
Current State of the Art and Prospects of T Cell-Redirecting Bispecific Antibodies in Multiple Myeloma

 ,
,  and
and 
Abstract
:1. Introduction
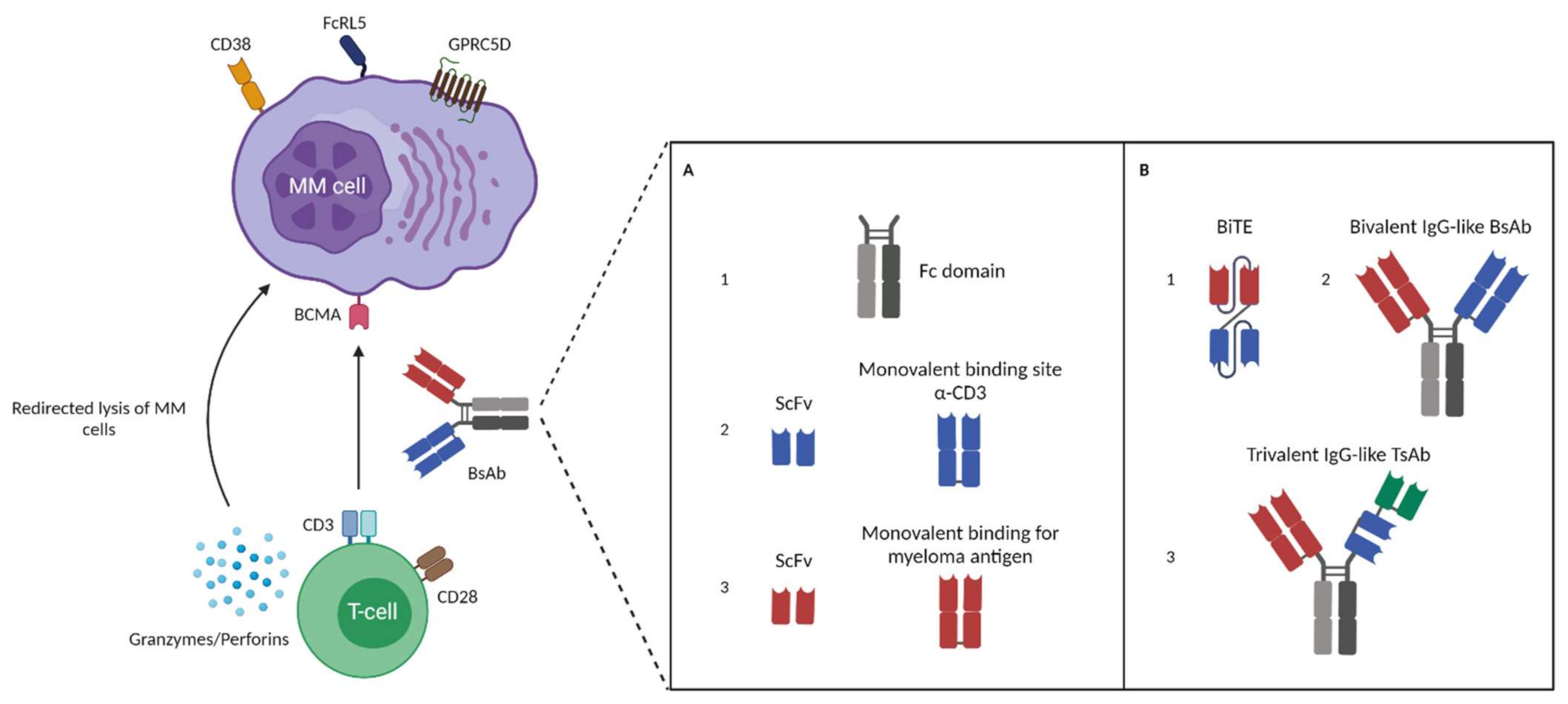
2. Bispecific Antibodies: Overview and Characteristics of Different BsAbs Formats
3. BsAb MM Targets and Their Potential as a Therapeutic Agent
3.1. B Cell Maturation Antigen (BCMA)
3.1.1. BsAbs Targeting BCMA Evaluated in Patients
Pacanalotamab (AMG 420)
Pavurutamab (AMG 701)
Elranatamab (PF-06863135)
Teclistamab (JNJ-64007957)
REGN5458
TNB-383B
Alnuctamab (CC-93269 or EM901)
3.2. CD38
3.2.1. BsAbs Targeting CD38 in the Clinic
AMG 424
GBR 1342 (ISB 1342)
3.3. B Lymphocyte Antigen CD19 (CD19)
3.3.1. BsAbs Targeting CD19 in the Clinic
Blinatumomab
3.4. Fc Receptor-Homolog 5 (FcRH5)
3.4.1. BsAbs Targeting FcRH5 in the Clinic
Cevostamab (BFCR4350A)
3.5. G Protein-Coupled Receptor Family C Group 5 Member D (GPRC5D)
3.5.1. BsAbs Targeting GPRC5D in the Clinic
Talquetamab (JNJ-64407564)
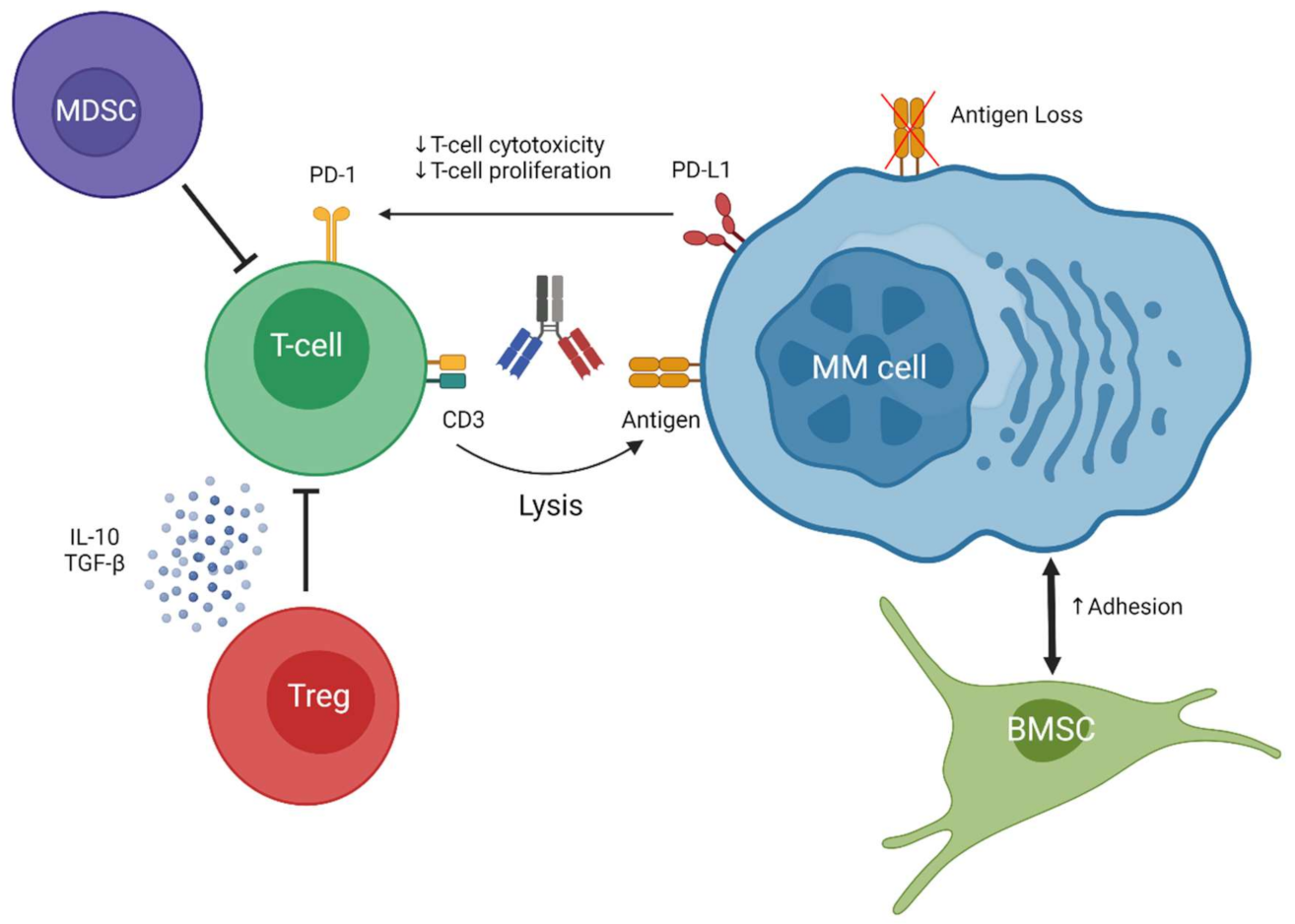
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Myeloma-Cancer Stat Facts. Available online: Seer.cancer.gov/statfacts/html/mulmy.html (accessed on 25 June 2021).
- Kurtin, S. Integrating emerging data into clinical practice: A case-based approach for multiple myeloma. J. Adv. Pract. Oncol. 2017, 8, 365–377. [Google Scholar] [PubMed]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 1–20. [Google Scholar] [CrossRef]
- Mehta, A. Multiple myeloma. Hematology 2015, 20, 58–59. [Google Scholar] [CrossRef]
- D’Agostino, M.; Bertamini, L.; Oliva, S.; Boccadoro, M.; Gay, F. Pursuing a curative approach in multiple myeloma: A review of new therapeutic strategies. Cancers 2019, 11, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood J. Am. Soc. Hematol. 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Dimopoulos, M.A.; White, D.J.; Benboubker, L.; Cook, G.; Leiba, M.; Ho, P.J.; Kim, K.; Takezako, N.; Moreau, P.; et al. Daratumumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: Extended follow-up of POLLUX, a randomized, open-label, phase 3 study. Leukemia 2020, 34, 1875–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Usmani, S.Z.; Weiss, B.M.; Plesner, T.; Bahlis, N.J.; Belch, A.; Lonial, S.; Lokhorst, H.M.; Voorhees, P.M.; Richardson, P.G.; Chari, A.; et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 2016, 128, 34–44. [Google Scholar] [CrossRef] [PubMed]
- White, D.; Grosicki, S.; Spicka, I.; Croneck, A.W.; Moreau, P.; Mateos, M.V.; Magen, H.; Belch, A.; Reece, D.; Beksac, M.; et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Nooka, A.K.; Kaufman, J.L.; Hofmeister, C.C.; Joseph, N.S.; Heffner, T.L.; Gupta, V.A.; Sullivan, H.C.; Neish, A.S.; Dhodapkar, M.V.; Lonial, S. Daratumumab in multiple myeloma. Cancer 2019, 125, 2364–2382. [Google Scholar] [CrossRef]
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.L.J.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2014, 28, 1122–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usmani, S.; Ahmadi, T.; Ng, Y.; Lam, A.; Desai, A.; Potluri, R.; Mehra, M. Analyses of real world data on overall survival in multiple myeloma patients with at least 3 prior lines of therapy including a PI and an IMiD, or double refractory to a PI and an IMiD. Oncologist 2016, 21, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Einsele, H.; Danhof, S. Bispecific antibodies: A new era of treatment for multiple myeloma. J. Clin. Med. 2020, 9, 2166. [Google Scholar] [CrossRef]
