
NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
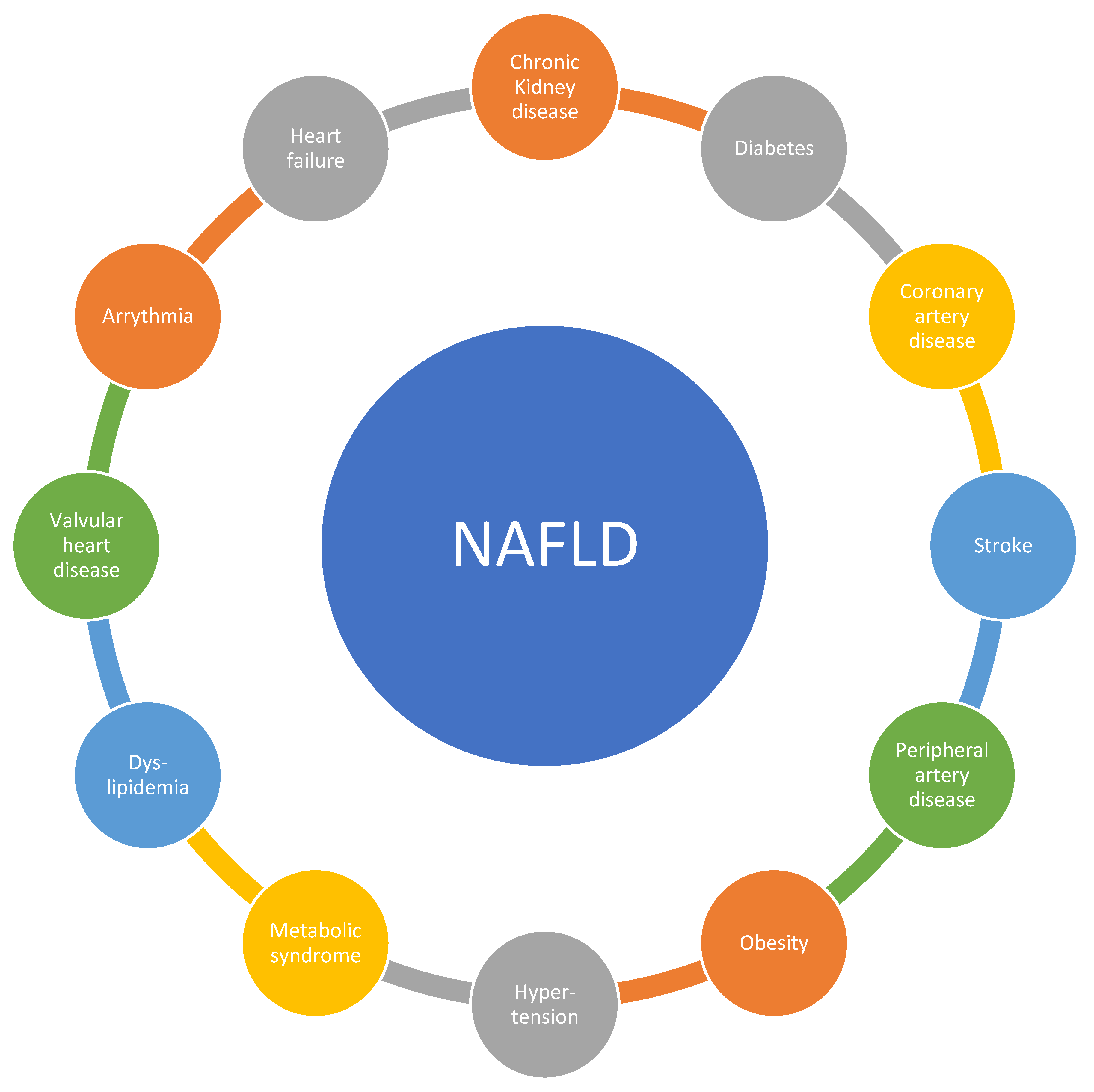
2. Epidemiological Data Linking NAFLD to Cardiovascular Disease (CVD)
2.1. Vascular Disease: Atherosclerosis Including Stroke, Peripheral Artery Disease, and Coronary Artery Disease
2.2. Metabolic Disease: Metabolic Syndrome, Dyslipidemia, Diabetes, and Obesity
2.3. Other Cardiovascular Disease Manifestations
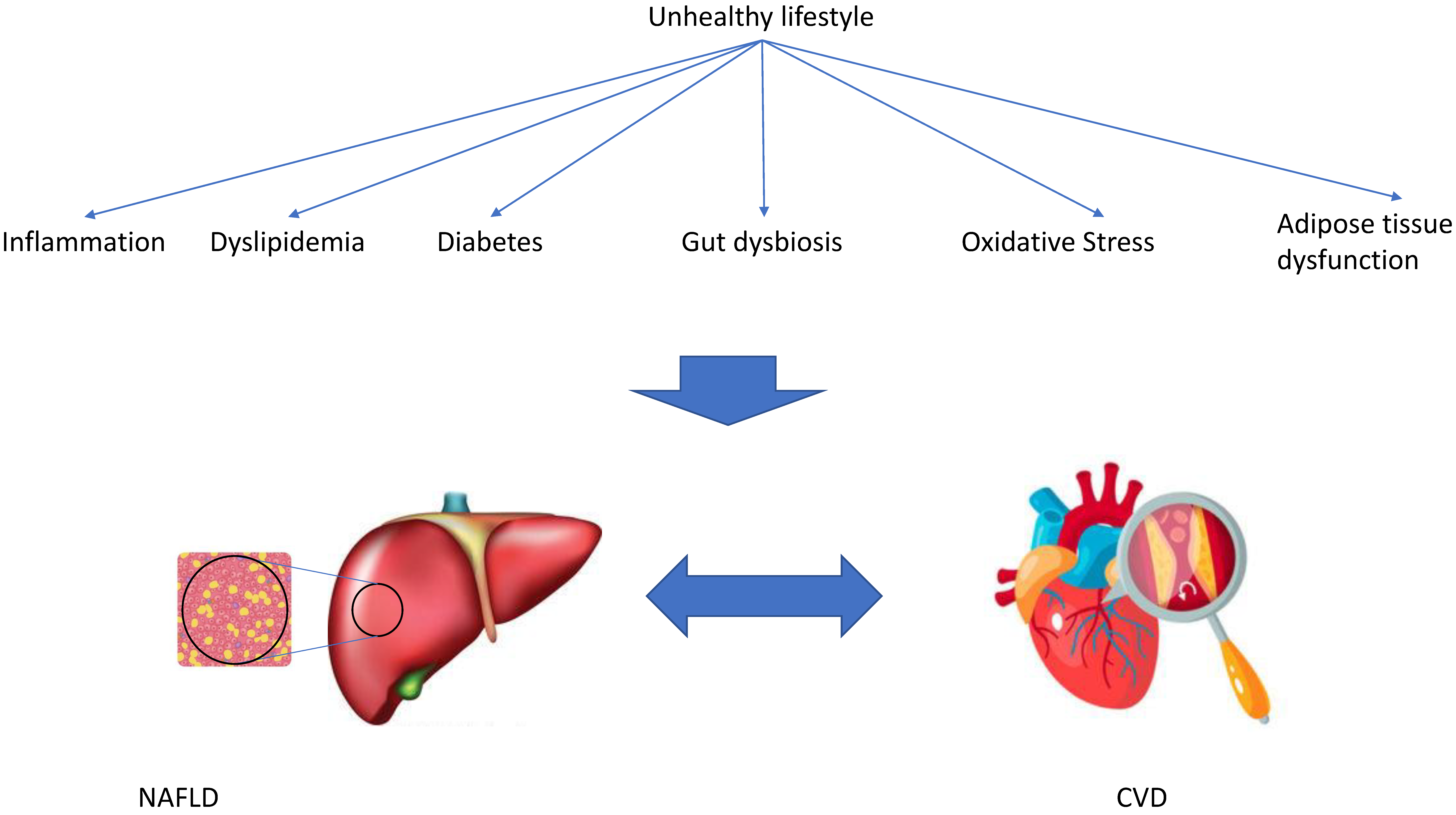
3. Possible Mechanisms That Might Explain the Association between NAFLD and CVD
3.1. Inflammation and Oxidative Stress
3.2. Gut Microbiota
3.3. Dyslipidemia and Adipose Tissue (Dys-)Function
3.4. Endothelial Dysfunction
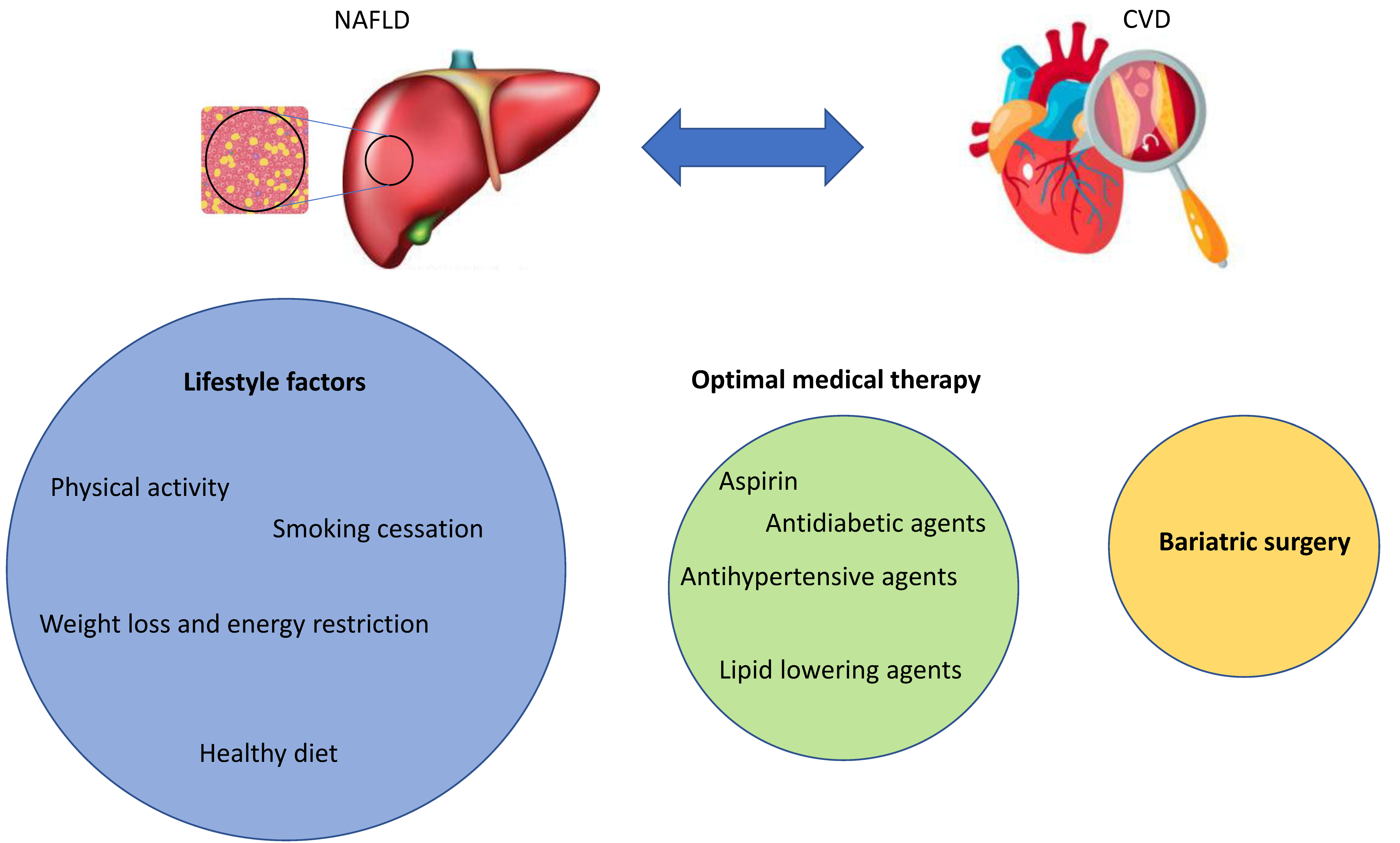
4. Current and Future Treatment Options for NAFLD Complicated by CVD
4.1. Lifestyle Interventions
4.1.1. Smoking Cessation
4.1.2. Weight Loss and Energy Restriction
4.1.3. Healthy Nutrition
4.1.4. Exercise
4.2. Medication
4.2.1. Aspirin
4.2.2. Lipid-Lowering Agents
4.2.3. Antihypertensive Agents
4.2.4. Antidiabetic Agents
- Thiazolidinediones (TZDs): TZDs are selective and potent PPARγ agonists by which they act as insulin sensitizers and, thus, synergistically with circulating insulin. Through this effect, TZD overcomes insulin resistance as a hallmark of NAFLD. Of the three compounds introduced to the market, only pioglitazone currently remains available after troglitazone was withdrawn due to severe hepatotoxicity and rosiglitazone was linked to adverse cardiovascular incidents. Pioglitazone was compared to vitamin E and a placebo in the PIVENS trial and given for 96 months at a dose of 30 mg daily. Elevated serum liver enzymes were reduced with both agents, and both were associated with significant reductions in hepatic steatosis (p = 0.005 for vitamin E and p < 0.001 for pioglitazone) and lobular inflammation (p = 0.02 for vitamin E and p = 0.004 for