
Angiotensin-(1-7)—A Potential Remedy for AKI: Insights Derived from the COVID-19 Pandemic


{kind=link}
{kind=link}
Abstract
:1. Introduction
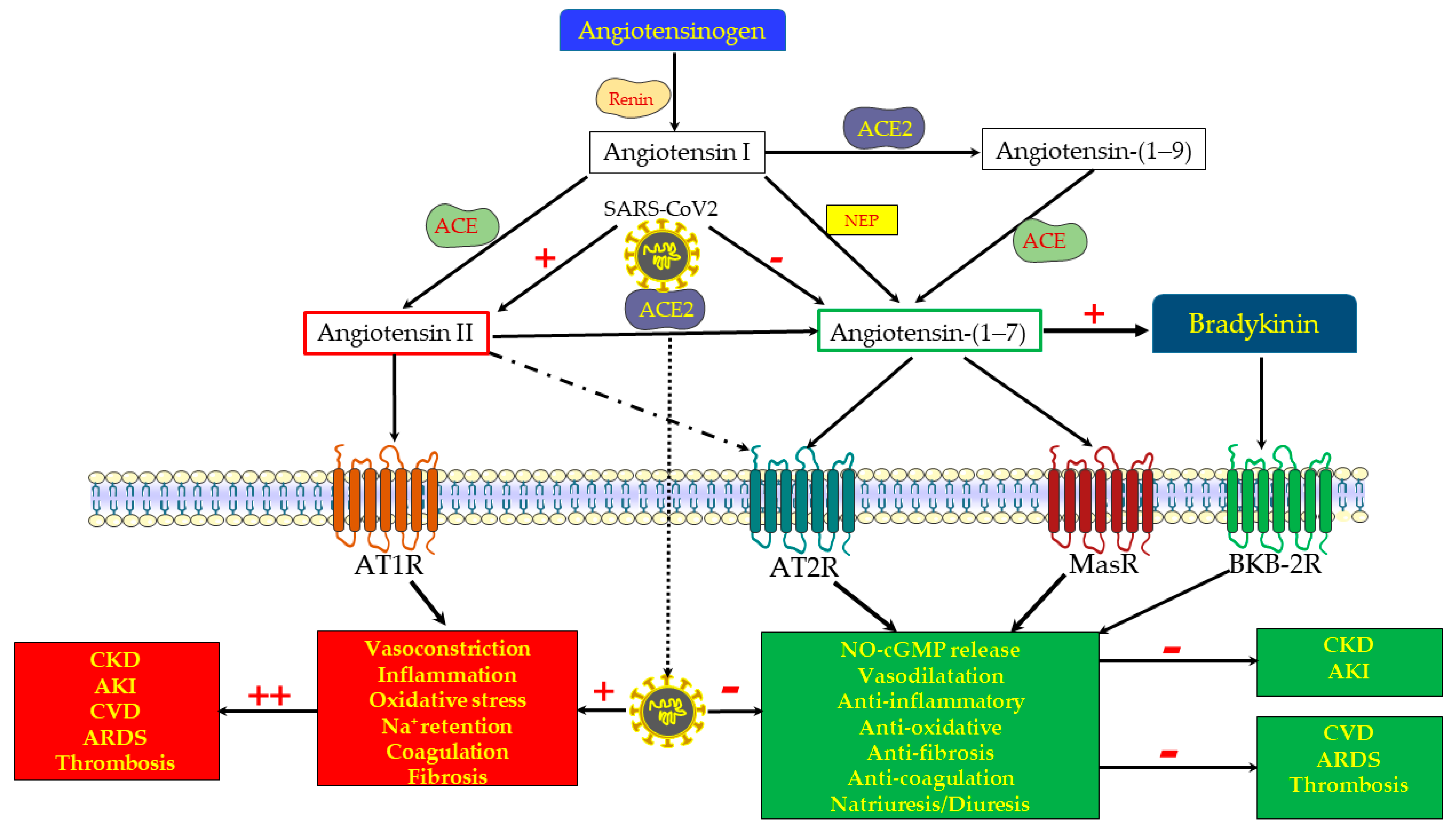
2. The RAS: Counteracting Harmful and Protective Pathways
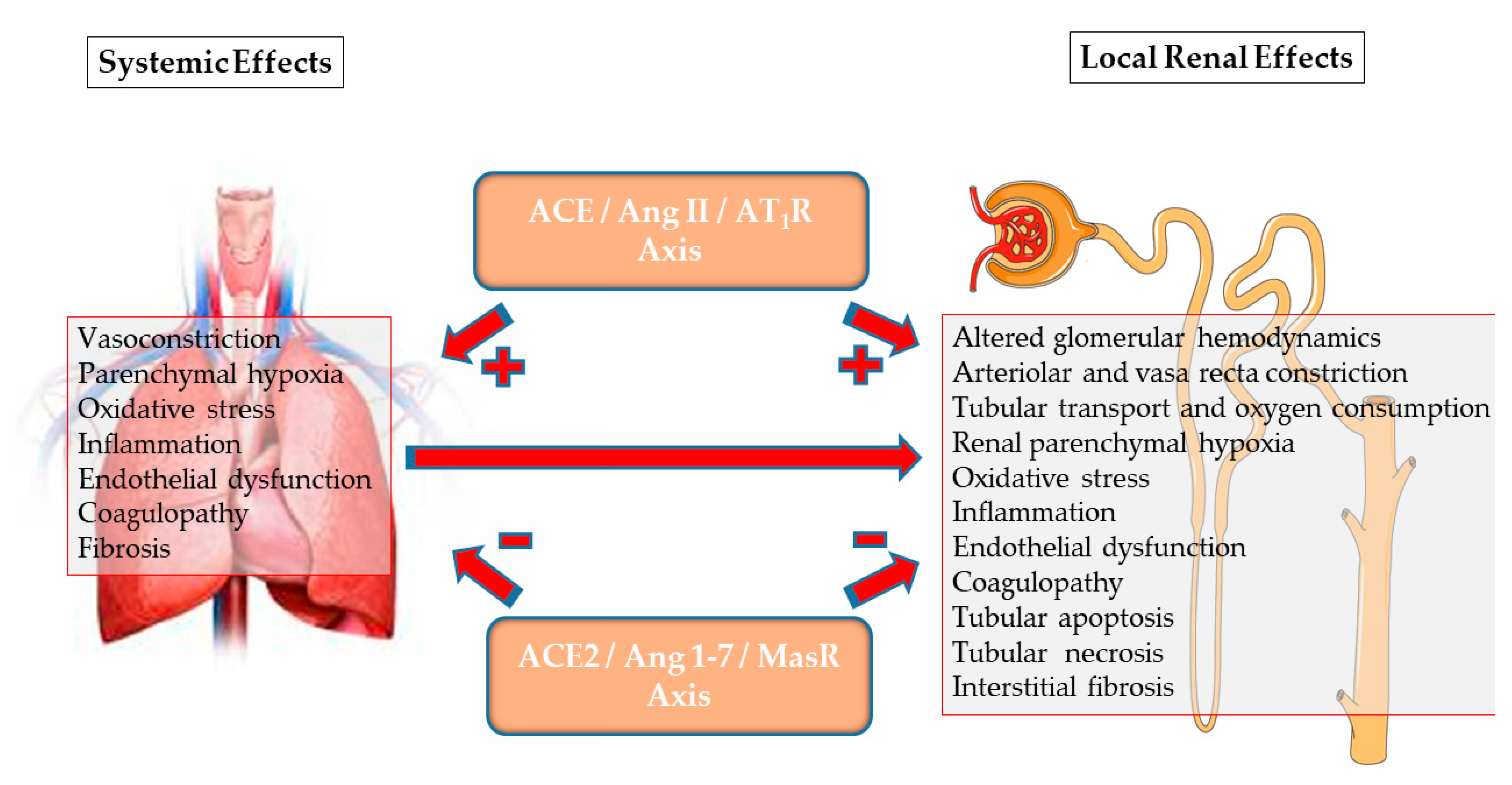
3. COVID-19 Disease: An Archetype of Imbalanced RAS
4. ACE2/Ang-(1-7)/MasR Axis and Renal Physiology
5. Alterations in ACE2/Ang-(1-7)/MasR Axis in Systemic and Renal Disorders
6. Activating Renal ACE2/Ang-(1-7)/asR: Plausible Therapeutic Interventions in Renal Diseases
7. Enhancing the ACE2/Ang-(1-7)/MasR Axis in the Management of AKI
8. Enigmatic Contradicting Findings
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Heyman, S.N.; Rosen, S.; Rosenberger, C. Critical assessment of experimental models of acute renal failure. In Critical Care Nephrology, 2nd ed.; Ronco, C., Bellomo, R., Kellum, J., Eds.; Saunders/Elsevier: Philadelphia, PA, USA, 2009; pp. 237–250. [Google Scholar]
- Heyman, S.N.; Rosenberger, C.; Rosen, S. Experimental ischemia–reperfusion: Biases and myths—The proximal vs. distal hypoxic tubular injury debate revisited. Kidney Int. 2010, 77, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.; Skorecki, K.; Heyman, S.N.; Kinaneh, S.; Armaly, Z. Covid-19 infection and mortality–A physiologist’s perspective enlight-ening clinical features and plausible interventional strategies. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, H1080–H1083. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.; Higazi, A.A.R.; Kinaneh, S.; Hmaud, S.; Khamaisi, I.; Skorecki, K.; Heyman, S.N. ACE2, CoVID-19 infection, inflam-mation and coagulopathy: Missing parts in the puzzle. Front. Physiol. 2020, 11, 574753. [Google Scholar] [CrossRef] [PubMed]