- Teoh, P.J.; Chng, W.J. CAR T-cell therapy in multiple myeloma: More room for improvement. Blood Cancer J. 2021, 11, 84. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Siegel, D.; Oriol, A.; Moreau, P.; Agha, I.Y.; Delforge, M.; Cavo, M.; Einsele, H.; Goldschmidt, H.; Weisel, K.; et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Lancman, G.; Richter, J.; Chari, A. Bispecifics, trispecifics, and other novel immune treatments in myeloma. Am. Soc. Hematol. 2020, 2020, 264–271. [Google Scholar] [CrossRef]
- Verkleij, C.; Broekmans, M.; van Duin, M.; Frerichs, K.A.; Kuiper, R.; de Jonge, A.V.; Kaiser, M.; Morgan, G.; Axel, A.; Boominathan, R.; et al. Preclinical activity and determinants of response of the GPRC5DxCD3 bispecific antibody talquetamab in multiple myeloma. Blood Adv. 2021, 5, 2196–2215. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Zuch de Zafra, C.L.; Fajardo, F.; Zhong, W.; Bernett, M.J.; Muchhal, U.S.; Moore, G.L.; Stevens, J.; Case, R.; Pearson, J.T.; Liu, S.; et al. Targeting multiple myeloma with AMG 424, a novel anti-CD38/CD3 bispecific T-cell-recruiting antibody optimized for cytotoxicity and cytokine release. Clin. Cancer Res. 2019, 25, 3921–3922. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, R.H. T cell anergy. Annu. Rev. Immunol. 2003, 21, 305–334. [Google Scholar] [CrossRef]
- Caraccio, C.; Krishna, S.; Phillips, D.J.; Schürch, C.M. Bispecific antibodies for multiple myeloma: A review of targets, drugs, clinical trials, and future directions. Front. Immunol. 2020, 11, 501. [Google Scholar] [CrossRef]
- Verkleij, C.; Frerichs, K.A.; Broekmans, M.; Absalah, S.; Maas-Bosman, P.; Kruyswijk, S.; Nijhof, I.S.; Mutis, T.; Zweegman, S.; van de Donk, N.W.C.J. T-cell redirecting bispecific antibodies targeting BCMA for the treatment of multiple myeloma. Oncotarget 2020, 11, 4076–4081. [Google Scholar] [CrossRef]
- Giralt, S.; Costa, L.J.; Maloney, D.; Krishnan, A.; Fei, M.; Antin, J.; Brunstein, C.; Geller, N.; Goodman, S.; Hari, P.; et al. Tandem Autologous-autologous versus autologous-allogeneic hematopoietic stem cell transplant for patients with multiple myeloma: Long-term follow-up results from the blood and marrow transplant clinical trials network 0102 trial. Biol. Blood Marrow Transplant. 2020, 26, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.C.J.; Usmani, S.Z.; Yong, K. Review CAR T-Cell Therapy for multiple myeloma: State of the art and prospects. Lancet Haematol. 2021, 8, e446–e461. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Saxena, A.; Sidhu, S.S.; Wu, D. Fc Engineering for developing therapeutic bispecific antibodies and novel scaffolds. Front. Immunol. 2017, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, M.; Brandl, C.; Zugmaier, G.; Hijazi, Y.; Bargou, R.C.; Topp, M.S.; Gökbuget, N.; Neumann, S.; Goebeler, M.; Viardot, A.; et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-Bispecific BiTE antibody blinatumomab. Blood 2012, 119, 6226–6233. [Google Scholar] [CrossRef]