pioglitazone). However, improvements in fibrosis scores were observed for neither active treatment. For the latter, however, a meta-analysis of five randomized controlled trials with pioglitazone in NAFLD patients [208] found an association with improved advanced fibrosis (OR (odds ratio), 3.15; 95% CI, 1.25–7.93; p = 0.01), fibrosis of any stage (OR, 1.66; 95% CI, 1.12–2.47; p = 0.01), and NASH resolution (OR, 3.22; 95% CI, 2.17–4.79; p < 0.001), even when given to subjects without diabetes [209]. Relevant disadvantages of pioglitazone in the treatment of T2DM in NAFLD patients include significant weight gain, fluid retention, and some evidence from observational studies demonstrating an increased risk of bladder cancer [210]. Pioglitazone is the current treatment of choice for NAFLD in subjects with T2DM.
- Sodium-glucose cotransporter-2 (SGLT-2) inhibitors: SGLT2 inhibitors are compounds that inhibit SGLT-2 proteins located in the renal tubules of the kidneys, which are responsible for the reabsorption of glucose, thereby increasing the loss of glucose via urinary excretion. There are three SGLT-2-selective inhibitors approved by the Food and Drug Administration (FDA), namely canagliflozin, dapagliflozin, and empagliflozin. In a recent systematic review of studies investigating SGLT-2 inhibitors in NAFLD patients, seven studies were reviewed, of which six demonstrated certain improvements by SGLT-2 inhibitors in NAFLD. Meanwhile, five studies showed a reduction in steatosis, and only one study suggested an improvement of NASH histology features [211]. Beyond these liver-related endpoints, SGLT-2 inhibitors also favorably impact weight, cardiovascular, and nephrological outcomes, rendering them a satisfactory and increasingly popular, albeit not ideal, group of agents to treat T2DM in NAFLD. Urinary tract infections limit their use in a relevant number of patients [212].
