
Functionally Heterogenous Macrophage Subsets in the Pathogenesis of Giant Cell Arteritis: Novel Targets for Disease Monitoring and Treatment

 , , , ,
, , , ,
Abstract
:1. Introduction
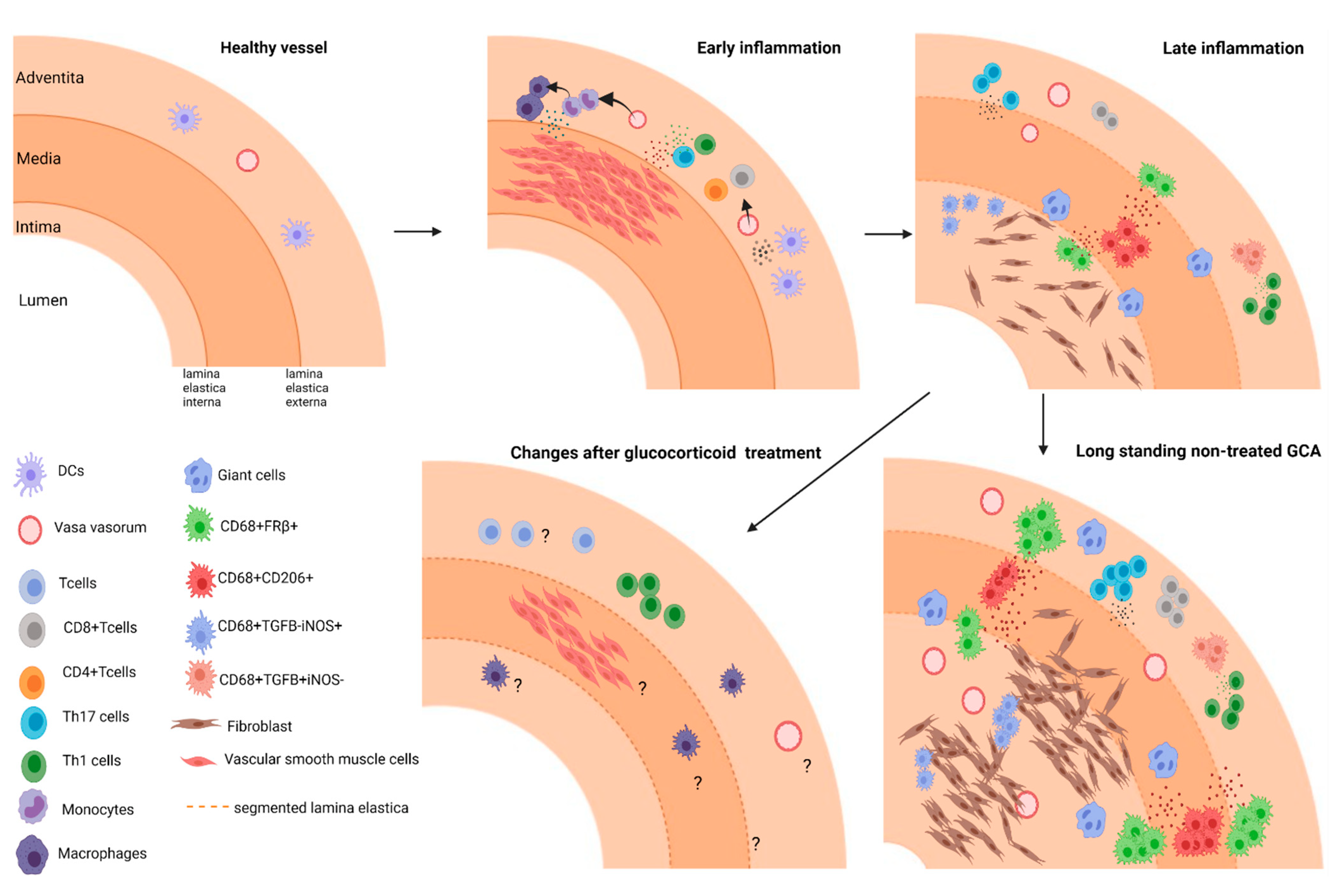
1.1. Pathogenesis of GCA
1.2. Monocytes in GCA
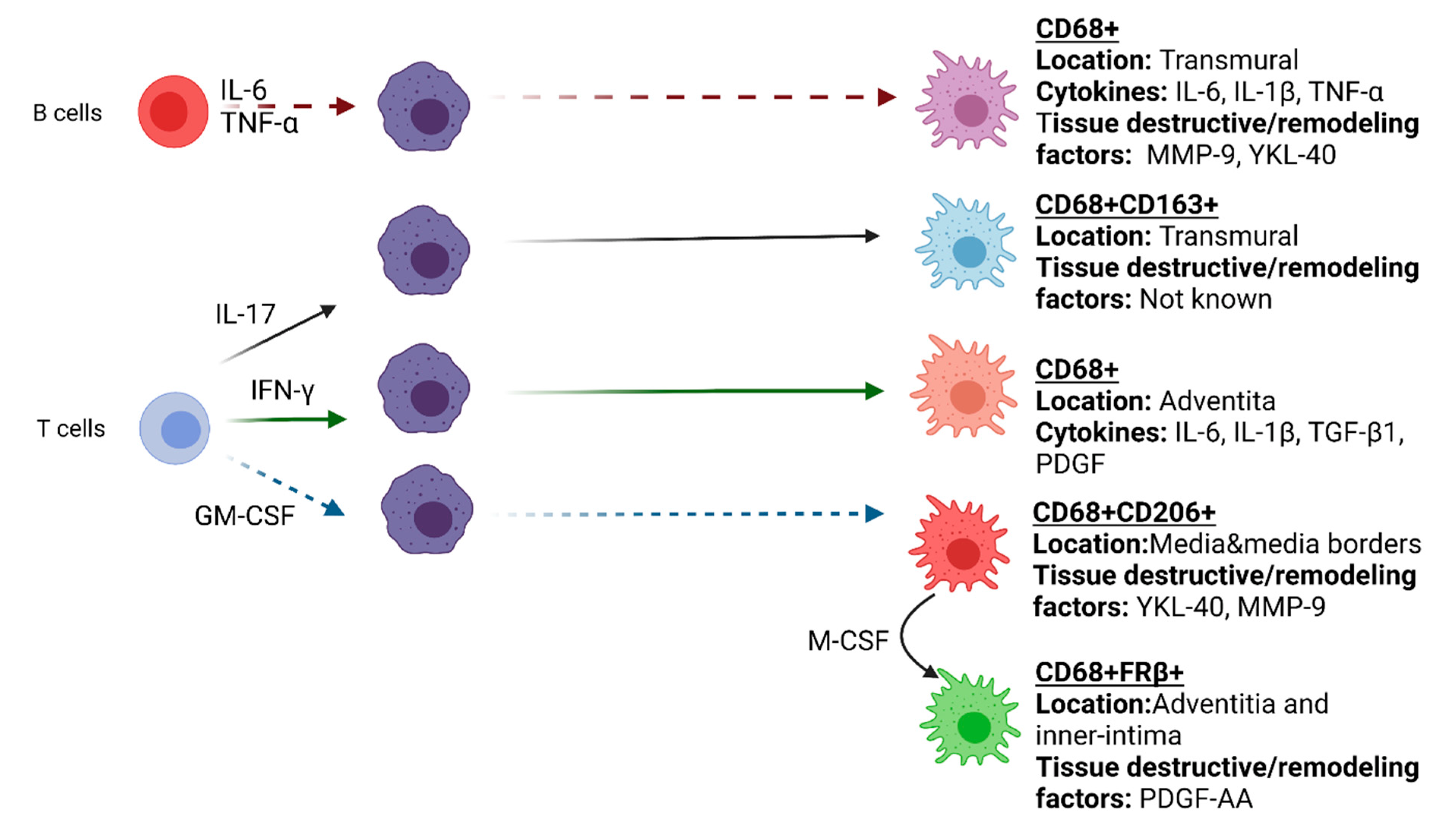
1.3. Macrophage Heterogeneity and Their Roles in the Vasculopathy of GCA
1.4. Macrophage Metabolism in GCA
1.5. Aging Macrophages
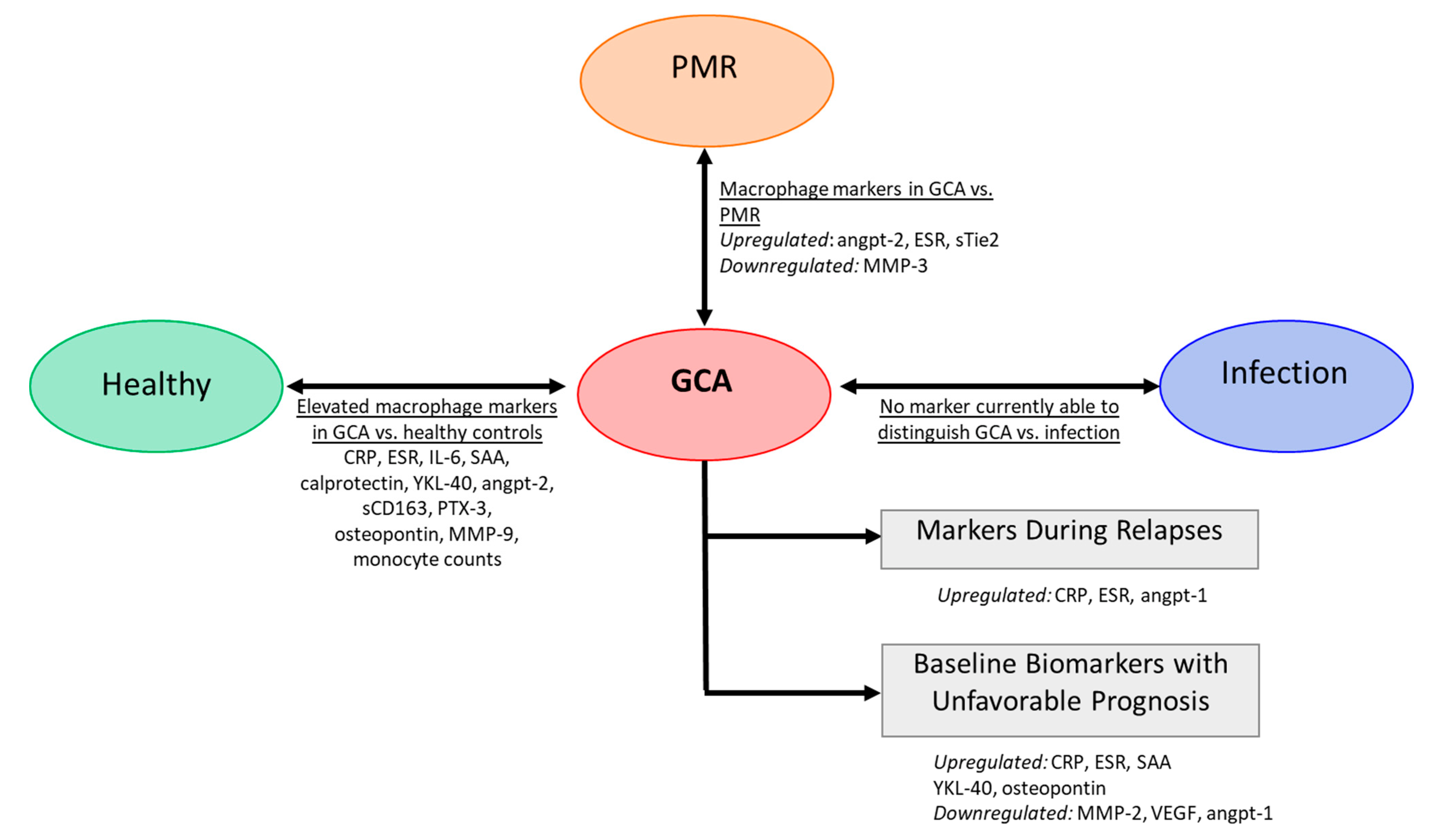
1.6. Macrophage Related Biomarkers as a Tool for GCA Diagnosis and Disease Monitoring
1.6.1. A. Serum Markers
1.6.2. B. Macrophage Targeted Imaging
1.7. Targeting Macrophages as Alternative Therapeutic Strategies for the Treatment of GCA
2. Future Perspectives
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van der Geest, K.S.M.; Sandovici, M.; Brouwer, E.; Mackie, S.L. Diagnostic Accuracy of Symptoms, Physical Signs, and Laboratory Tests for Giant Cell Arteritis: A Systematic Review and Meta-analysis. JAMA Intern. Med. 2020, 180, 1295–1304. [Google Scholar] [CrossRef]
- Dejaco, C.; Brouwer, E.; Mason, J.C.; Buttgereit, F.; Matteson, E.L.; Dasgupta, B. Giant cell arteritis and polymyalgia rheumatica: Current challenges and opportunities. Nat. Rev. Rheumatol. 2017, 13, 578–592. [Google Scholar] [CrossRef]
- Agard, C.; Barrier, J.-H.; Dupas, B.; Ponge, T.; Mahr, A.; Fradet, G.; Chevalet, P.; Masseau, A.; Batard, E.; Pottier, P.; et al. Aortic involvement in recent-onset giant cell (temporal) arteritis: A case-control prospective study using helical aortic computed tomodensitometric scan. Arthritis Rheum. 2008, 59, 670–676. [Google Scholar] [CrossRef]
- Koster, M.J.; Matteson, E.L.; Warrington, K.J. Large-vessel giant cell arteritis: Diagnosis, monitoring and management. Rheumatology 2018, 57, ii32–ii42. [Google Scholar] [CrossRef] [Green Version]
- Prieto-González, S.; Arguis, P.; García-Martínez, A.; Espígol-Frigolé, G.; Tavera-Bahillo, I.; Butjosa, M.; Sánchez, M.; Hernández-Rodríguez, J.; Grau, J.M.; Cid, M.C. Large vessel involvement in biopsy-proven giant cell arteritis: Prospective study in 40 newly diagnosed patients using CT angiography. Ann. Rheum. Dis. 2012, 71, 1170–1176. [Google Scholar] [CrossRef]
- Schmidt, W.A.; Seifert, A.; Gromnica-Ihle, E.; Krause, A.; Natusch, A. Ultrasound of proximal upper extremity arteries to increase the diagnostic yield in large-vessel giant cell arteritis. Rheumatology 2008, 47, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Hunder, G.G.; Bloch, D.A.; Michel, B.A.; Stevens, M.B.; Arend, W.P.; Calabrese, L.H.; Edworthy, S.M.; Fauci, A.S.; Leavitt, R.Y.; Lie, J.T. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990, 33, 1122–1128. [Google Scholar] [CrossRef]
- Dejaco, C.; Duftner, C.; Buttgereit, F.; Matteson, E.L.; Dasgupta, B. The spectrum of giant cell arteritis and polymyalgia rheumatica: Revisiting the concept of the disease. Rheumatology 2017, 56, 506–515. [Google Scholar] [CrossRef] [Green Version]
- Hellmich, B.; Agueda, A.; Monti, S.; Buttgereit, F.; De Boysson, H.; Brouwer, E.; Cassie, R.; Cid, M.C.; Dasgupta, B.; Dejaco, C.; et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann. Rheum. Dis. 2020, 79, 19–130. [Google Scholar] [CrossRef] [Green Version]
- Luqmani, R.; Lee, E.; Singh, S.; Gillett, M.; Schmidt, W.A.; Bradburn, M.; Dasgupta, B.; Diamantopoulos, A.P.; Forrester-Barker, W.; Hamilton, W.; et al. The Role of Ultrasound Compared to Biopsy of Temporal Arteries in the Diagnosis and Treatment of Giant Cell Arteritis (TABUL): A diagnostic accuracy and cost-effectiveness study. Health Technol. Assess. 2016, 20, 1–238. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, W.A. Ultrasound in the diagnosis and management of giant cell arteritis. Rheumatology 2018, 57, ii22–ii31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttgereit, F.; Matteson, E.L.; Dejaco, C.; Dasgupta, B. Prevention of glucocorticoid morbidity in giant cell arteritis. Rheumatology 2018, 57, ii11–ii21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broder, M.S.; Sarsour, K.; Chang, E.; Collinson, N.; Tuckwell, K.; Napalkov, P.; Klearman, M. Corticosteroid-related adverse events in patients with giant cell arteritis: A claims-based analysis. Semin. Arthritis Rheum. 2016, 46, 246–252. [Google Scholar] [CrossRef]
- Restuccia, G.; Boiardi, L.; Cavazza, A.; Catanoso, M.; Macchioni, P.; Muratore, F.; Cimino, L.; Aldigeri, R.; Crescentini, F.; Pipitone, N.; et al. Flares in Biopsy-Proven Giant Cell Arteritis in Northern Italy: Characteristics and Predictors in a Long-Term Follow-Up Study. Medicine 2016, 95, e3524. [Google Scholar] [CrossRef]
- Maleszewski, J.J.; Younge, B.R.; Fritzlen, J.T.; Hunder, G.G.; Goronzy, J.J.; Warrington, K.J.; Weyand, C.M. Clinical and pathological evolution of giant cell arteritis: A prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod. Pathol. 2017, 30, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, W.A.; Moll, A.; Seifert, A.; Schicke, B.; Gromnica-Ihle, E.; Krause, A. Prognosis of large-vessel giant cell arteritis. Rheumatology 2008, 47, 1406–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Sleen, Y.; Graver, J.C.; Abdulahad, W.H.; van der Geest, K.S.M.; Boots, A.M.H.; Sandovici, M.; Brouwer, E. Leukocyte Dynamics Reveal a Persistent Myeloid Dominance in Giant Cell Arteritis and Polymyalgia Rheumatica. Front. Immunol. 2019, 10, 1981. [Google Scholar] [CrossRef]