- Pratz, K.W.; Gojo, I.; Gocke, C.; Matsui, W.; Huff, C.A. Blinatumomab induced response of multiply refractory multiple myeloma in the context of secondary pre-B cell acute lymphoblastic leukemia. Ann. Hematol. Oncol. 2017, 4, 1174. [Google Scholar] [CrossRef] [Green Version]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C. Anti–B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic receptor co-stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- Harrison, S.J.; Minnema, M..; Lee, H..; Spencer, A.; Kapoor, P.; Madduri, D.; Larsen, J.; Ailawadhi, S.; Kaufman, J.L.; Raab, M.S.; et al. A phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE® (Bispecific T-Cell Engager) molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). In Proceedings of the 62nd American Society of Hematology Annual Meeting and Exposition, San Diego, CA, USA, 5 December 2020; 2020; p. 653. [Google Scholar]
- Bahlis, N.J.; Raje, N.S.; Costello, C.; Dholaria, B.R.; Solh, M.M.; Levy, M.Y.; Tomasson, M.H.; Dube, H.; Liu, F.; Liao, K.H.; et al. Efficacy and safety of elranatamab (PF-06863135), a B-Cell Maturation Antigen (BCMA)-CD3 bispecific antibody, in patients with relapsed or refractory Multiple Myeloma (MM). J. Clin. Oncol. 2021, 39, 8006. [Google Scholar] [CrossRef]
- Krishnan, A.Y.; Garfall, A.L.; Mateos, M.; van de Donk, N.W.C.J.; Nahi, H.F.; San-Miguel, J.; Oriol, A.; Rosiñol, L.; Chari, A.; Bhutani, M.; et al. Updated phase 1 results of teclistamab, a B-Cell Maturation Antigen (BCMA) × CD3 bispecific antibody, in relapsed/refractory Multiple Myeloma (MM). J. Clin. Oncol. 2021, 39, 8007. [Google Scholar] [CrossRef]
- Van de Donk, N.W.C.J.; Garfall, A.L.; Mateos, M.-A.; Krishnan, A.Y.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Rosiñol, L.; Chari, A.; Bhutani, M.; et al. Teclistamab, a B-Cell Maturation Antigen (BCMA) × CD3 bispecific antibody, in patients with relapsed/refractory multiple myeloma: Updated phase 1 results. In Proceedings of the European Hematology Association 2021, Vienna, Austria, 11 June 2021; p. S193. [Google Scholar]
- Madduri, D.; Rosko, A.; Brayer, J.; Zonder, J.; Bensinger, W.I.; Li, J.; Xu, L.; Adriaens, L.; Chokshi, D.; Zhang, W.; et al. REGN5458, a BCMA × CD3 bispecific monoclonal antibody, induces deep and durable responses in patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 41–42. [Google Scholar] [CrossRef]
- Rodriguez, C.; D’Souza, A.; Shah, N.; Voorhees, P.M.; Buelow, B.; Vij, R.; Kumar, S.K. Initial results of a phase I study of TNB-383B, a BCMA × CD3 Bispecific T-Cell redirecting antibody, in Relapsed/Refractory Multiple Myeloma. In Proceedings of the 62nd American Socitety of Hematology Annual Meeting and Exposition, San Diego, CA, USA, 5 December 2020; p. 293. [Google Scholar]
- Costa, L.; Wong, S.; Bermudez, A.; de la Rubia, J.; Mateos, M.V.; Ocio, E.M.; Rodriguez Otero, P.; San Miguel, J.; Li, S.; Sarmiento, R.; et al. Interim Results from the First Phase 1 Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T-Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM). In Proceedings of the European Hematology Association 2020, Frankfurt, Germany, 12 June 2020; p. S205. [Google Scholar]
- Richter, J.R.; Landgren, C.O.; Kauh, J.S.; Back, J.; Salhi, Y.; Reddy, V.; Bayever, E.; Berdeja, J.G. Phase 1, multicenter, open-label study of single-agent bispecific antibody t-cell engager GBR 1342 in Relapsed/Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, TPS3132. [Google Scholar] [CrossRef]
- Cohen, A.D.; Harrison, S.J.; Krishnan, A.; Fonseca, R.; Forsberg, P.A.; Spencer, A.; Berdeja, J.G.; Laubach, J.P.; Li, M.; Choeurng, V.; et al. Initial clinical activity and safety of BFCR4350A, a FcRH5/CD3 T-cell-engaging bispecific antibody, in Relapsed/Refractory Multiple Myeloma. In Proceedings of the 62nd American Socitety of Hematology Annual Meeting and Exposition, American Socitety of Hematology, San Diego, CA, USA, 5 December 2020; p. 292. [Google Scholar]