- Glucagon-like peptide 1 (GLP-1) agonists. GLP-1 is an antihyperglycemic hormone that induces pancreatic β cells to secrete insulin, and it was demonstrated that patients with NAFLD reveal insufficient GLP-1 secretion [213]. Thus, pharmaceuticals that offset this imbalance to improve glucose homeostasis are attractive candidates for treating T2DM in the context of NAFLD. Indeed, GLP-1 agonists have recently gained much attention based on clinical data from trials that provide strong evidence for a powerful effect of GLP-1 agonism on hepatic endpoints in patients with NAFLD and also a reduction in CV endpoints.
4.3. Surgery
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Targher, G.; Day, C.P.; Bonora, E. Risk of Cardiovascular Disease in Patients with Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Association of non-alcoholic fatty liver disease with major adverse cardiovascular events: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 33386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söderberg, C.; Stål, P.; Askling, J.; Glaumann, H.; Lindberg, G.; Marmur, J.; Hultcrantz, R. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology 2010, 51, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanova, M.; Younossi, Z.M. Independent Association between Nonalcoholic Fatty Liver Disease and Cardiovascular Disease in the US Population. Clin. Gastroenterol. Hepatol. 2012, 10, 646–650. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Health Estimates 2016: Disease Burden by Cause, Age, Sex, by Country and by Region, 2000–2016; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease. Circulation 2019, 74, 1376–1414. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics—2019 update: A report from the American heart association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [Green Version]
- Bellentani, S.; Marino, M. Epidemiology and natural history of non-alcoholic fatty liver disease (NAFLD). Ann. Hepatol. 2009, 8, S4–S8. [Google Scholar] [CrossRef]
- Scorletti, E.; Calder, P.C.; Byrne, C.D. Non-alcoholic fatty liver disease and cardiovascular risk: Metabolic aspects and novel treatments. Endocrinology 2011, 40, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Lentz, J.S. Fatty liver hepatitis (steatohepatitis) and obesity: An autopsy study with analysis of risk factors. Hepatology 1990, 12, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Wojcik-Cichy, K.; Koslinska-Berkan, E.; Piekarska, A. The influence of NAFLD on the risk of atherosclerosis and cardiovascular diseases. Clin. Exp. Hepatol. 2018, 4, 1–6. [Google Scholar] [CrossRef]
- Morrison, A.E.; Zaccardi, F.; Khunti, K.; Davies, M.J. Causality between non-alcoholic fatty liver disease and risk of cardiovascular disease and type 2 diabetes: A meta-analysis with bias analysis. Liver Int. 2019, 39, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Moscatiello, S.; Di Domizio, S.; Forlani, G. Obesity-associated liver disease. J. Clin. Endocrinol. Metab. 2008, 93, S74–S80. [Google Scholar] [CrossRef]
- Kotronen, A.; Yki-Jarvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 27–38. [Google Scholar] [CrossRef]
- De Alwis, N.M.; Day, C.P. Non-alcoholic fatty liver disease: The mist gradually clears. J. Hepatol. 2008, 48, S104–S112. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.; Wong, G.L.; Yeung, J.C.; Fung, C.Y.; Chan, J.K.; Chang, Z.H.; Kwan, C.T.; Lam, H.W.; Limquiaco, J.; Chim, A.M.; et al. Long-term clinical outcomes after fatty liver screening in patients undergoing coronary angiogram: A prospective cohort study. Hepatology 2016, 63, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Treeprasertsuk, S.; Leverage, S.; Adams, L.A.; Lindor, K.D.; St Sauver, J.; Angulo, P. The Framingham risk score and heart disease in nonalcoholic fatty liver disease. Liver Int. 2012, 32, 945–950. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.C.; Kim, D.J.; Huh, K.B. Association between nonalcoholic fatty liver disease and carotid intima-media thickness according to the presence of metabolic syndrome. Atherosclerosis 2009, 204, 521–525. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: A systematic review. J. Hepatol. 2008, 49, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Salvi, P.; Ruffini, R.; Agnoletti, D.; Magnani, E.; Pagliarani, G.; Comandini, G.; Praticò, A.; Borghi, C.; Benetos, A.; Pazzi, P. Increased arterial stiffness in nonalcoholic fatty liver disease: The Cardio-GOOSE study. J. Hypertens. 2010, 28, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, A.; Motoyama, M.; Matsuda, T.; Miyanishi, T. Brachial-ankle Pulse Wave Velocity in Japanese University Students. Intern. Med. 2005, 44, 696–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motamed, N.; Rabiee, B.; Poustchi, H.; Dehestani, B.; Hemasi, G.R.; Khonsari, M.R.; Maadi, M.; Saeedian, F.S.; Zamani, F. Non-alcoholic fatty liver disease (NAFLD) and 10-year risk of cardiovascular diseases. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ampuero, J.; Gallego-Duran, R.; Romero-Gomez, M. Association of NAFLD with subclinical atherosclerosis and coronary-artery disease: Meta-analysis. Rev. Esp. Enferm. Dig. 2015, 107, 10–16. [Google Scholar] [PubMed]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, N.; Preiss, D.; Sattar, N. Liver enzymes, nonalcoholic fatty liver disease, and incident cardiovascular disease: A narrative review and clinical perspective of prospective data. Hepatology 2010, 52, 1156–1161. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Coupland, C.; Robson, J.; Brindle, P. Derivation, validation, and evaluation of a new QRISK model to estimate lifetime risk of cardiovascular disease: Cohort study using QResearch database. BMJ 2010, 341, c6624. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Non-alcoholic fatty liver disease and risk of incident acute myocardial infarction and stroke: Findings from matched cohort study of 18 million European adults. BMJ 2019, 367, l5367. [Google Scholar] [CrossRef] [Green Version]
- Sattar, N.; Forrest, E.; Preiss, D. Non-alcoholic fatty liver disease. BMJ 2014, 349, g4596. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Forlani, G.; Cerrelli, F.; Manini, R.; Natale, S.; Baraldi, L.; Ermini, G.; Savorani, G.; Zocchi, D.; Melchionda, N. WHO and ATPIII proposals for the definition of the metabolic syndrome in patients with Type 2 diabetes. Diabetes Med. 2004, 21, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Jung, H.S.; Cho, J.; Zhang, Y.; Yun, K.E.; Lazo, M.; Pastor-Barriuso, R.; Ahn, J.; Kim, C.W.; Rampal, S.; et al. Metabolically Healthy Obesity and the Development of Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2016, 111, 1133–1140. [Google Scholar] [CrossRef]
- Chen, F.; Esmaili, S.; Rogers, G.B.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A Distinct Entity Shaped by Differential Metabolic Adaptation. Hepatology 2020, 71, 1213–1227. [Google Scholar] [CrossRef]
