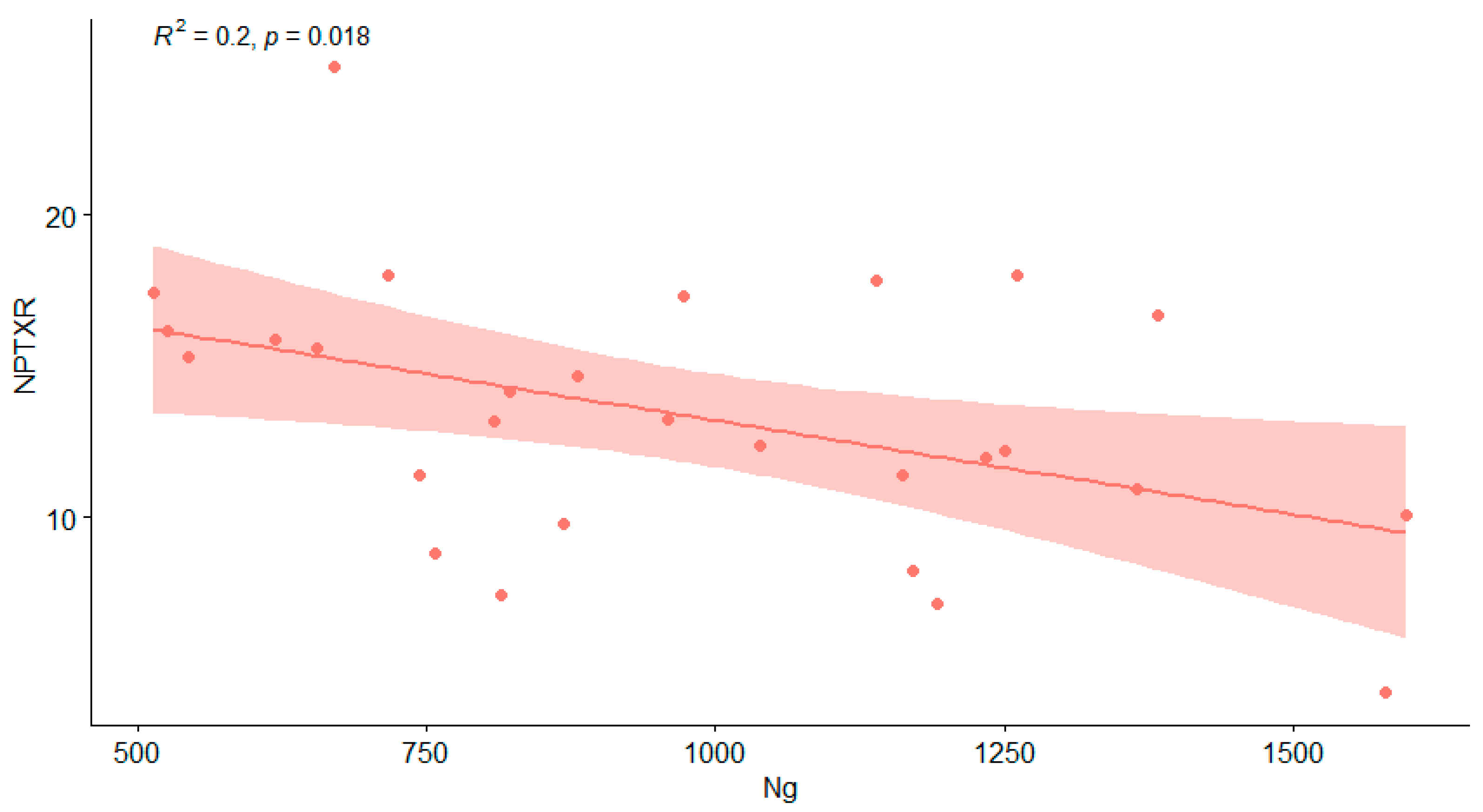
| Ng | - | NPTXR | −0.40 | * | 0.038 |
| Ng | - | pTau181 | 0.38 | * | 0.044 |
| Aβ42 | - | Aβ42/40 | 0.52 | ** | 0.004 |
| Tau | - | pTau181 | 0.88 | *** | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dulewicz, M.; Kulczyńska-Przybik, A.; Słowik, A.; Borawska, R.; Mroczko, B. Neurogranin and Neuronal Pentraxin Receptor as Synaptic Dysfunction Biomarkers in Alzheimer’s Disease. J. Clin. Med. 2021, 10, 4575. https://doi.org/10.3390/jcm10194575
Dulewicz M, Kulczyńska-Przybik A, Słowik A, Borawska R, Mroczko B. Neurogranin and Neuronal Pentraxin Receptor as Synaptic Dysfunction Biomarkers in Alzheimer’s Disease. Journal of Clinical Medicine. 2021; 10(19):4575. https://doi.org/10.3390/jcm10194575
Chicago/Turabian StyleDulewicz, Maciej, Agnieszka Kulczyńska-Przybik, Agnieszka Słowik, Renata Borawska, and Barbara Mroczko. 2021. "Neurogranin and Neuronal Pentraxin Receptor as Synaptic Dysfunction Biomarkers in Alzheimer’s Disease" Journal of Clinical Medicine 10, no. 19: 4575. https://doi.org/10.3390/jcm10194575
APA StyleDulewicz, M., Kulczyńska-Przybik, A., Słowik, A., Borawska, R., & Mroczko, B. (2021). Neurogranin and Neuronal Pentraxin Receptor as Synaptic Dysfunction Biomarkers in Alzheimer’s Disease. Journal of Clinical Medicine, 10(19), 4575. https://doi.org/10.3390/jcm10194575