- Krishnan, A.Y.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.C.J.; Rodríguez-Otero, P.; Askari, P.; Mateos, M.; Minnema, M..; Costa, L.C.; Verona, R.; et al. Talquetamab, A G Protein-Coupled Receptor Family Group 5 Member D (GPRC5D) x CD3 Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma: Updated Results of a Phase 1, First-in-Human Study. In Proceedings of the European Hematology Association, Vienna, Austria, 11 June 2021; 2021; p. S191. [Google Scholar]
- Velasquez, M.P.; Bonifant, C.L.; Gottschalk, S. Redirecting T cells to hematological malignancies with bispecific antibodies. Blood 2018, 131, 30–38. [Google Scholar] [CrossRef]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by Anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Coquery, C.M.; Erickson, L.D. Regulatory roles of the tumor necrosis factor receptor BCMA. Crit. Rev. Immunol. 2012, 32, 287–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, D.T.; Kalled, S.L.; Ellyard, J.I.; Ambrose, C.; Bixler, S.A.; Thien, M.; Brink, R.; Mackay, F.; Hodgkin, P.D.; Tangye, S.G. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J. Clin. Investig. 2003, 112, 286–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Anderson, K.C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef] [Green Version]
- Hatzoglou, A.; Roussel, J.; Bourgeade, M.F.; Rogier, E.; Madry, C.; Inoue, J.; Devergne, O.; Tsapis, A. TNF receptor family member BCMA (B Cell Maturation) associates with TNF Receptor-Associated Factor (TRAF) 1, TRAF2, and TRAF3 and Activates NF-Kappa B, Elk-1, c-Jun N-Terminal kinase, and P38 mitogen-activated protein kinase. J. Immunol. 2000, 165, 1322–1330. [Google Scholar] [CrossRef] [Green Version]
- Mackay, F.; Ambrose, C. The TNF family members BAFF and APRIL: The growing complexity. Cytokine Growth Factor Rev. 2003, 14, 311–324. [Google Scholar] [CrossRef]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in multiple myeloma: Potential uses of BCMA-Based immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Dylan, T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. Serum B-Cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br. J. Haematol. 2012, 158, 727–738. [Google Scholar] [CrossRef]
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-cell maturation antigen: A novel biomarker to predict outcomes for multiple myeloma patients. Plasma Cell Disord. 2017, 102, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Palma, B.D.; Marchica, V.; Catarozzo, M.T.; Giuliani, N.; Accardi, F. Monoclonal and bispecific anti-BCMA antibodies in multiple myeloma. J. Clin. Med. 2020, 9, 3022. [Google Scholar] [CrossRef]
- Hipp, S.; Tai, Y.T.; Blanset, D.; Deegen, P.; Wahl, J.; Thomas, O.; Rattel, B.; Adam, P.J.; Anderson, K.C.; Friedrich, M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in Vitro and in Vivo. Leukemia 2017, 31, 1743–1751. [Google Scholar] [CrossRef]
- Cho, S.F.; Lin, L.; Xing, L.; Wen, K.; Yu, T.; Wahl, J.; Matthes, K.; Munshi, N.; Anderson, K.C.; Arvedson, T.; et al. AMG 701, a half-life extended Anti-BCMA BiTE®; potently induces T cell-redirected lysis of human multiple myeloma cells and can be combined with IMiDs to overcome the immunosuppressive bone marrow microenvironment. Clin. Lymphoma Myeloma Leuk. 2019, 19, e54. [Google Scholar] [CrossRef]