- Jover, J.A.; Hernández-García, C.; Morado, I.C.; Vargas, E.; Bañares, A.; Fernández-Gutiérrez, B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2001, 134, 106–114. [Google Scholar] [CrossRef]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; et al. Trial of Tocilizumab in Giant-Cell Arteritis. N. Engl. J. Med. 2017, 377, 317–328. [Google Scholar] [CrossRef]
- Reichenbach, S.; Adler, S.; Bonel, H.; Cullmann, J.L.; Kuchen, S.; Bütikofer, L.; Seitz, M.; Villiger, P.M. Magnetic resonance angiography in giant cell arteritis: Results of a randomized controlled trial of tocilizumab in giant cell arteritis. Rheumatology 2018, 57, 982–986. [Google Scholar] [CrossRef] [Green Version]
- Gloor, A.D.; Yerly, D.; Adler, S.; Reichenbach, S.; Kuchen, S.; Seitz, M.; Villiger, P.M. Immuno-monitoring reveals an extended subclinical disease activity in tocilizumab-treated giant cell arteritis. Rheumatology 2018, 57, 1795–1801. [Google Scholar] [CrossRef] [Green Version]
- González-Gay, M.A.; Amoli, M.M.; Garcia-Porrua, C.; Ollier, W.E. r Genetic markers of disease susceptibility and severity in giant cell arteritis and polymyalgia rheumatica. Semin. Arthritis Rheum. 2003, 33, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Peña, D.; Remuzgo-Martínez, S.; Ocejo-Vinyals, J.G.; Atienza-Mateo, B.; Genre, F.; Muñoz-Jimenez, A.; Ortiz-Sanjuán, F.; Romero-Yuste, S.; Moriano, C.; Galindez-Agirregoikoa, E.; et al. The presence of both HLA-DRB1[*]04:01 and HLA-B[*]15:01 increases the susceptibility to cranial and extracranial giant cell arteritis. Clin. Exp. Rheumatol. 2021, 39, 21–26. [Google Scholar]
- Rhee, R.L.; Grayson, P.C.; Merkel, P.A.; Tomasson, G. Infections and the risk of incident giant cell arteritis: A population-based, case-control study. Ann. Rheum. Dis. 2017, 76, 1031–1035. [Google Scholar] [CrossRef]
- Hoffman, G.S.; Getz, T.M.; Padmanabhan, R.; Villa-Forte, A.; Clifford, A.H.; Funchain, P.; Sankunny, M.; Perry, J.D.; Blandford, A.; Kosmorsky, G.; et al. The Microbiome of Temporal Arteries. Pathog. Immun. 2019, 4, 21–38. [Google Scholar] [CrossRef]
- Getz, T.M.; Hoffman, G.S.; Padmanabhan, R.; Villa-Forte, A.; Roselli, E.E.; Blackstone, E.; Johnston, D.; Pettersson, G.; Soltesz, E.; Svensson, L.G.; et al. Microbiomes of Inflammatory Thoracic Aortic Aneurysms Due to Giant Cell Arteritis and Clinically Isolated Aortitis Differ From Those of Non-Inflammatory Aneurysms. Pathog. Immun. 2019, 4, 105–123. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.S.; Manzo, V.E.; Pedamallu, C.S.; Duke, F.; Cai, D.; Bienfang, D.C.; Padera, R.F.; Meyerson, M.; Docken, W.P. In search of a candidate pathogen for giant cell arteritis: Sequencing-based characterization of the giant cell arteritis microbiome. Arthritis Rheumatol. 2014, 66, 1939–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyand, C.M.; Watanabe, R.; Zhang, H.; Akiyama, M.; Berry, G.J.; Goronzy, J.J. Cytokines, growth factors and proteases in medium and large vessel vasculitis. Clin. Immunol. 2019, 206, 33–41. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, J.; Kurban, M.; Abbas, O. Elastophagocytosis: Underlying mechanisms and associated cutaneous entities. J. Am. Acad. Dermatol. 2014, 70, 934–944. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Bosco, M.C. Macrophage polarization: Reaching across the aisle? J. Allergy Clin. Immunol. 2019, 143, 1348–1350. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Corbera-Bellalta, M.; Audia, S.; Planas-Rigol, E.; Martin, L.; Cid, M.C.; Bonnotte, B. Recent advances in our understanding of giant cell arteritis pathogenesis. Autoimmun. Rev. 2017, 16, 833–844. [Google Scholar] [CrossRef]
- Ciccia, F.; Rizzo, A.; Ferrante, A.; Guggino, G.; Croci, S.; Cavazza, A.; Salvarani, C.; Triolo, G. New insights into the pathogenesis of giant cell arteritis. Autoimmun. Rev. 2017, 16, 675–683. [Google Scholar] [CrossRef] [PubMed]
- van Sleen, Y.; Jiemy, W.F.; Pringle, S.; van der Geest, K.S.M.; Abdulahad, W.H.; Sandovici, M.; Brouwer, E.; Heeringa, P.; Boots, A.M.H. A Distinct Macrophage Subset Mediating Tissue Destruction and Neovascularization in Giant Cell Arteritis: Implication of the YKL-40-IL-13 Receptor α2 Axis. Arthritis Rheumatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Hofer, T.P.J. Toward a refined definition of monocyte subsets. Front. Immunol. 2013, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- van Sleen, Y.; Wang, Q.; van der Geest, K.S.M.; Westra, J.; Abdulahad, W.H.; Heeringa, P.; Boots, A.M.H.; Brouwer, E. Involvement of Monocyte Subsets in the Immunopathology of Giant Cell Arteritis. Sci. Rep. 2017, 7, 6553. [Google Scholar] [CrossRef]
- Matsumoto, K.; Suzuki, K.; Yoshimoto, K.; Seki, N.; Tsujimoto, H.; Chiba, K.; Takeuchi, T. Significant association between clinical characteristics and changes in peripheral immuno-phenotype in large vessel vasculitis. Arthritis Res. Ther. 2019, 21, 304. [Google Scholar] [CrossRef] [Green Version]