- Iacobini, C.; Pugliese, G.; Blasetti Fantauzzi, C.; Federici, M.; Menini, S. Metabolically healthy versus metabolically unhealthy obesity. Metabolism 2019, 92, 51–60. [Google Scholar] [CrossRef]
- Younes, R.; Bugianesi, E. NASH in Lean Individuals. Semin. Liver Dis. 2019, 39, 086–095. [Google Scholar] [CrossRef] [Green Version]
- Gentile, C.L.; Weir, T.L.; Cox-York, K.A.; Wei, Y.; Wang, D.; Reese, L.; Moran, G.; Estrada, A.; Mulligan, C.; Pagliassotti, M.J.; et al. The role of visceral and subcutaneous adipose tissue fatty acid composition in liver pathophysiology associated with NAFLD. Adipocyte 2015, 4, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Graffy, P.M.; Pickhardt, P.J. Quantification of hepatic and visceral fat by CT and MR imaging: Relevance to the obesity epidemic, metabolic syndrome and NAFLD. Br. J. Radiol. 2016, 89, 20151024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, S.; Tariq, R.; Provenza, J.; Satapathy, S.K.; Faisal, K.; Choudhry, A.; Friedman, S.L.; Singal, A.K. Prevalence and Profile of Nonalcoholic Fatty Liver Disease in Lean Adults: Systematic Review and Meta-Analysis. Hepatol. Commun. 2020, 4, 953–972. [Google Scholar] [CrossRef] [PubMed]
- Golabi, P.; Paik, J.; Fukui, N.; Locklear, C.T.; de Avilla, L.; Younossi, Z.M. Patients with Lean Nonalcoholic Fatty Liver Disease Are Metabolically Abnormal and Have a Higher Risk for Mortality. Clin. Diabetes 2018, 37, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stols-Gonçalves, D.; Hovingh, G.K.; Nieuwdorp, M.; Holleboom, A.G. NAFLD and Atherosclerosis: Two Sides of the Same Dysmetabolic Coin? Trends Endocrinol. Metab. 2019, 30, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.P.; Rawal, K.; Bagchi, A.K.; Akolkar, G.; Bernardes, N.; Dias, D.D.S.; Gupta, S.; Singal, P.K. Insulin resistance: An additional risk factor in the pathogenesis of cardiovascular disease in type 2 diabetes. Heart Fail. Rev. 2016, 21, 11–23. [Google Scholar] [CrossRef]
- Balakumar, P.; Maung-U, K.; Jagadeesh, G. Prevalence and prevention of cardiovascular disease and diabetes mellitus. Pharmacol. Res. 2016, 113, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.-W.; Lin, C.-L.; Liao, K.-F. Association between diabetes mellitus and hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2019, 31, 898–899. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Lauridsen, B.K.; Stender, S.; Kristensen, T.S.; Kofoed, K.F.; Køber, L.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta-analysis of 279,013 individuals. Eur. Heart J. 2018, 39, 385–393. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Mantovani, A.; Tilg, H.; Targher, G. Risk of cardiomyopathy and cardiac arrhythmias in patients with nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 425–439. [Google Scholar] [CrossRef]
- Mantovani, A.; Dauriz, M.; Sandri, D.; Bonapace, S.; Zoppini, G.; Tilg, H.; Byrne, C.D.; Targher, G. Association between non-alcoholic fatty liver disease and risk of atrial fibrillation in adult individuals: An updated meta-analysis. Liver Int. 2019, 39, 758–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M. Atrial Fibrillation and Heart Failure with Preserved Ejection Fraction in Patients with Nonalcoholic Fatty Liver Disease. Am. J. Med. 2020, 133, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Cohney, S.; De Michieli, F.; Pinach, S.; Saba, F.; Gambino, R. Fatty Liver and Chronic Kidney Disease: Novel Mechanistic Insights and Therapeutic Opportunities. Diabetes Care 2016, 39, 1830–1845. [Google Scholar] [CrossRef] [Green Version]
- Targher, G.; Chonchol, M.; Byrne, C.D. CKD and Nonalcoholic Fatty Liver Disease. Am. J. Kidney Dis. 2014, 64, 638–652. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef]
- Thorand, B.; Löwel, H.; Schneider, A.; Kolb, H.; Meisinger, C.; Fröhlich, M.; Koenig, W. C-reactive protein as a predictor for incident diabetes mellitus among middle-aged men: Results from the MONICA Augsburg cohort study, 1984–1998. Arch. Intern. Med. 2003, 163, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Kubes, P.; Mehal, W.Z. Sterile Inflammation in the Liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer Cells Trigger Nonalcoholic Steatohepatitis Development in Diet-induced Mouse Model through Tumor Necrosis Factor-α Production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Macrophages in Nonalcoholic Fatty Liver Disease: A Role Model of Pathogenic Immunometabolism. Semin. Liver Dis. 2017, 37, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, K.; McKenzie, A.L.; Kränkel, N.; Von Schacky, C.; Worm, N.; Nixdorff, U.; Lechner, B.; Scherr, J.; Weingärtner, O.; Krauss, R.M. High-Risk Atherosclerosis and Metabolic Phenotype: The Roles of Ectopic Adiposity, Atherogenic Dyslipidemia, and Inflammation. Metab. Syndr. Relat. Disord. 2020, 18, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [Green Version]
- Hirase, T.; Node, K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am. J. Physiol. Circ. Physiol. 2012, 302, H499–H505. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Guo, S.; Zhang, S.; Liu, A.; Shi, L.; Zhang, Y. Matrine attenuates endoplasmic reticulum stress and mitochondrion dysfunction in nonalcoholic fatty liver disease by regulating SERCA pathway. J. Transl. Med. 2018, 16, 319. [Google Scholar] [CrossRef]
- O’Rourke, R.W.; Metcalf, M.D.; White, A.E.; Madala, A.; Winters, B.R.; Maizlin, I.I.; Jobe, B.A.; Roberts, C.T., Jr.; Slifka, M.K.; Marks, D.L. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int. J. Obes. 2009, 33, 978–990. [Google Scholar]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Tourniaire, F.; Romier-Crouzet, B.; Lee, J.H.; Marcotorchino, J.; Gouranton, E.; Salles, J.; Malezet, C.; Astier, J.; Darmon, P.; Blouin, E.; et al. Chemokine Expression in Inflamed Adipose Tissue is Mainly Mediated by NF-κB. PLoS ONE 2013, 8, e66515. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar] [PubMed]
- Lagathu, C.; Yvan-Charvet, L.; Bastard, J.P.; Maachi, M.; Quignard-Boulangé, A.; Capeau, J.; Caron, M. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia 2006, 49, 2162–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, J.; Kiefer, F.W.; Zeyda, M.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; Zlabinger, G.J.; Stulnig, T.M. CC Chemokine and CC Chemokine Receptor Profiles in Visceral and Subcutaneous Adipose Tissue Are Altered in Human Obesity. J. Clin. Endocrinol. Metab. 2008, 93, 3215–3221. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Du Plessis, J.; van Pelt, J.; Korf, H.; Mathieu, C.; van der Schueren, B.; Lannoo, M.; Oyen, T.; Topal, B.; Fetter, G.; Nayler, S.; et al. Association of Adipose Tissue Inflammation with Histologic Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 635–648.e14. [Google Scholar] [CrossRef] [Green Version]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in NAFLD—Revisited After a Decade. Hepatology 2020. [Google Scholar] [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Fricker, Z.P.; Pedley, A.; Massaro, J.M.; Vasan, R.S.; Hoffmann, U.; Benjamin, E.J.; Long, M.T. Liver Fat is Associated with Markers of Inflammation and Oxidative Stress in Analysis of Data From the Framingham Heart Study. Clin. Gastroenterol. Hepatol. 2019, 17, 1157–1164.e4. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, M.; Simons, N.; Stehouwer, C.D.A.; Isaacs, A. Non-alcoholic fatty liver disease and cardiovascular disease: Assessing the evidence for causality. Diabetology 2020, 63, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismaiel, A.; Dumitraşcu, D.L. Cardiovascular Risk in Fatty Liver Disease: The Liver-Heart Axis—Literature Review. Front. Med. 2019, 6, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouvas, N.; Kontogiannis, C.; Georgiopoulos, G.; Spartalis, M.; Tsilimigras, D.I.; Spartalis, E.; Kapelouzou, A.; Kosmopoulos, M.; Chatzidou, S. The complex crosstalk between inflammatory cytokines and ventricular arrhythmias. Cytokine 2018, 111, 171–177. [Google Scholar] [CrossRef]