- Goldstein, R.L.; Goyos, A.; Li, C.M.; Deegen, P.; Bogner, P.; Sternjak, A.; Thomas, O.; Klinger, M.; Wahl, J.; Friedrich, M.; et al. AMG 701 Induces cytotoxicity of multiple myeloma cells and depletes plasma cells in cynomolgus monkeys. Blood Adv. 2020, 4, 4180–4194. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Lin, L.; Xing, L.; Li, Y.; Wen, K.; Yu, T.; Hsieh, P.A.; Munshi, N.; Wahl, J.; Matthes, K.; et al. The immunomodulatory drugs lenalidomide and pomalidomide enhance the potency of AMG 701 in multiple myeloma preclinical models. Blood Adv. 2020, 4, 4195–4207. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.H.; Kuo, T.C.; Zhang, Y.; Chen, A.; Geng, T.; Aschenbrenner, L.; Kamperschroer, C.; Pascua, E.; Chen, W.; Delaria, K.; et al. Preclinical efficacy and safety comparison of CD3 Bispecific and ADC modalities targeting BCMA for the treatment of multiple myeloma. Mol. Cancer Ther. 2019, 18, 2008–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesokhin, A.M.; Levy, M.Y.; Dalovisio, A.P.; Bahlis, N.; Solh, M.; Sebag, M.; Jakubowiak, A.; Jethava, Y.S.; Costello, C.L.; Chu, M.P.; et al. Preliminary safety, efficacy, pharmacokinetics, and pharmacodynamics of subcutaneously (SC) administered PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 bispecific antibody, in patients with Relapsed/Refractory Multiple Myeloma (RRMM). In Proceedings of the 62nd American Socitety of Hematology Annual Meeting and Exposition, San Diego, CA, USA, 5 December 2020; Volume 6, p. 3206. [Google Scholar]
- Costello, C.; Raje, N.; Bahlis, N.; Dholaria, B.; Solh, M.; Levy, M.; Tomasson, M.; Dube, H.; Damore, M.; Liao, K.; et al. Magnetismm-1: Phase 1 Study of Elranatamab (Pf-06863135), a B-Cell Maturation Antigen (BCMA) Targeted CD3-Engaging Bispecific Antibody, for Patients with Relapsed or Refractory Multiple Myeloma (MM). In Proceedings of the European Hematology Association 2021, Vienna, Austria; 9 June 2021; p. S192.
- Frerichs, K.A.; Broekmans, M.E.C.; Soto, J.A.M.; van Kessel, B.; Heymans, M.W.; Holthof, L.C.; Verkleij, C.P.M.; Boominathan, R.; Vaidya, B.; Sendecki, J.; et al. Preclinical activity of JNJ-7957, a Novel BCMAxCD3 bispecific antibody for the treatment of multiple myeloma, is potentiated by daratumumab. Clin. Cancer Res. 2020, 26, 2203–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillarisetti, K.; Powers, G.; Luistro, L.; Babich, A.; Baldwin, E.; Li, Y.; Zhang, X.; Mendonça, M.; Majewski, N.; Nanjunda, R.; et al. Teclistamab Is an Active T Cell–redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020, 4, 4538–4549. [Google Scholar] [CrossRef]
- Girgis, S.; Shetty, S.; Jiao, T.; Amuzie, C.; Weinstock, D.; Grimme Watson, R.; Ford, J.; Pillarisetti, K.; Baldwin, E.; Bellew, K. exploratory pharmacokinetic/pharmacodynamic and tolerability study of BCMAxCD3 in cynomolgus monkeys. Blood 2016, 128, 5668. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Rosinol, L.; Chari, A.; Bhutani, M.; Karlin, L.; et al. Teclistamab, a B-Cell maturation antigen × CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): A multicentre, open-label, single-arm, phase 1 study. Lancet 2021, 398, 665–674. [Google Scholar] [CrossRef]
- Dilillo, D.J.; Olson, K.; Mohrs, K.; Meagher, T.C.; Bray, K.; Sineshchekova, O.; Startz, T.; Kuhnert, J.; Retter, M.W.; Godin, S.; et al. A BCMAxCD3 bispecific T cell-engaging antibody demonstrates robust antitumor efficacy similar to that of Anti-BCMA CAR T Cells. Blood Adv. 2021, 5, 1291–1304. [Google Scholar] [CrossRef]