- Dayyani, F.; Belge, K.-U.; Frankenberger, M.; Mack, M.; Berki, T.; Ziegler-Heitbrock, L. Mechanism of glucocorticoid-induced depletion of human CD14+ CD16+ monocytes. J. Leukoc. Biol. 2003, 74, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.C.; Hoffman, M.P.; Hernández-Rodríguez, J.; Segarra, M.; Elkin, M.; Sánchez, M.; Vilardell, C.; García-Martínez, A.; Pla-Campo, M.; Grau, J.M.; et al. Association between increased CCL2 (MCP-1) expression in lesions and persistence of disease activity in giant-cell arteritis. Rheumatology 2006, 45, 1356–1363. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Maeda, T.; Zhang, H.; Berry, G.J.; Zeisbrich, M.; Brockett, R.; Greenstein, A.E.; Tian, L.; Goronzy, J.J.; Weyand, C.M. MMP (Matrix Metalloprotease)-9-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ. Res. 2018, 123, 700–715. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Boots, A.M.H.; Steenbakkers, P.G.A.; Elewaut, D.; Bos, E.; Verheijden, G.F.M.; Verbruggen, G.; Miltenburg, A.M.M.; Rijnders, A.W.M.; Veys, E.M.; et al. Human cartilage gp-39, CD16+ monocytes in peripheral blood and synovium: Correlation with joint destruction in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1233–1243. [Google Scholar] [CrossRef] [Green Version]
- Campos, T.M.; Passos, S.T.; Novais, F.O.; Beiting, D.P.; Costa, R.S.; Queiroz, A.; Mosser, D.; Scott, P.; Carvalho, E.M.; Carvalho, L.P. Matrix metalloproteinase 9 production by monocytes is enhanced by TNF and participates in the pathology of human cutaneous Leishmaniasis. PLoS Negl. Trop. Dis. 2014, 8, e3282. [Google Scholar] [CrossRef] [Green Version]
- Jiemy, W.F.; van Sleen, Y.; van der Geest, K.S.; Ten Berge, H.A.; Abdulahad, W.H.; Sandovici, M.; Boots, A.M.; Heeringa, P.; Brouwer, E. Distinct macrophage phenotypes skewed by local granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) are associated with tissue destruction and intimal hyperplasia in giant cell arteritis. Clin. Transl. Immunol. 2020, 9, e1164. [Google Scholar] [CrossRef]
- Shirai, T.; Hilhorst, M.; Harrison, D.G.; Goronzy, J.J.; Weyand, C.M. Macrophages in vascular inflammation--From atherosclerosis to vasculitis. Autoimmunity 2015, 48, 139–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laria, A.; Lurati, A.; Marrazza, M.; Mazzocchi, D.; Re, K.A.; Scarpellini, M. The macrophages in rheumatic diseases. J. Inflamm. Res. 2016, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Di Benedetto, P.; Ruscitti, P.; Vadasz, Z.; Toubi, E.; Giacomelli, R. Macrophages with regulatory functions, a possible new therapeutic perspective in autoimmune diseases. Autoimmun. Rev. 2019, 18, 102369. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Anders, H.J. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maeyer, R.P.H.; Chambers, E.S. The impact of ageing on monocytes and macrophages. Immunol. Lett. 2021, 230, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.D.; Björnsson, J.; Bartley, G.B.; Goronzy, J.J.; Weyand, C.M. Interferon-gamma-producing T cells in giant cell vasculitis represent a minority of tissue-infiltrating cells and are located distant from the site of pathology. Am. J. Pathol. 1996, 148, 1925–1933. [Google Scholar]
- Ciccia, F.; Alessandro, R.; Rizzo, A.; Raimondo, S.; Giardina, A.; Raiata, F.; Boiardi, L.; Cavazza, A.; Guggino, G.; De Leo, G.; et al. IL-33 is overexpressed in the inflamed arteries of patients with giant cell arteritis. Ann. Rheum. Dis. 2013, 72, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. Immune mechanisms in medium and large-vessel vasculitis. Nat. Rev. Rheumatol. 2013, 9, 731–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyand, C.M.; Wagner, A.D.; Björnsson, J.; Goronzy, J.J. Correlation of the topographical arrangement and the functional pattern of tissue-infiltrating macrophages in giant cell arteritis. J. Clin. Investig. 1996, 98, 1642–1649. [Google Scholar] [CrossRef]
- Wagner, A.D.; Goronzy, J.J.; Weyand, C.M. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. Evidence for two components of the disease. J. Clin. Investig. 1994, 94, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaniego, R.; Palacios, B.S.; Domiguez-Soto, Á.; Vidal, C.; Salas, A.; Matsuyama, T.; Sánchez-Torres, C.; de la Torre, I.; Miranda-Carús, M.E.; Sánchez-Mateos, P.; et al. Macrophage uptake and accumulation of folates are polarization-dependent in vitro and in vivo and are regulated by activin A. J. Leukoc. Biol. 2014, 95, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Puig-Kröger, A.; Sierra-Filardi, E.; Domínguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gómez-Aguado, F.; Ratnam, M.; Sánchez-Mateos, P.; Corbí, A.L. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef] [Green Version]
- Kittan, N.A.; Allen, R.M.; Dhaliwal, A.; Cavassani, K.A.; Schaller, M.; Gallagher, K.A.; Carson, W.F., 4th; Mukherjee, S.; Grembecka, J.; Cierpicki, T.; et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS ONE 2013, 8, e78045. [Google Scholar] [CrossRef] [Green Version]