- Widjaja, A.A.; Singh, B.K.; Adami, E.; Viswanathan, S.; Dong, J.; D’Agostino, G.A.; Ng, B.; Lim, W.; Tan, J.; Paleja, B.S.; et al. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology 2019, 157, 777–792.e14. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Eslam, M.; Sarin, S.K.; Wong, V.W.-S.; Fan, J.-G.; Kawaguchi, T.; Ahn, S.H.; Zheng, M.-H.; Shiha, G.; Yilmaz, Y.; Gani, R.; et al. The Asian Pacific Association for the Study of the Liver clinical practice guidelines for the diagnosis and management of metabolic associated fatty liver disease. Hepatol. Int. 2020, 14, 1–31. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Shiha, G.; Alswat, K.; Al Khatry, M.; I Sharara, A.; Örmeci, N.; Waked, I.; Benazzouz, M.; Al-Ali, F.; Hamed, A.E.; Hamoudi, W.; et al. Nomenclature and definition of metabolic-associated fatty liver disease: A consensus from the Middle East and north Africa. Lancet Gastroenterol. Hepatol. 2021, 6, 57–64. [Google Scholar] [CrossRef]
- Shiha, G.; Korenjak, M.; Eskridge, W.; Casanovas, T.; Velez-Moller, P.; Högström, S.; Richardson, B.; Munoz, C.; Sigurðardóttir, S.; Coulibaly, A.; et al. Redefining fatty liver disease: An international patient perspective. Lancet Gastroenterol. Hepatol. 2021, 6, 73–79. [Google Scholar] [CrossRef]
- Mendez-Sanchez, N.; Arrese, M.; Gadano, A.; Oliveira, C.P.; Fassio, E.; Arab, J.P.; Chávez-Tapia, N.C.; Dirchwolf, M.; Torre, A.; Ridruejo, E.; et al. The Latin American Association for the Study of the Liver (ALEH) position statement on the redefinition of fatty liver disease. Lancet Gastroenterol. Hepatol. 2021, 6, 65–72. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.; Harrison, S.A.; Brunt, E.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a premature change in terminology. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, J.; Wang, M.; Kumar, R.; Liu, Y.; Liu, S.; Wu, Y.; Wang, X.; Zhu, Y. Comparison of MAFLD and NAFLD diagnostic criteria in real world. Liver Int. 2020, 40, 2082–2089. [Google Scholar] [CrossRef]
- Targher, G. Concordance of MAFLD and NAFLD diagnostic criteria in “real-world” data. Liver. Int. 2020, 40, 2879–2880. [Google Scholar] [CrossRef]
- Huang, J.; Kumar, R.; Wang, M.; Zhu, Y.; Lin, S. MAFLD criteria overlooks a number of patients with severe steatosis: Is it clinically relevant? J. Hepatol. 2020, 73, 1265–1267. [Google Scholar] [CrossRef]
- Zhang, L.; She, Z.-G.; Li, H.-L.; Zhang, X.-J. Non-alcoholic fatty liver disease: A metabolic burden promoting atherosclerosis. Clin. Sci. 2020, 134, 1775–1799. [Google Scholar] [CrossRef]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rodriguez, E.; Egea-Zorrilla, A.; Plaza-Díaz, J.; Jerónimo, A.-V.; Muñoz-Quezada, S.; Tercedor-Sánchez, L.; Abadía-Molina, F. The Gut Microbiota and Its Implication in the Development of Atherosclerosis and Related Cardiovascular Diseases. Nutrition 2020, 12, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.W.; Bäckhed, F.; Landmesser, U.; Hazen, S.L. Intestinal Microbiota in Cardiovascular Health and Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2089–2105. [Google Scholar] [CrossRef] [PubMed]
- Indias, I.E.; Queipo-Ortuño, M.I.; Tinahones, F.J.; Queipo-Ortuno, M.I. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front. Microbiol. 2014, 5, 190. [Google Scholar]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Henao-Mejia, J.; Elinav, E.; Jin, C.-C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nat. Cell Biol. 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.; Rivera, L.; Furness, L.R.J.B.; Angus, C.L.P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Clément, K. The gut microbiome, diet, and links to cardiometabolic and chronic disorders. Nat. Rev. Nephrol. 2015, 12, 169–181. [Google Scholar] [CrossRef]
- Hoyles, L.; Fernández-Real, J.M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Sharpton, S.R.; Ajmera, V.; Loomba, R. Emerging Role of the Gut Microbiome in Nonalcoholic Fatty Liver Disease: From Composition to Function. Clin. Gastroenterol. Hepatol. 2019, 17, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.G.; Kim, S.M.; Caussy, C.; Fu, T.; Guo, J.; Bassirian, S.; Singh, S.; Madamba, E.V.; Bettencourt, R.; Richards, L.; et al. A Universal Gut-Microbiome-Derived Signature Predicts Cirrhosis. Cell. Metab. 2020, 32, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Lang, S.; Demir, M.; Martin, A.; Jiang, L.; Zhang, X.; Duan, Y.; Gao, B.; Wisplinghoff, H.; Kasper, P.; Roderburg, C.; et al. Intestinal Virome Signature Associated with Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 159, 1839–1852. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal Barrier Dysfunction, LPS Translocation, and Disease Development. J. Endocr. Soc. 2020, 4, bvz039. [Google Scholar] [CrossRef] [Green Version]
- Verdugo, J.P.A.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 321–350. [Google Scholar]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Arslan, N. Obesity, fatty liver disease and intestinal microbiota. World J. Gastroenterol. 2014, 20, 16452–16463. [Google Scholar] [CrossRef]
- Turnbaugh, P.J. Microbiology: Fat, bile and gut microbes. Nature 2012, 487, 47–48. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Zhou, X.; Wang, M.; Zhou, L.; Wang, Z.; Wang, M.; Deng, J.; Wang, Y.; Zhou, Z.; Zhang, Y.; et al. Gut microbe-derived metabolite trimethylamine N-oxide activates the cardiac autonomic nervous system and facilitates ischemia-induced ventricular arrhythmia via two different pathways. EBioMedicine 2019, 44, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-M.; Liu, Y.; Zhou, R.-F.; Chen, X.-L.; Wang, C.; Tan, X.-Y.; Wang, L.-J.; Zheng, R.-D.; Zhang, H.-W.; Ling, W.-H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef]
- Tan, X.; Liu, Y.; Long, J.; Chen, S.; Liao, G.; Wu, S.; Li, C.; Wang, L.; Ling, W.; Zhu, H. Trimethylamine N-Oxide Aggravates Liver Steatosis through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900257. [Google Scholar] [CrossRef]