- Basso, N.; Terragno, N.A. History About the Discovery of the Renin-Angiotensin System. Hypertension 2001, 38, 1246–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Anders, H.-J.; Gaikwad, A.B. Fiend and friend in the renin angiotensin system: An insight on acute kidney injury. Biomed. Pharmacother. 2019, 110, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Silver, S.A.; Beaubien-Souligny, W.; Shah, P.S.; Harel, S.; Blum, D.; Kishibe, T.; Meraz-Munoz, A.; Wald, R.; Harel, Z. The Prevalence of Acute Kidney Injury in Patients Hospitalized With COVID-19 Infection: A Systematic Review and Meta-analysis. Kidney Med. 2021, 3, 83–98.e1. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Cooper, M.E.; De Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.-H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of Losartan on Renal and Cardiovascular Outcomes in Patients with Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, S.; Pitt, B.; Davis, C.E.; Hood, W.B.; Cohn, J.N. Effect of enalapril on survival in patients with re-duced left ventricular ejection fractions and congestive heart failure. N. Engl. J. Med. 1991, 325, 293–302. [Google Scholar]
- Yusuf, S.; Pitt, B.; Davis, C.E.; Hood, W.B.; Cohn, J.N. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N. Engl. J. Med. 1992, 327, 685–691. [Google Scholar] [CrossRef] [Green Version]
- KDIGO Work Group: KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease: Sum-mary of recommendation statements. Kidney Int. Suppl. 2012, 3, 5–14.