- Tomita, T.; Imakawa, K. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in giant cell arteritis: An immunocytochemical study. Pathology 1998, 30, 40–50. [Google Scholar] [CrossRef]
- Rodríguez-Pla, A.; Bosch-Gil, J.A.; Rosselló-Urgell, J.; Huguet-Redecilla, P.; Stone, J.H.; Vilardell-Tarres, M. Metalloproteinase-2 and -9 in giant cell arteritis: Involvement in vascular remodeling. Circulation 2005, 112, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Nikkari, S.T.; Höyhtyä, M.; Isola, J.; Nikkari, T. Macrophages contain 92-kd gelatinase (MMP-9) at the site of degenerated internal elastic lamina in temporal arteritis. Am. J. Pathol. 1996, 149, 1427–1433. [Google Scholar]
- Albano-Aluquin, S.; Malysz, J.; Aluquin, V.R.; Ratnam, M.; Olsen, N. An immunohistochemical analysis of folate receptor beta expression and distribution in giant cell arteritis—A pilot study. Am. J. Clin. Exp. Immunol. 2017, 6, 107–114. [Google Scholar] [PubMed]
- Erbel, C.; Akhavanpoor, M.; Okuyucu, D.; Wangler, S.; Dietz, A.; Zhao, L.; Stellos, K.; Little, K.M.; Lasitschka, F.; Doesch, A.; et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J. Immunol. 2014, 193, 4344–4355. [Google Scholar] [CrossRef] [Green Version]
- Jiemy, W.F.; Reitsema, R.; Kwant, A.; Abdulahad, W.; Boots, A.; Heeringa, P.; Brouwer, E. POS0112 CD8+ T-CELL infiltration is associated with lesional GM-CSF overexpression in GCA. Ann. Rheum. Dis. 2021, 80, 267. [Google Scholar] [CrossRef]
- Van Der Geest, K.S.M.; Abdulahad, W.H.; Chalan, P.; Rutgers, A.; Horst, G.; Huitema, M.G.; Roffel, M.P.; Roozendaal, C.; Kluin, P.M.; Bos, N.A.; et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol. 2014, 66, 1927–1938. [Google Scholar] [CrossRef] [PubMed]
- Graver, J.C.; Sandovici, M.; Diepstra, A.; Boots, A.M.H.; Brouwer, E. Artery tertiary lymphoid organs in giant cell arteritis are not exclusively located in the media of temporal arteries. Ann. Rheum. Dis. 2018, 77, 2–3. [Google Scholar] [CrossRef]
- Ciccia, F.; Rizzo, A.; Maugeri, R.; Alessandro, R.; Croci, S.; Guggino, G.; Cavazza, A.; Raimondo, S.; Cannizzaro, A.; Iacopino, D.G.; et al. Ectopic expression of CXCL13, BAFF, April and LT-β is associated with artery tertiary lymphoid organs in giant cell arteritis. Ann. Rheum. Dis. 2017, 76, 235–243. [Google Scholar] [CrossRef]
- Graver, J.C.; Jiemy, W.F.; Altulea, D.; Boots, A.; Heeringa, P.; Abdulahad, W.; Brouwer, E.; Sandovici, M. OP0062 Cytokine producing B cells skew macrophages towards a pro-inflammatory phenotype in giant cell arteritis. Ann. Rheum. Dis. 2021, 80, 33–34. [Google Scholar] [CrossRef]
- Kostic, M.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I. Granulocyte-macrophage colony-stimulating factor as a mediator of autoimmunity in multiple sclerosis. J. Neuroimmunol. 2018, 323, 1–9. [Google Scholar] [CrossRef]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef] [PubMed]
- Artyomov, M.N.; Sergushichev, A.; Schilling, J.D. Integrating immunometabolism and macrophage diversity. Semin. Immunol. 2016, 28, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Batista-Gonzalez, A.; Vidal, R.; Criollo, A.; Carreño, L.J. New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front. Immunol. 2020, 10, 2993. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Saraber, D.L. Metabolic regulation of macrophages in tissues. Cell. Immunol. 2018, 330, 54–59. [Google Scholar] [CrossRef]
- Kang, S.; Kumanogoh, A. The spectrum of macrophage activation by immunometabolism. Int. Immunol. 2020, 32, 467–473. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J. A critical role for citrate metabolism in LPS signalling. Biochem. J. 2011, 438, e5–e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, J.; Segarra, M.; Vilardell, C.; Sánchez, M.; García-Martínez, A.; Esteban, M.J.; Queralt, C.; Grau, J.M.; Urbano-Márquez, A.; Palacín, A.; et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology 2004, 43, 294–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittner, H.L.; Kaiser, M.; Brack, A.; Szweda, L.I.; Goronzy, J.J.; Weyand, C.M. Tissue-destructive macrophages in giant cell arteritis. Circ. Res. 1999, 84, 1050–1058. [Google Scholar] [CrossRef]
- Tavakoli, S.; Short, J.D.; Downs, K.; Nguyen, H.N.; Lai, Y.; Zhang, W.; Jerabek, P.; Goins, B.; Sadeghi, M.M.; Asmis, R. Differential Regulation of Macrophage Glucose Metabolism by Macrophage Colony-stimulating Factor and Granulocyte-Macrophage Colony-stimulating Factor: Implications for (18)F FDG PET Imaging of Vessel Wall Inflammation. Radiology 2017, 283, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.R.; Gu, G.J.; Jung, D.; Kim, Y.W.; Na, J.; Woo, J.S.; Cho, J.Y.; Youn, H.; Seok, S.H. GM-CSF Induces Inflammatory Macrophages by Regulating Glycolysis and Lipid Metabolism. J. Immunol. 2016, 197, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- van Beek, A.A.; Van den Bossche, J.; Mastroberardino, P.G.; de Winther, M.P.J.; Leenen, P.J.M. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Appay, V.; Campisi, J.; Frasca, D.; Fülöp, T.; Sauce, D.; Larbi, A.; Weinberger, B.; Cossarizza, A. Aging of the immune system: Focus on inflammation and vaccination. Eur. J. Immunol. 2016, 46, 2286–2301. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.V.; Liao, Y.J.; Kim, J.W.; Goronzy, J.J.; Weyand, C.M. Giant cell arteritis: Immune and vascular aging as disease risk factors. Arthritis Res. Ther. 2011, 13, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, J.; Ramirez, R.; Soriano, S.; Alvarez de Lara, M.A.; Rodriguez, M.; Martin-Malo, A.; Aljama, P. Monocytes from dialysis patients exhibit characteristics of senescent cells: Does it really mean inflammation? Contrib. Nephrol. 2005, 149, 208–218. [Google Scholar] [CrossRef]