- Arias, N.; Arboleya, S.; Allison, J.; Kaliszewska, A.; Higarza, S.G.; Gueimonde, M.; Arias, J.L. The Relationship between Choline Bioavailability from Diet, Intestinal Microbiota Composition, and Its Modulation of Human Diseases. Nutrition 2020, 12, 2340. [Google Scholar]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.; Van Der Velden, S.; Ríos-Morales, M.; Van Faassen, M.J.; Loreti, M.G.; De Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Vieira-Silva, S.; Falony, G.; Belda, E.; Nielsen, T.; Aron-Wisnewsky, J.; Chakaroun, R.; Forslund, S.K.; Assmann, K.; Valles-Colomer, M.; Nguyen, T.T.D.; et al. Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nat. Cell Biol. 2020, 581, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Wang, X.; Ren, X.; Liu, Y. Changes of intestinal bacterial microbiota in coronary heart disease complicated with nonalcoholic fatty liver disease. BMC Genom. 2019, 20, 862. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halcox, J.P.J.; Banegas, J.R.; Roy, C.; Dallongeville, J.; De Backer, G.G.; Guallar, E.; Perk, J.; Hajage, D.; Henriksson, K.M.; Borghi, C. Prevalence and treatment of atherogenic dyslipidemia in the primary prevention of cardiovascular disease in Europe: EURIKA, a cross-sectional observational study. BMC Cardiovasc. Disord. 2017, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.G. Hepatic glucose and lipid metabolism. Diabetologia 2016, 59, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725.e6. [Google Scholar] [CrossRef]
- Patterson, R.E.; Kalavalapalli, S.; Williams, C.M.; Nautiyal, M.; Mathew, J.T.; Martinez, J.; Reinhard, M.K.; McDougall, D.J.; Rocca, J.R.; Yost, R.A.; et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E484–E494. [Google Scholar] [CrossRef] [Green Version]
- Chatrath, H.; Vuppalanchi, R.; Chalasani, N. Dyslipidemia in Patients with Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2012, 32, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.W.; Yu, J.H.; Jin, Y.J.; Suh, Y.J.; Lee, J.W. Correlation between the small dense LDL level and nonalcoholic fatty liver disease: Possibility of a new biomarker. Medicine 2020, 99, e21162. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Fuchs, M.; Idowu, M.O.; Luketic, V.A.; Boyett, S.; Sargeant, C.; Stravitz, R.T.; Puri, P.; Matherly, S.; Sterling, R.K.; et al. Severity of Nonalcoholic Fatty Liver Disease and Progression to Cirrhosis Are Associated with Atherogenic Lipoprotein Profile. Clin. Gastroenterol. Hepatol. 2015, 13, 1000–1008.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imajo, K.; Hyogo, H.; Yoneda, M.; Honda, Y.; Kessoku, T.; Tomeno, W.; Ogawa, Y.; Taguri, M.; Mawatari, H.; Nozaki, Y.; et al. LDL-Migration Index (LDL-MI), an Indicator of Small Dense Low-Density Lipoprotein (sdLDL), Is Higher in Non-Alcoholic Steatohepatitis than in Non-Alcoholic Fatty Liver: A Multicenter Cross-Sectional Study. PLoS ONE 2014, 9, e115403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, M.; Berneis, K.; Corrado, E.; Novo, S. The significance of low-density-lipoproteins size in vascular diseases. Int. Angiol. 2006, 25, 4–9. [Google Scholar] [PubMed]
- Saeed, A.; Feofanova, E.V.; Yu, B.; Sun, W.; Virani, S.S.; Nambi, V.; Coresh, J.; Guild, C.S.; Boerwinkle, E.; Ballantyne, C.M.; et al. Remnant-Like Particle Cholesterol, Low-Density Lipoprotein Triglycerides, and Incident Cardiovascular Disease. J. Am. Coll. Cardiol. 2018, 72, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Dissociation between APOC3 variants, hepatic triglyceride content and insulin resistance. Hepatology 2010, 53, 467–474. [Google Scholar] [CrossRef]
- Dittrich, J.; Beutner, F.; Teren, A.; Thiery, J.; Burkhardt, R.; Scholz, M.; Ceglarek, U. Plasma levels of apolipoproteins C-III, A-IV, and E are independently associated with stable atherosclerotic cardiovascular disease. Atherosclerosis 2019, 281, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Zewinger, S.; Reiser, J.; Jankowski, V.; Al Ansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2019, 21, 30–41. [Google Scholar] [CrossRef]
- Afonso, M.S.; Lottenberg, A.M.; Koike, M.K.; Cintra, D.E.; Ferreira, F.D.; Nunes, V.S.; Castilho, G.; Gioielli, L.A.; Bombo, R.P.; Catanozi, S.; et al. Dietary interesterified fat enriched with palmitic acid induces atherosclerosis by impairing macrophage cholesterol efflux and eliciting inflammation. J. Nutr. Biochem. 2016, 32, 91–100. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, R.; Xiong, Y.; Du, F.; Zhu, S. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, S.; Panera, N.; Mina, M.; Gnani, D.; De Stefanis, C.; Crudele, A.; Rychlicki, C.; Petrini, S.; Bruscalupi, G.; Agostinelli, L.; et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 2015, 6, 41434–41452. [Google Scholar] [CrossRef] [Green Version]
- Verrijken, A.; Francque, S.; Mertens, I.; Prawitt, J.; Caron, S.; Hubens, G.; Van Marck, E.; Staels, B.; Michielsen, P.; Van Gaal, L. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2014, 59, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.N.W.; Treskes, P.; Martin, S.F.; Manning, J.R.; Dunbar, D.R.; Rogers, S.M.; Le Bihan, T.; Lockman, K.A.; Morley, S.D.; Hayes, P.C.; et al. Fibrinogen production is enhanced in an in-vitro model of non-alcoholic fatty liver disease: An isolated risk factor for cardiovascular events? Lipids Heal. Dis. 2015, 14, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.-J.; Wang, P.W.; Hu, T.H. Association of Adiponectin Gene Polymorphism with Nonalcoholic Fatty Liver Disease in Taiwanese Patients with Type 2 Diabetes. PLoS ONE 2015, 10, e0127521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-L.; Sui, J.-Q.; Lu, L.-L.; Zhang, N.-N.; Xu, X.; Dong, Q.-Y.; Xin, Y.; Xuan, S. Gene polymorphisms associated with non-alcoholic fatty liver disease and coronary artery disease: A concise review. Lipids Health Dis. 2016, 15, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, E.P.; Dhindsa, D.S.; Lee, S.K.; Sandesara, P.B.; Chalasani, N.P.; Sperling, L.S. Nonalcoholic Fatty Liver Disease and the Heart: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 948–963. [Google Scholar] [CrossRef]
- Francque, S.M.; van der Graaff, D.; Kwanten, W.J. Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications. J. Hepatol. 2016, 65, 425–443. [Google Scholar] [CrossRef] [Green Version]
- Villanova, N.; Moscatiello, S.; Ramilli, S.; Bugianesi, E.; Magalotti, D.; Vanni, E.; Zoli, M.; Marchesini, G. Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 2005, 42, 473–480. [Google Scholar] [CrossRef]
- Hawksworth, D.J.; Burnett, A.L. Nonalcoholic Fatty Liver Disease, Male Sexual Dysfunction, and Infertility: Common Links, Common Problems. Sex. Med. Rev. 2020, 8, 274–285. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Jung, H.S.; Chang, Y.; Kwon, M.J.; Sung, E.; Yun, K.E.; Cho, Y.K.; Shin, H.; Ryu, S. Smoking and the Risk of Non-Alcoholic Fatty Liver Disease: A Cohort Study. Am. J. Gastroenterol. 2019, 114, 453–463. [Google Scholar] [CrossRef]