- Bumma, N.; Quek, R.G.W.; Brayer, J.; Zonder, J.A.; Hoffman, J.E.; Bensinger, W.I.; Dhodapkar, M.V.; Lentzsch, S.; Cooper, D.; Maly, J.J.; et al. Quality of life with REGN5458 in patients with Relapsed/Refractory Multiple Myeloma (RRMM): Patient-reported outcomes (Pros) from the first-in-human phase 1/2 trial. In Proceedings of the European Hematology Association 2021, Vienna, Austria, 9 June 2021; 2021; p. EP1163. [Google Scholar]
- Trinklein, N.D.; Pham, D.; Schellenberger, U.; Buelow, B.; Boudreau, A.; Choudhry, P.; Clarke, S.C.; Dang, K.; Harris, K.E.; Iyer, S.; et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. MAbs 2019, 11, 639–652. [Google Scholar] [CrossRef]
- Buelow, B.; Pham, D.; Clarke, S.; Force Aldred, S.; Dang, K.; Pratap, P.; Ugamraj, H.; Harris, K.; Trinklein, N.; Schellenberger, U.; et al. Development of a fully human T cell engaging bispecific antibody for the treatment of multiple myeloma. J. Clin. Oncol. 2017, 35, 8017. [Google Scholar] [CrossRef]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; de la Rubia, J.; Mateos, M.V.; Ocio, E.M.; Rodríguez-Otero, P.; San-Miguel, J.; Li, S.; Sarmiento, R.; et al. First clinical study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim results of a phase 1 multicenter trial. Blood 2019, 134, 143. [Google Scholar] [CrossRef]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.N.H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; van de Donk, N.W.C.J.; et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- Deaglio, S.; Vaisitti, T.; Billington, R.; Bergui, L.; Omede’, P.; Genazzani, A.A.; Malavasi, F. CD38/CD19: A lipid raft-dependent signaling complex in human B Cells. Blood 2007, 109, 5390–5398. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-Ribosyl Cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402. [Google Scholar] [PubMed]
- Doucey, M.-A.; Pouleau, B.; Estoppey, C.; Stutz, C.; Croset, A.; Laurendon, A.; Monney, T.; Pluess, M.; Ries-Fecourt, C.; Macoin, J.; et al. ISB 1342: A first-in-class CD38 T Cell engager for the treatment of relapsed refractory multiple myeloma. J. Clin. Oncol. 2021, 39, 8044. [Google Scholar] [CrossRef]
- Ishikawa, H.; Tsuyama, N.; Mahmoud, M.S.; Fujii, R.; Abroun, S.; Liu, S.; Li, F.J.; Kawano, M.M. CD19 Expression and growth inhibition of tumours in human multiple myeloma. Leuk. Lymphoma 2002, 43, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B Cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Poe, J.C.; Minard-colin, V.; Evgueni, I.; Haas, K.M.; Tedder, T.F. A C-Myc and surface CD19 signaling amplification loop promotes B Cell lymphoma development and progression in mice. J Immunol 2012, 189, 2318–2325. [Google Scholar] [CrossRef] [Green Version]
- Nerreter, T.; Letschert, S.; Götz, R.; Doose, S.; Einsele, H.; Sauer, M.; Hudecek, M. Super-Resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat. Commun. 2019, 10, 3137. [Google Scholar] [CrossRef] [PubMed]
- Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; Arslan, Ö.; Sanz, M.A.; Bergeron, J.; Demirkan, F.; et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Benjamin, J.E.; Stein, A.S. The role of blinatumomab in patients with relapsed/refractory acute lymphoblastic leukemia. Ther. Adv. Hematol. 2016, 7, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfred Garfall, M. Pilot Study of Blinatumomab in Combination with Salvage Autologous Stem Cell Transplantation for Patients with Refractory Multiple Myeloma. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03173430 (accessed on 15 July 2021).