- Merino, A.; Buendia, P.; Martin-Malo, A.; Aljama, P.; Ramirez, R.; Carracedo, J. Senescent CD14+CD16+ monocytes exhibit proinflammatory and proatherosclerotic activity. J. Immunol. 2011, 186, 1809–1815. [Google Scholar] [CrossRef] [Green Version]
- Seidler, S.; Zimmermann, H.W.; Bartneck, M.; Trautwein, C.; Tacke, F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 2010, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Coit, P.; De Lott, L.B.; Nan, B.; Elner, V.M.; Sawalha, A.H. DNA methylation analysis of the temporal artery microenvironment in giant cell arteritis. Ann. Rheum. Dis. 2016, 75, 1196–1202. [Google Scholar] [CrossRef]
- Wang, Q.; Westra, J.; van der Geest, K.S.M.; Moser, J.; Bijzet, J.; Kuiper, T.; Lorencetti, P.G.; Joosten, L.A.B.; Netea, M.G.; Heeringa, P.; et al. Reduced levels of cytosolic DNA sensor AIM2 are associated with impaired cytokine responses in healthy elderly. Exp. Gerontol. 2016, 78, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Pryshchep, O.; Ma-Krupa, W.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation 2008, 118, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.; Molloy, E.S. The role of toll like receptors in giant cell arteritis. Rheumatology 2016, 55, 1921–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, L.Á.; López-Hoyos, M.; Mata, C.; Fontalba, A.; Alen, J.C.; Marín, M.J.; Fernández-Luna, J.L.; Balbín, J.A.; Zaldunbide, M.A.; Blanco, R.; et al. Expression and function of toll-like receptors in peripheral blood mononuclear cells of patients with polymyalgia rheumatica and giant cell arteritis. Ann. Rheum. Dis. 2011, 70, 1677–1683. [Google Scholar] [CrossRef] [PubMed]
- Bardi, M.; Diamantopoulos, A.P. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice summary. Radiol. Med. 2019, 124, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Van Der Geest, K.S.M.; Borg, F.; Kayani, A.; Paap, D.; Gondo, P.; Schmidt, W.; Luqmani, R.A.; Dasgupta, B. Novel ultrasonographic Halo Score for giant cell arteritis: Assessment of diagnostic accuracy and association with ocular ischaemia. Ann. Rheum. Dis. 2020, 79, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-González, S.; Depetris, M.; García-Martínez, A.; Espígol-Frigolé, G.; Tavera-Bahillo, I.; Corbera-Bellata, M.; Planas-Rigol, E.; Alba, M.A.; Hernández-Rodríguez, J.; Grau, J.M.; et al. Positron emission tomography assessment of large vessel inflammation in patients with newly diagnosed, biopsy-proven giant cell arteritis: A prospective, case-control study. Ann. Rheum. Dis. 2014, 73, 1388–1392. [Google Scholar] [CrossRef] [Green Version]
- Nienhuis, P.H.; Sandovici, M.; Glaudemans, A.W.; Slart, R.H.; Brouwer, E. Visual and semiquantitative assessment of cranial artery inflammation with FDG-PET/CT in giant cell arteritis. Semin. Arthritis Rheum. 2020, 50, 616–623. [Google Scholar] [CrossRef]
- Aschwanden, M.; Schegk, E.; Imfeld, S.; Staub, D.; Rottenburger, C.; Berger, C.T.; Daikeler, T. Vessel wall plasticity in large vessel giant cell arteritis: An ultrasound follow-up study. Rheumatology 2019, 58, 792–797. [Google Scholar] [CrossRef]
- van der Geest, K.S.M.; Treglia, G.; Glaudemans, A.W.J.M.; Brouwer, E.; Sandovici, M.; Jamar, F.; Gheysens, O.; Slart, R.H.J.A. Diagnostic value of [18F]FDG-PET/CT for treatment monitoring in large vessel vasculitis: A systematic review and meta-analysis. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3886–3902. [Google Scholar] [CrossRef] [PubMed]
- Burja, B.; Feichtinger, J.; Lakota, K.; Thallinger, G.G.; Sodin-Semrl, S.; Kuret, T.; Rotar, Ž.; Ješe, R.; Žigon, P.; Čučnik, S.; et al. Utility of serological biomarkers for giant cell arteritis in a large cohort of treatment-naïve patients. Clin. Rheumatol. 2019, 38, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Geest, K.S.M.; Sandovici, M.; van Sleen, Y.; Sanders, J.-S.; Bos, N.A.; Abdulahad, W.H.; Stegeman, C.A.; Heeringa, P.; Rutgers, A.; Kallenberg, C.G.M.; et al. Review: What Is the Current Evidence for Disease Subsets in Giant Cell Arteritis? Arthritis Rheumatol. 2018, 70, 1366–1376. [Google Scholar] [CrossRef] [Green Version]
- van Sleen, Y.; Sandovici, M.; Abdulahad, W.H.; Bijzet, J.; van der Geest, K.S.M.; Boots, A.M.H.; Brouwer, E. Markers of angiogenesis and macrophage products for predicting disease course and monitoring vascular inflammation in giant cell arteritis. Rheumatology 2019, 58, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Tombetti, E.; Hysa, E.; Mason, J.C.; Cimmino, M.A.; Camellino, D. Blood Biomarkers for Monitoring and Prognosis of Large Vessel Vasculitides. Curr. Rheumatol. Rep. 2021, 23, 17. [Google Scholar] [CrossRef]