- Boden, G. High- or Low-Carbohydrate Diets: Which is better for Weight Loss, Insulin Resistance, and Fatty Livers? Gastroenterology 2009, 136, 1490–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perreault, L.; Pan, Q.; Mather, K.J.; Watson, K.E.; Hamman, R.F.; Kahn, S.E. Effect of regression from prediabetes to normal glucose regulation on long-term reduction in diabetes risk: Results from the Diabetes Prevention Program Outcomes Study. Lancet 2012, 379, 2243–2251. [Google Scholar] [CrossRef] [Green Version]
- Lazo, M.; Solga, S.F.; Horska, A.; Bonekamp, S.; Diehl, A.M.; Brancati, F.L.; Wagenknecht, L.E.; Pi-Sunyer, F.X.; Kahn, S.E.; Clark, J.M.; et al. Effect of a 12-Month Intensive Lifestyle Intervention on Hepatic Steatosis in Adults with Type 2 Diabetes. Diabetes Care 2010, 33, 2156–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, F.; George, J. The Role of Diet and Nutritional Intervention for the Management of Patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef]
- Dunn, W.; Sanyal, A.J.; Brunt, E.M.; Unalp-Arida, A.; Donohue, M.; McCullough, A.J.; Schwimmer, J.B. Modest alcohol consumption is associated with decreased prevalence of steatohepatitis in patients with non-alcoholic fatty liver disease (NAFLD). J. Hepatol. 2012, 57, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.K.; Greenson, J.K.; Conjeevaram, H.S. Effect of lifetime alcohol consumption on the histological severity of non-alcoholic fatty liver disease. Liver Int. 2013, 34, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Liangpunsakul, S.; Chalasani, N. What Should We Recommend to Our Patients with NAFLD Regarding Alcohol Use? Am. J. Gastroenterol. 2012, 107, 976–978. [Google Scholar] [CrossRef] [Green Version]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.-R.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Saab, S.; Mallam, D.; Cox, G.A.; Tong, M.J. Impact of coffee on liver diseases: A systematic review. Liver Int. 2014, 34, 495–504. [Google Scholar] [CrossRef]
- Pelliccia, A.; Sharma, S.; Gati, S.; Bäck, M.; Börjesson, M.; Caselli, S.; Collet, J.-P.; Corrado, D.; Drezner, J.A.; Halle, M. 2020 ESC Guidelines on sports cardiology and exercise in patients with cardiovascular disease: The Task Force on sports cardiology and exercise in patients with cardiovascular disease of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 42, 17–96. [Google Scholar]
- Keating, S.E.; Hackett, D.A.; George, J.; Johnson, N.A. Exercise and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 57, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, E.; Negri, C.; Targher, G.; Faccioli, N.; Lanza, M.; Zoppini, G.; Zanolin, E.; Schena, F.; Bonora, E.; Moghetti, P. Both resistance training and aerobic training reduce hepatic fat content in type 2 diabetic subjects with nonalcoholic fatty liver disease (the RAED2 Randomized Trial). Hepatology 2013, 58, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Roman, J.H.; Patel, S.S. Why Do Lifestyle Recommendations Fail in Most Patients with Nonalcoholic Fatty Liver Disease? Gastroenterol. Clin. N. Am. 2020, 49, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Ismaiel, A.; Dumitrascu, D.L. How to Reduce Cardiovascular Risk in Nonalcoholic Fatty Liver Disease. Am. J. Ther. 2020. [Google Scholar] [CrossRef]
- Jiang, Z.G.; Feldbrügge, L.; Tapper, E.B.; Popov, Y.; Ghaziani, T.; Afdhal, N.; Robson, S.C.; Mukamal, K.J. Aspirin use is associated with lower indices of liver fibrosis among adults in the United States. Aliment. Pharmacol. Ther. 2016, 43, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Henson, J.; Osganian, S.; Masia, R.; Chan, A.T.; Chung, R.T.; Corey, K.E. Daily Aspirin Use Associated with Reduced Risk For Fibrosis Progression in Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 2776–2784.e4. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Endo, H.; Takahashi, H.; Iwasaki, T.; Inamori, M.; Abe, Y.; Kobayashi, N.; et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut 2008, 57, 1583–1591. [Google Scholar] [CrossRef]
- Russo, M.W.; Pierson, J.; Narang, T.; Montegudo, A.; Eskind, L.; Gulati, S. Coronary Artery Stents and Antiplatelet Therapy in Patients with Cirrhosis. J. Clin. Gastroenterol. 2012, 46, 339–344. [Google Scholar] [CrossRef]
- Li, C.-J.; Yang, Z.-H.; Shi, X.-L.; Liu, D.-L. Effects of aspirin and enoxaparin in a rat model of liver fibrosis. World J. Gastroenterol. 2017, 23, 6412–6419. [Google Scholar] [CrossRef]
- Leonardi, F.; Maria, N.; Villa, E. Anticoagulation in cirrhosis: A new paradigm? Clin. Mol. Hepatol. 2017, 23, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Cholesterol Treatment Trialists. Efficacy and safety of LDL-lowering therapy among men and women: Meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015, 385, 1397–1405. [Google Scholar] [CrossRef]
- Björnsson, E.S.; Jacobsen, E.I.; Kalaitzakis, E. Hepatotoxicity associated with statins: Reports of idiosyncratic liver injury post-marketing. J. Hepatol. 2012, 56, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Labenz, C.; Huber, Y.; Kalliga, E.; Nagel, M.; Ruckes, C.; Straub, B.K.; Galle, P.R.; Wörns, M.-A.; Anstee, Q.M.; Schuppan, D.; et al. Predictors of advanced fibrosis in non-cirrhotic non-alcoholic fatty liver disease in Germany. Aliment. Pharmacol. Ther. 2018, 48, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Labenz, C.; Prochaska, J.H.; Huber, Y.; Nagel, M.; Straub, B.K.; Wild, P.; Galle, P.R.; Schattenberg, J.M. Cardiovascular Risk Categories in Patients with Nonalcoholic Fatty Liver Disease and the Role of Low-Density Lipoprotein Cholesterol. Hepatol. Commun. 2019, 3, 1472–1481. [Google Scholar] [CrossRef] [Green Version]
- Athyros, V.G.; Tziomalos, K.; Gossios, T.D.; Griva, T.; Anagnostis, P.; Kargiotis, K.; Pagourelias, E.D.; Theocharidou, E.; Karagiannis, A.; Mikhailidis, D.P. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: A post-hoc analysis. Lancet 2010, 376, 1916–1922. [Google Scholar] [CrossRef]
- Athyros, V.G.; Kakafika, A.I.; Papageorgiou, A.A.; Tziomalos, K.; Skaperdas, A.; Pagourelias, E.; Pirpasopoulou, A.; Karagiannis, A.; Mikhailidis, D.P. Atorvastatin decreases triacylglycerol-associated risk of vascular events in coronary heart disease patients. Lipids 2007, 42, 999–1009. [Google Scholar] [CrossRef]
- Tikkanen, M.J.; Fayyad, R.; Faergeman, O.; Olsson, A.G.; Wun, C.-C.; Laskey, R.; Kastelein, J.J.; Holme, I.; Pedersen, T.R. Effect of intensive lipid lowering with atorvastatin on cardiovascular outcomes in coronary heart disease patients with mild-to-moderate baseline elevations in alanine aminotransferase levels. Int. J. Cardiol. 2013, 168, 3846–3852. [Google Scholar] [CrossRef]
- Kargiotis, K.; Athyros, V.G.; Giouleme, O.; Katsiki, N.; Katsiki, E.; Anagnostis, P.; Boutari, C.; Doumas, M.; Karagiannis, A.; Mikhailidis, D.P. Resolution of non-alcoholic steatohepatitis by rosuvastatin monotherapy in patients with metabolic syndrome. World J. Gastroenterol. 2015, 21, 7860–7868. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Mannisto, V.; Mancina, R.M.; Pipitone, R.M.; Karja, V.; Maggioni, M.; Kakela, P.; Wiklund, O.; Mozzi, E.; et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef]