- Wilson, T.J.; Fuchs, A.; Colonna, M. Cutting edge: Human FcRL4 and FcRL5 Are receptors for IgA and IgG. J. Immunol. 2012, 188, 4741–4745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, A.; Damdinsuren, B.; Ise, T.; Dement-Brown, J.; Li, H.; Nagata, S.; Tolnay, M. Human Fc receptor-like 5 binds intact IgG via mechanisms distinct from that of Fc-receptors. J. Immunol. 2011, 190, 5739–5746. [Google Scholar] [CrossRef]
- Elkins, K.; Zheng, B.; Go, M.A.; Slaga, D.; Du, C.; Scales, S.J.; Yu, S.F.; McBride, J.; De Tute, R.; Rawstron, A.; et al. FcRL5 as a target of antibody-drug conjugates for the treatment of multiple myeloma. Mol. Cancer Ther. 2012, 11, 2222–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ise, T.; Nagata, S.; Kreitman, R.J.; Wilson, W.H.; Wayne, A.S.; Stetler-Stevenson, M.; Bishop, M.R.; Scheinberg, D.A.; Rassenti, L.; Kipps, T.J.; et al. Elevation of soluble CD307 (IRTA2/FcRH5) Protein in the blood and expression on malignant cells of patients with multiple myeloma, chronic lymphocytic leukemia, and mantle cell lymphoma. Leukemia 2007, 21, 169–174. [Google Scholar] [CrossRef]
- Sawyer, J.R.; Tricot, G.; Mattox, S.; Jagannath, S.; Barlogie, B. Jumping translocations of chromosome 1q in multiple myeloma: Evidence for a mechanism involving decondensation of pericentromeric heterochromatin. Blood 1998, 91, 1732–1741. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Miller, I.; Takizawa, J.; Palanisamy, N.; Rao, P.H.; Iida, S.; Tagawa, S.; Taniwaki, M.; Russo, J.; Neri, A.; et al. IRTA1 and IRTA2, Novel immunoglobulin superfamily receptors expressed in B cells and involved in chromosome 1q21 abnormalities in B Cell malignancy. Immunity 2001, 14, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Bluemel, C.; Hausmann, S.; Fluhr, P.; Sriskandarajah, M.; Stallcup, W.B.; Baeuerle, P.A.; Kufer, P. Epitope distance to the target cell membrane and antigen size determine the potency of T Cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 2010, 59, 1197–1209. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Edavettal, S.; Mendonça, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-cell-redirecting bispecific G-protein-coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood 2020, 135, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Kochi, Y.; Nakai, W.; Mizuno, H.; Baba, T.; Habu, K.; Sawada, N.; Tsunoda, H.; Shima, T.; Miyawaki, K.; et al. Anti-GPRC5D/CD3 bispecific T-cell–redirecting antibody for the treatment of multiple myeloma. Mol. Cancer Ther. 2019, 18, 1555–1565. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef] [PubMed]
- Atamaniuk, J.; Gleiss, A.; Porpaczy, E.; Kainz, B.; Grunt, T.W.; Raderer, M.; Hilgarth, B.; Drach, J.; Ludwig, H.; Gisslinger, H.; et al. Overexpression of G protein-coupled receptor 5D in the bone marrow is associated with poor prognosis in patients with multiple myeloma. Eur. J. Clin. Investig. 2012, 42, 953–960. [Google Scholar] [CrossRef]
- Inoue, S.; Nambu, T.; Shimomura, T. The RAIG family member, GPRC5D, is associated with hard-keratinized structures. J. Investig. Dermatol. 2004, 122, 565–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truger, M.; Düll, J.; Zhou, X.; Heimeshoff, L.; Ruckdeschel, A.; John, M.; Riedel, A.; Hüper, S.; Peter, J.; Haertle, L.; et al. Single and double hit events in genes encoding for immunotherapy targets in multiple myeloma. In Proceedings of the European Hematology Association 2021, Vienna, Austria, 9 June 2021; p. S175. [Google Scholar]
- Meermeier, E.W.; Welsh, S.J.; Sharik, M.E.; Du, M.T.; Garbitt, V.M.; Riggs, D.L.; Shi, C.; Stein, C.K.; Bergsagel, M.; Chau, B.; et al. Tumor burden limits bispecific antibody efficacy through T Cell exhaustion averted by concurrent cytotoxic therapy. Blood Cancer Discov. 2021, 2, 354–369. [Google Scholar] [CrossRef]
- Sumiyoshi, T.; Nakamura, R.; Lear, S.; Wilson, D.; Choeurng, V.; Vaze, A.; Trudel, S.; Spencer, A.; Cohen, A.D.; Fonseca, R.; et al. Fcrh5 target expression in patients with Relapsed Refractory Multiple Myeloma (RRMM) treated with cevostamab in an ongoing phase I dose escalation study. In Proceedings of the European Hematology Association 2021, Vienna, Austria, 9 June 2021; 2021; p. EP965. [Google Scholar]
- Lameris, R.; Shahine, A.; Pellicci, D.G.; Uldrich, A.P.; Gras, S.; Le Nours, J.; Groen, R.W.J.; Vree, J.; Reddiex, S.J.J.; Quiñones-Parra, S.M.; et al. A single-domain bispecific antibody targeting CD1d and the NKT T-cell receptor induces a potent antitumor response. Nat. Cancer 2020, 1, 1054–1065. [Google Scholar] [CrossRef]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with Γδ T Cells: Many paths ahead of us. Cell. Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef]
- Demaria, O.; Gauthier, L.; Debroas, G.; Vivier, E. Natural killer cell engagers in cancer immunotherapy: Next generation of immuno-oncology treatments. Eur. J. Immunol. 2021, 51, 1934–1942. [Google Scholar] [CrossRef]