- Johansen, J.S.; Baslund, B.; Garbarsch, C.; Hansen, M.; Stoltenberg, M.; Lorenzen, I.; Price, P.A. YKL-40 in giant cells and macrophages from patients with giant cell arteritis. Arthritis Rheum. 1999, 42, 2624–2630. [Google Scholar] [CrossRef]
- Baldini, M.; Maugeri, N.; Ramirez, G.A.; Giacomassi, C.; Castiglioni, A.; Prieto-González, S.; Corbera-Bellalta, M.; Di Comite, G.; Papa, I.; Dell’antonio, G.; et al. Selective up-regulation of the soluble pattern-recognition receptor pentraxin 3 and of vascular endothelial growth factor in giant cell arteritis: Relevance for recent optic nerve ischemia. Arthritis Rheum. 2012, 64, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.; Younge, B.; Björnsson, J.; Goronzy, J.J.; Weyand, C.M. Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells. Am. J. Pathol. 1999, 155, 765–774. [Google Scholar] [CrossRef]
- Sorbi, D.; French, D.L.; Nuovo, G.J.; Kew, R.R.; Arbeit, L.A.; Gruber, B.L. Elevated levels of 92-kd type IV collagenase (matrix metalloproteinase 9) in giant cell arteritis. Arthritis Rheum. 1996, 39, 1747–1753. [Google Scholar] [CrossRef]
- Grossman, C.; Yassin, N.; Avivi, C.; Bornstein, G.; Ben-Zvi, I.; Barshack, I. Cytokine expression in temporal arteries: Comparative analysis between patients with biopsy-positive giant cell arteritis, biopsy-negative giant cell arteritis and biopsy-negative without arteritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 1), 122–129. [Google Scholar] [PubMed]
- Prieto-González, S.; Terrades-García, N.; Corbera-Bellalta, M.; Planas-Rigol, E.; Miyabe, C.; Alba, M.A.; Ponce, A.; Tavera-Bahillo, I.; Murgia, G.; Espígol-Frigolé, G.; et al. Serum osteopontin: A biomarker of disease activity and predictor of relapsing course in patients with giant cell arteritis. Potential clinical usefulness in tocilizumab-treated patients. RMD Open 2017, 3, e000570. [Google Scholar] [CrossRef]
- Desbois, A.-C.; Cacoub, P.; Leroyer, A.S.; Tellier, E.; Garrido, M.; Maciejewski-Duval, A.; Comarmond, C.; Barete, S.; Arock, M.; Bruneval, P.; et al. Immunomodulatory role of Interleukin-33 in large vessel vasculitis. Sci. Rep. 2020, 10, 6405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, S.; Nunokawa, T.; Kobayashi, S.; Kamei, S.; Yokogawa, N.; Takizawa, Y.; Shimada, K.; Sugii, S.; Setoguchi, K. MMP-3 can distinguish isolated PMR from PMR with GCA: A retrospective study regarding PMR and GCA in Japan. Mod. Rheumatol. 2016, 26, 259–264. [Google Scholar] [CrossRef] [PubMed]
- van Sleen, Y.; Boots, A.M.H.; Abdulahad, W.H.; Bijzet, J.; Sandovici, M.; van der Geest, K.S.M.; Brouwer, E. High angiopoietin-2 levels associate with arterial inflammation and long-term glucocorticoid requirement in polymyalgia rheumatica. Rheumatology 2020, 59, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Muratore, F.; Boiardi, L.; Restuccia, G.; Cavazza, A.; Catanoso, M.; Macchioni, P.; Spaggiari, L.; Cimino, L.; Aldigeri, R.; Pipitone, N.; et al. Relapses and long-term remission in large vessel giant cell arteritis in northern Italy: Characteristics and predictors in a long-term follow-up study. Semin. Arthritis Rheum. 2020, 50, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; et al. Glucocorticoid Dosages and Acute-Phase Reactant Levels at Giant Cell Arteritis Flare in a Randomized Trial of Tocilizumab. Arthritis Rheumatol. 2019, 71, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Stellingwerff, M.D.; Brouwer, E.; Lensen, K.-J.D.F.; Rutgers, A.; Arends, S.; van der Geest, K.S.M.; Glaudemans, A.W.J.M.; Slart, R.H.J.A. Different Scoring Methods of FDG PET/CT in Giant Cell Arteritis: Need for Standardization. Medicine 2015, 94, e1542. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, B.D.; Gormsen, L.C.; Hansen, I.T.; Keller, K.K.; Therkildsen, P.; Hauge, E.-M. Three days of high-dose glucocorticoid treatment attenuates large-vessel 18F-FDG uptake in large-vessel giant cell arteritis but with a limited impact on diagnostic accuracy. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1119–1128. [Google Scholar] [CrossRef]
- Dunphy, M.P.S.; Freiman, A.; Larson, S.M.; Strauss, H.W. Association of vascular 18F-FDG uptake with vascular calcification. J. Nucl. Med. 2005, 46, 1278–1284. [Google Scholar]
- Bural, G.G.; Torigian, D.A.; Basu, S.; Bozkurt, M.F.; Houseni, M.; Alavi, A. Atherosclerotic inflammatory activity in the aorta and its correlation with aging and gender as assessed by 18F-FDG-PET. Hell. J. Nucl. Med. 2013, 16, 164–168. [Google Scholar] [CrossRef]