- Averna, M. The effect of ezetimibe on NAFLD. Atheroscler. Suppl. 2015, 17, 27–34. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H.; Hobbs, H.H. Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, D.M.; Morris, P.B.; Ballantyne, C.M.; Birtcher, K.K.; Daly, D.D., Jr.; DePalma, S.M.; Minissian, M.B.; Orringer, C.E.; Smith, S.C., Jr. 2016 ACC Expert Consensus Decision Pathway on the Role of Non-Statin Therapies for LDL-Cholesterol Lowering in the Management of Atherosclerotic Cardiovascular Disease Risk: A Report of the American College of Cardiology Task Force on Clinical Expert Consensus Documents. J. Am. Coll. Cardiol. 2016, 68, 92–125. [Google Scholar] [PubMed]
- Theocharidou, E.; Papademetriou, M.; Reklou, A.; Sachinidis, A.; Boutari, C.; Giouleme, O. The Role of PCSK9 in the Pathogenesis of Non-alcoholic Fatty Liver Disease and the Effect of PCSK9 Inhibitors. Curr. Pharm. Des. 2019, 24, 3654–3657. [Google Scholar] [CrossRef]
- Cameron, J.; Ranheim, T.; Kulseth, M.A.; Leren, T.P.; Berge, K.E. Berberine decreases PCSK9 expression in HepG2 cells. Atheroscler. 2008, 201, 266–273. [Google Scholar] [CrossRef]
- Yan, H.-M.; Xia, M.-F.; Wang, Y.; Chang, X.-X.; Yao, X.-Z.; Rao, S.-X.; Zeng, M.-S.; Tu, Y.-F.; Feng, R.; Jia, W.-P.; et al. Efficacy of Berberine in Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0134172. [Google Scholar] [CrossRef] [Green Version]
- Aneni, E.C.; Oni, E.T.; Martin, S.S.; Blaha, M.J.; Agatston, A.S.; Feldman, T.; Veledar, E.; Conçeicao, R.D.; Carvalho, J.A.; Santos, R.D.; et al. Blood pressure is associated with the presence and severity of nonalcoholic fatty liver disease across the spectrum of cardiometabolic risk. J. Hypertens. 2015, 33, 1207–1214. [Google Scholar] [CrossRef]
- Ryoo, J.-H.; Suh, Y.J.; Shin, H.C.; Cho, Y.K.; Choi, J.-M.; Park, S.K. Clinical association between non-alcoholic fatty liver disease and the development of hypertension. J. Gastroenterol. Hepatol. 2014, 29, 1926–1931. [Google Scholar] [CrossRef]
- Vasunta, R.-L.; Kesäniemi, Y.A.; Ylitalo, A.S.; Ukkola, O. High ambulatory blood pressure values associated with non-alcoholic fatty liver in middle-aged adults. J. Hypertens. 2012, 30, 2015–2019. [Google Scholar] [CrossRef]
- Muthiah, M.D.; Sanyal, A.J. Current management of non-alcoholic steatohepatitis. Liver Int. 2020, 40, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisseleva, T.; A Brenner, D. Anti-fibrogenic strategies and the regression of fibrosis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 305–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, H.; Wu, W.; Ye, J.; Fang, D.; Shi, D.; Li, L. Clinical application of angiotensin receptor blockers in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. Oncotarget 2018, 9, 24155–24167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Jarvis, H.; Craig, D.; O Barker, R.; Spiers, G.; Stow, D.; Anstee, Q.M.; Hanratty, B. Metabolic risk factors and incident advanced liver disease in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of population-based observational studies. PLoS Med. 2020, 17, e1003100. [Google Scholar] [CrossRef] [PubMed]
- Schulte, L.; Scheiner, B.; Voigtländer, T.; Koch, S.; Schweitzer, N.; Marhenke, S.; Ivanyi, P.; Manns, M.P.; Rodt, T.; Hinrichs, J.B.; et al. Treatment with metformin is associated with a prolonged survival in patients with hepatocellular carcinoma. Liver Int. 2019, 39, 714–726. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef]
- Bril, F.; Kalavalapalli, S.; Clark, V.C.; Lomonaco, R.; Soldevila-Pico, C.; Liu, I.-C.; Orsak, B.; Tio, F.; Cusi, K. Response to Pioglitazone in Patients with Nonalcoholic Steatohepatitis With vs. Without Type 2 Diabetes. Clin. Gastroenterol. Hepatol. 2018, 16, 558–566.e2. [Google Scholar] [CrossRef]
- Ripamonti, E.; Azoulay, L.; Abrahamowicz, M.; Platt, R.W.; Suissa, S. A systematic review of observational studies of the association between pioglitazone use and bladder cancer. Diabet. Med. 2019, 36, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, J.A.; Guirguis, E.; Thornby, K.-A. A Systematic Review of Newer Antidiabetic Agents in the Treatment of Nonalcoholic Fatty Liver Disease. Ann. Pharmacother. 2021, 55, 65–79. [Google Scholar] [CrossRef]
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42, S90–S102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernsmeier, C.; Meyer-Gerspach, A.C.; Blaser, L.S.; Jeker, L.; Steinert, R.E.; Heim, M.H.; Beglinger, C. Glucose-Induced Glucagon-Like Peptide 1 Secretion Is Deficient in Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2014, 9, e87488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Newsome, P.N.; Francque, S.; Harrison, S.; Ratziu, V.; Van Gaal, L.; Calanna, S.; Hansen, M.; Linder, M.; Sanyal, A.J. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment. Pharmacol. Ther. 2019, 50, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluud, L.L.; Knop, F.K.; Vilsbøll, T. Effects of lixisenatide on elevated liver transaminases: Systematic review with individual patient data meta-analysis of randomised controlled trials on patients with type 2 diabetes. BMJ Open 2014, 4, e005325. [Google Scholar] [CrossRef]
- Cusi, K.; Sattar, N.; García-Pérez, L.-E.; Pavo, I.; Yu, M.; Robertson, K.E.; Karanikas, C.A.; Haupt, A. Dulaglutide decreases plasma aminotransferases in people with Type 2 diabetes in a pattern consistent with liver fat reduction: A post hoc analysis of the AWARD programme. Diabet. Med. 2018, 35, 1434–1439. [Google Scholar] [CrossRef]
- Tacelli, M.; Celsa, C.; Magro, B.; Giannetti, A.; Pennisi, G.; Spatola, F.; Petta, S. Antidiabetic Drugs in NAFLD: The Accomplishment of Two Goals at Once? Pharmacy 2018, 11, 121. [Google Scholar] [CrossRef] [Green Version]
- Laursen, T.L.; Hagemann, C.A.; Wei, C.; Kazankov, K.; Thomsen, K.L.; Knop, F.K.; Grønbæk, H. Bariatric surgery in patients with non-alcoholic fatty liver disease—From pathophysiology to clinical effects. World J. Hepatol. 2019, 11, 138–149. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niederseer, D.; Wernly, B.; Aigner, E.; Stickel, F.; Datz, C. NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations. J. Clin. Med. 2021, 10, 467. https://doi.org/10.3390/jcm10030467
Niederseer D, Wernly B, Aigner E, Stickel F, Datz C. NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations. Journal of Clinical Medicine. 2021; 10(3):467. https://doi.org/10.3390/jcm10030467
Chicago/Turabian StyleNiederseer, David, Bernhard Wernly, Elmar Aigner, Felix Stickel, and Christian Datz. 2021. "NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations" Journal of Clinical Medicine 10, no. 3: 467. https://doi.org/10.3390/jcm10030467
APA StyleNiederseer, D., Wernly, B., Aigner, E., Stickel, F., & Datz, C. (2021). NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations. Journal of Clinical Medicine, 10(3), 467. https://doi.org/10.3390/jcm10030467