- Casneuf, T.; Xu, X.S.; Adams, H.C.; Axel, A.E.; Chiu, C.; Khan, I.; Ahmadi, T.; Yan, X.; Lonial, S.; Plesner, T.; et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 2017, 1, 2105–2114. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Targets | Drug Name | Design | Trial Type (Phase) | Enrolled Patients | Overall Response Rate | Minimal Residual Disease | Cytokine Release Syndrome | Clinical Trial Identifier/Reference |
|---|---|---|---|---|---|---|---|---|
| BCMA x CD3 | Pacanalotamab (AMG 420) | BiTE | 1 | 42 | 31% (13 of 42 patients, IV); 70% (7 of 10 patients) at the MTD (400 µg/day) | 71% (5 of 7 patients) at the MTD | 38% (16 of 42 patients): Grade 1 = 28%; Grade 2 = 5%; Grade 3 = 2% | NCT02514239 [34] |
| BCMA x CD3 | Pavurutamab (AMG 701) | Half-life extended BiTE (scFvs plus Fc domain) | 1 | 85 | 26% (21 of 82 patients, IV); 83% (5 of 6 patients) at the most recent evaluable cohort | 85% (6 of 7 patients) across all cohorts | 65% (55 of 85 patients): Grade 1 = 27%; Grade 2 = 28%; Grade 3 = 9% | NCT03287908 [36] |
| BCMA x CD3 | Elranatamab (PF-06863135) | IgG2a Fc region | 1 | 30 (SC administration) | 70% at SC doses ≥215 µg/kg (14 of 20 patients); 83% (5 of 6 patients) at the RP2D (1000 µg/kg) | 100% (3 of 3 patients) across all cohorts | 73.3%: Grade 1 = 57%; Grade 2 = 17% (no events >grade 2) | NCT03269136 [37] |
| BCMA x CD3 | Teclistamab (JNJ-64007957) | IgG4 Fc region | 1 | 157 total 40 at the RP2D | 65% (26 of 40 patients) at the RP2D (SC 1500 µg/kg) | 100% (6 of 6 evaluable patients treated at the RP2D); 69% (18 of 26 patients) across both IV and SC cohorts | 70% (28 of 40 patients) at the RP2D: Grade 1 = 45%; Grade 2 = 25% (no events >grade 2) | NCT03145181 [38,39] |
| BCMA x CD3 | REGN5458 | VelociBiTM Fc region | 1 | 49 | 39% (19 of 49 patients, IV); 63% in dose-level 6 (8 patients) | 57% (4 of 7 patients) across all cohorts | 39% (19 of 49 patients): Grade 1 = 33%; Grade 2 = 6% (no events >grade 2) | NCT03761108 [40] |
| BCMA x CD3 | REGN5459 | VelociBiTM Fc region | 1 | N/A | N/A | N/A | N/A | NCT04083534 |
| BCMA x CD3 | TNB-383B | IgG4 Fc region | 1 | 58 | 46.5% (27 of 58 patients, IV); 80% (12 of 15 patients) at IV doses ≥ 40 mg | 75% (3 of 4 patients) across all cohorts | 80% (12 of 15 patients treated at IV doses ≥ 40 mg): Grade 1 = 46.7%; Grade 2 = 33.3% (no events >grade 2) | NCT03933735 [41] |
| BCMA x CD3 | CC-93269 | IgG1-based Fc region | 1 | 30 | 43% (13 of 30 patients, IV); 89% (8 of 9 patients) at highest dose of 10 mg | 92% (12 of 13 patients) across all cohorts | 77% (23 of 30 patients): Grade 1 = 50%; Grade 2 = 23%; grade ≥3 = 3% | NCT03486067 [42] |
| CD38 x CD3 | AMG424 * | Fc region | 1 | 27 | N/A | N/A | N/A | NCT03445663 [24] |
| CD38 x CD3 | GBR 1342 | BEAT® platform | 1 | N/A | N/A | N/A | N/A | NCT03309111 [43] |
| CD19 x CD3 | Blinatumomab ** | BiTE | 1 | 6 | N/A | N/A | N/A | NCT03173430 [33] |
| FcRH5 x CD3 | Cevostamab | IgG1-based Fc region | 1 | 53 | 53% (18 of 34 patients, IV) at doses ≥3.6/20 mg | 86% (6 of 7 evaluable patients) across all cohorts | 76% (40 of 53 patients): Grade 1 = 18%; Grade 2 = 40%; Grade 3 = 2% | NCT03275103 [44] |
| GPRC5D x CD3 | Talquetamab | IgG4 Fc region | Phase 1 | Total of 184 patients with 30 participants treated at the RP2D | 70% (21 of 30 patients) at the RP2D (SC 405 µg/kg, with 10.0 and 60.0 µg/kg step-up doses) | 67% (4 of 6 patients) across both IV and SC cohorts including 1 patient from the RP2D cohort | 73% (22 of 30 patients) at the RP2D: Grade 1 = 60%; Grade 2 = 10%; Grade 3 = 3% | NCT03399799 [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosny, M.; Verkleij, C.P.M.; van der Schans, J.; Frerichs, K.A.; Mutis, T.; Zweegman, S.; van de Donk, N.W.C.J. Current State of the Art and Prospects of T Cell-Redirecting Bispecific Antibodies in Multiple Myeloma. J. Clin. Med. 2021, 10, 4593. https://doi.org/10.3390/jcm10194593
Hosny M, Verkleij CPM, van der Schans J, Frerichs KA, Mutis T, Zweegman S, van de Donk NWCJ. Current State of the Art and Prospects of T Cell-Redirecting Bispecific Antibodies in Multiple Myeloma. Journal of Clinical Medicine. 2021; 10(19):4593. https://doi.org/10.3390/jcm10194593
Chicago/Turabian StyleHosny, Mashhour, Christie P. M. Verkleij, Jort van der Schans, Kristine A. Frerichs, Tuna Mutis, Sonja Zweegman, and Niels W. C. J. van de Donk. 2021. "Current State of the Art and Prospects of T Cell-Redirecting Bispecific Antibodies in Multiple Myeloma" Journal of Clinical Medicine 10, no. 19: 4593. https://doi.org/10.3390/jcm10194593
APA StyleHosny, M., Verkleij, C. P. M., van der Schans, J., Frerichs, K. A., Mutis, T., Zweegman, S., & van de Donk, N. W. C. J. (2021). Current State of the Art and Prospects of T Cell-Redirecting Bispecific Antibodies in Multiple Myeloma. Journal of Clinical Medicine, 10(19), 4593. https://doi.org/10.3390/jcm10194593