- Jiemy, W.F.; Heeringa, P.; Kamps, J.A.A.M.; van der Laken, C.J.; Slart, R.H.J.A.; Brouwer, E. Positron emission tomography (PET) and single photon emission computed tomography (SPECT) imaging of macrophages in large vessel vasculitis: Current status and future prospects. Autoimmun. Rev. 2018, 17, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Farrah, T.E.; Basu, N.; Dweck, M.; Calcagno, C.; Fayad, Z.A.; Dhaun, N. Advances in Therapies and Imaging for Systemic Vasculitis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1520–1541. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.J.; Isakow, W.; Byers, D.E.; Engle, J.T.; Griffin, E.A.; Kemp, D.; Brody, S.L.; Gropler, R.J.; Miller, J.P.; Chu, W.; et al. Imaging pulmonary inducible nitric oxide synthase expression with PET. J. Nucl. Med. 2015, 56, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Verweij, N.J.F.; Yaqub, M.; Bruijnen, S.T.G.; Pieplenbosch, S.; ter Wee, M.M.; Jansen, G.; Chen, Q.; Low, P.S.; Windhorst, A.D.; Lammertsma, A.A.; et al. First in man study of [18F]fluoro-PEG-folate PET: A novel macrophage imaging technique to visualize rheumatoid arthritis. Sci. Rep. 2020, 10, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzel, T.; Müller, C.; Groehn, V.; Müller, A.; Reber, J.; Fischer, C.R.; Krämer, S.D.; Schibli, R.; Ametamey, S.M. Radiosynthesis and preclinical evaluation of 3′-Aza-2′-[(18)F]fluorofolic acid: A novel PET radiotracer for folate receptor targeting. Bioconjug. Chem. 2013, 24, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Moisio, O.; Palani, S.; Virta, J.; Elo, P.; Liljenbäck, H.; Tolvanen, T.; Käkelä, M.; Miner, M.G.; Herre, E.A.; Marjamäki, P.; et al. Radiosynthesis and preclinical evaluation of [(68)Ga]Ga-NOTA-folate for PET imaging of folate receptor β-positive macrophages. Sci. Rep. 2020, 10, 13593. [Google Scholar] [CrossRef]
- Schniering, J.; Benešová, M.; Brunner, M.; Haller, S.; Cohrs, S.; Frauenfelder, T.; Vrugt, B.; Feghali-Bostwick, C.; Schibli, R.; Distler, O.; et al. (18)F-AzaFol for Detection of Folate Receptor-β Positive Macrophages in Experimental Interstitial Lung Disease-A Proof-of-Concept Study. Front. Immunol. 2019, 10, 2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvola, J.M.U.; Li, X.-G.; Virta, J.; Marjamäki, P.; Liljenbäck, H.; Hytönen, J.P.; Tarkia, M.; Saunavaara, V.; Hurme, S.; Palani, S.; et al. Aluminum fluoride-18 labeled folate enables in vivo detection of atherosclerotic plaque inflammation by positron emission tomography. Sci. Rep. 2018, 8, 9720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichendorff, S.; Svendsen, P.; Bender, D.; Keiding, S.; Christensen, E.I.; Deleuran, B.; Moestrup, S.K. Biodistribution and PET imaging of a novel [68Ga]-anti-CD163-antibody conjugate in rats with collagen-induced arthritis and in controls. Mol. Imaging Biol. 2015, 17, 87–93. [Google Scholar] [CrossRef]
- Kiugel, M.; Hellberg, S.; Käkelä, M.; Liljenbäck, H.; Saanijoki, T.; Li, X.-G.; Tuomela, J.; Knuuti, J.; Saraste, A.; Roivainen, A. Evaluation of [(68)Ga]Ga-DOTA-TCTP-1 for the Detection of Metalloproteinase 2/9 Expression in Mouse Atherosclerotic Plaques. Molecules 2018, 23, 3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Pan, D.; Cheng, C.; Zhang, A.; Ma, C.; Wang, L.; Zhang, D.; Liu, H.; Jiang, H.; Wang, T.; et al. Targeting of MMP2 activity in malignant tumors with a 68Ga-labeled gelatinase inhibitor cyclic peptide. Nucl. Med. Biol. 2015, 42, 939–944. [Google Scholar] [CrossRef]
- Tahara, N.; Mukherjee, J.; de Haas, H.J.; Petrov, A.D.; Tawakol, A.; Haider, N.; Tahara, A.; Constantinescu, C.C.; Zhou, J.; Boersma, H.H.; et al. 2-deoxy-2-[18F]fluoro-D-mannose positron emission tomography imaging in atherosclerosis. Nat. Med. 2014, 20, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kim, S.; Seo, H.S.; Lee, Y.J.; Eo, J.S.; Jeong, J.M.; Lee, B.; Kim, J.Y.; Park, Y.M.; Jeong, M. Novel PET Imaging of Atherosclerosis with 68Ga-Labeled NOTA-Neomannosylated Human Serum Albumin. J. Nucl. Med. 2016, 57, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Blykers, A.; Schoonooghe, S.; Xavier, C.; D’hoe, K.; Laoui, D.; D’Huyvetter, M.; Vaneycken, I.; Cleeren, F.; Bormans, G.; Heemskerk, J.; et al. PET Imaging of Macrophage Mannose Receptor-Expressing Macrophages in Tumor Stroma Using 18F-Radiolabeled Camelid Single-Domain Antibody Fragments. J. Nucl. Med. 2015, 56, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Locke, L.W.; Mayo, M.W.; Yoo, A.D.; Williams, M.B.; Berr, S.S. PET imaging of tumor associated macrophages using mannose coated 64Cu liposomes. Biomaterials 2012, 33, 7785–7793. [Google Scholar] [CrossRef]
- Xavier, C.; Blykers, A.; Laoui, D.; Bolli, E.; Vaneyken, I.; Bridoux, J.; Baudhuin, H.; Raes, G.; Everaert, H.; Movahedi, K.; et al. Clinical Translation of [(68)Ga]Ga-NOTA-anti-MMR-sdAb for PET/CT Imaging of Protumorigenic Macrophages. Mol. Imaging Biol. 2019, 21, 898–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamare, F.; Hinz, R.; Gaemperli, O.; Pugliese, F.; Mason, J.C.; Spinks, T.; Camici, P.G.; Rimoldi, O.E. Detection and quantification of large-vessel inflammation with 11C-(R)-PK11195 PET/CT. J. Nucl. Med. 2011, 52, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.C.; Sarsour, K.; Collinson, N.; Tuckwell, K.; Musselman, D.; Klearman, M.; Napalkov, P.; Jick, S.S.; Stone, J.H.; Meier, C.R. Incidence of outcomes potentially associated with corticosteroid therapy in patients with giant cell arteritis. Semin. Arthritis Rheum. 2017, 46, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.C.; Sarsour, K.; Collinson, N.; Tuckwell, K.; Musselman, D.; Klearman, M.; Napalkov, P.; Jick, S.S.; Stone, J.H.; Meier, C.R. Serious adverse effects associated with glucocorticoid therapy in patients with giant cell arteritis (GCA): A nested case–control analysis. Semin. Arthritis Rheum. 2017, 46, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Brack, A.; Rittner, H.L.; Younge, B.R.; Kaltschmidt, C.; Weyand, C.M.; Goronzy, J.J. Glucocorticoid-mediated repression of cytokine gene transcription in human arteritis-SCID chimeras. J. Clin. Investig. 1997, 99, 2842–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Younge, B.R.; Olshen, R.A.; Goronzy, J.J.; Weyand, C.M. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation 2010, 121, 906–915. [Google Scholar] [CrossRef] [Green Version]
- Sultan, H.; Smith, S.V.; Lee, A.G.; Chévez-Barrios, P. Pathologic Markers Determining Prognosis in Patients With Treated or Healing Giant Cell Arteritis. Am. J. Ophthalmol. 2018, 193, 45–53. [Google Scholar] [CrossRef]
- Schönau, V.; Roth, J.; Tascilar, K.; Corte, G.; Manger, B.; Rech, J.; Schmidt, D.; Cavallaro, A.; Uder, M.; Crescentini, F.; et al. Resolution of vascular inflammation in patients with new-onset giant cell arteritis: Data from the RIGA study. Rheumatology 2021, 60, 3851–3861. [Google Scholar] [CrossRef]
- Slart, R.H.J.A.; Glaudemans, A.W.J.M.; Brouwer, E.; van der Geest, K.S.M. Therapy response evaluation in large-vessel vasculitis: A new role for [18F]FDG-PET/CT? Rheumatology 2021, 60, 3494–3495. [Google Scholar] [CrossRef]
- Unizony, S.; Arias-Urdaneta, L.; Miloslavsky, E.; Arvikar, S.; Khosroshahi, A.; Keroack, B.; Stone, J.R.; Stone, J.H. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res. 2012, 64, 1720–1729. [Google Scholar] [CrossRef]
- Monin, L.; Gaffen, S.L. Interleukin 17 family cytokines: Signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb. Perspect. Biol. 2018, 10, a028522. [Google Scholar] [CrossRef]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.C.; Muralidharan, S.; Corbera-Bellalta, M.; Espigol-Frigole, G.; Marco Hernandez, J.; Denuc, A.; Rios-Garces, R.; Terrades-Garcia, N.; Paolini, J.F.; D’andrea, A. FRI0010 GM-CSFR pathway is implicated in pathogenic inflammatory mechanisms in giant cell arteritis. Ann. Rheum. Dis. 2020, 79, 576. [Google Scholar] [CrossRef]
- Cid, M.C.; Unizony, S.; Pupim, L.; Fang, F.; Pirrello, J.; Ren, A.; Samant, M.; Zhou, T.; Paolini, J.F. OP0059 mavrilimumab (anti GM-CSF receptor α monoclonal antibody) reduces risk of flare and increases sustained remission in a phase 2 trial of patients with giant cell arteritis. Ann. Rheum. Dis. 2021, 80, 31–32. [Google Scholar] [CrossRef]
- Harrington, R.; Al Nokhatha, S.A.; Conway, R. Biologic Therapies for Giant Cell Arteritis. Biol. Targets Ther. 2021, 15, 17–29. [Google Scholar] [CrossRef]
- Yoshida, K.; Sakamoto, N.; Kurosaka, D. Improvement in Polymyalgia Rheumatica Associated With Improved Control of Diabetes Mellitus: A Case Series. Ann. Intern. Med. 2021, 174, 274–276. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 13280. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Tracer Target Defined in GCA | References | Radiotracers | Status | References |
|---|---|---|---|---|
| iNOS | [53] | [18F]NOS | Clinical | [123] |
| FRβ | [44] | [18F]PEG-folate | Clinical | [124] |
| 3′-aza-2′-[18F]fluorofolicacid | Preclinical | [125] | ||
| [68Ga]NOTA-folate | Preclinical | [126] | ||
| [18F]AzaFol-based PET/CT | Preclinical | [127] | ||
| [18F]FOL | Preclinical | [128] | ||
| CD163 | [104] | [68Ga]anti-CD163-antibody | Preclinical | [129] |
| MMP2/MM9 | [58,59] | [68Ga]DOTA-TCTP-1 | Preclinical | [130] |
| [68Ga]NOTA-C6 | Preclinical | [131] | ||
| CD206 | [44] | [18F]FDM | Preclinical | [132] |
| [68Ga]NOTA-MSA | Preclinical | [133] | ||
| [18F]FB-anti-MMR | Preclinical | [134] | ||
| [64Cu]MAN-LIPs | Preclinical | [135] | ||
| [68Ga]NOTA-anti-MMR-sdAb | Clinical | [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esen, I.; Jiemy, W.F.; van Sleen, Y.; van der Geest, K.S.M.; Sandovici, M.; Heeringa, P.; Boots, A.M.H.; Brouwer, E. Functionally Heterogenous Macrophage Subsets in the Pathogenesis of Giant Cell Arteritis: Novel Targets for Disease Monitoring and Treatment. J. Clin. Med. 2021, 10, 4958. https://doi.org/10.3390/jcm10214958
Esen I, Jiemy WF, van Sleen Y, van der Geest KSM, Sandovici M, Heeringa P, Boots AMH, Brouwer E. Functionally Heterogenous Macrophage Subsets in the Pathogenesis of Giant Cell Arteritis: Novel Targets for Disease Monitoring and Treatment. Journal of Clinical Medicine. 2021; 10(21):4958. https://doi.org/10.3390/jcm10214958
Chicago/Turabian StyleEsen, Idil, William F. Jiemy, Yannick van Sleen, Kornelis S.M. van der Geest, Maria Sandovici, Peter Heeringa, Annemieke M. H. Boots, and Elisabeth Brouwer. 2021. "Functionally Heterogenous Macrophage Subsets in the Pathogenesis of Giant Cell Arteritis: Novel Targets for Disease Monitoring and Treatment" Journal of Clinical Medicine 10, no. 21: 4958. https://doi.org/10.3390/jcm10214958
APA StyleEsen, I., Jiemy, W. F., van Sleen, Y., van der Geest, K. S. M., Sandovici, M., Heeringa, P., Boots, A. M. H., & Brouwer, E. (2021). Functionally Heterogenous Macrophage Subsets in the Pathogenesis of Giant Cell Arteritis: Novel Targets for Disease Monitoring and Treatment. Journal of Clinical Medicine, 10(21), 4958. https://doi.org/10.3390/jcm10214958