- KDIGO Work Group: KDIGO clinical practice guideline for the management of blood pressure in chronic kidney disease: Summary of recommendation statements. Kidney Int. Suppl. 2012, 2, 341–342. [CrossRef] [Green Version]
- Weir, M.R.; Lakkis, J.I.; Jaar, B.; Rocco, M.V.; Choi, M.J.; Kramer, H.J.; Ku, E. Use of renin-angiotensin system blockade in ad-vanced CKD: An NKF-KDOQI controversies report. Am. J. Kidney Dis. 2018, 72, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Maschio, G.; Alberti, D.; Janin, G.; Locatelli, F.; Mann, J.F.; Motolese, M.; Ponticelli, C.; Ritz, E.; Zucchelli, P. Effect of the angioten-sin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting-Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N. Engl. J. Med. 1996, 334, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Linz, W.; Wiemer, G.; Schaper, J.; Nagasawa, K.; Gohlke, P.; Unger, T. Angiotensin converting enzyme inhibitors, left ventricular hypertrophy and fibrosis. Mol. Cell. Biochem. 1995, 147, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, A.; Gagliardini, E.; Sangalli, F.; Bonomelli, M.; Piccinelli, M.; Benigni, A.; Remuzzi, G. ACE inhibition reduces glomerulo-sclerosis and regenerates glomerular tissue in a model of progressive renal disease. Kidney Int. 2006, 69, 1124–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangalore, S.; Fakheri, R.; Wandel, S.; Toklu, B.; Wandel, J.; Messerli, F.H. Renin angiotensin system inhibitors for patients with stable coronary artery disease without heart failure: Systematic review and meta-analysis of randomized trials. BMJ 2017, 356, j4. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; Kolkhof, P.; Bakris, G.; Bauersachs, J.; Haller, H.; Wada, T.; Zannad, F. Steroidal and non-steroidal mineralocorticoid receptor antagonists in cardiorenal medicine. Eur. Heart J. 2021, 42, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Colafella, K.M.M.; Hilliard, L.M.; Denton, K.M. Epochs in the depressor/pressor balance of the renin–angiotensin system. Clin. Sci. 2016, 130, 761–771. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Campagnole-Santos, M.J.; Andrade, S.P. Angiotensin-(1-7): An update. Regul. Pept. 2000, 91, 45–62. [Google Scholar] [CrossRef]
- Verano-Braga, T.; Martins, A.L.V.; Motta-Santos, D.; Campagnole-Santos, M.J.; Santos, R.A.S. ACE2 in the renin-angiotensin sys-tem. Clin. Sci. 2020, 134, 3063–3078. [Google Scholar] [CrossRef] [PubMed]
- Domenig, O.; Manzel, A.; Bader, M.; Motta-Santos, D.; Santos, R.A.; Elased, K.M.; Säemann, M.D.; Linker, R.A.; Poglitsch, M.; Grobe, N.; et al. Neprilysin is a mediator of alternative renin-angiotensin-system activation in the murine and human kidney. Sci. Rep. 2016, 6, srep33678. [Google Scholar] [CrossRef] [Green Version]
- Gironacci, M.M.; Adamo, H.P.; Corradi, G.; Santos, R.A.; Ortiz, P.; Carretero, O.A. Angiotensin (1-7) induces MAS receptor internalization. Hypertension 2011, 58, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Burns, W.C.; Toffoli, B.; Pickering, R.; Sakoda, M.; Tsorotes, D.; Grixti, E.; Velkoska, E.; Burrell, L.M.; Johnston, C.; et al. Angiotensin-converting enzyme 2 regulates renal atrial natriuretic peptide through angiotensin-(1-7). Clin. Sci. 2012, 123, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A. Angiotensin-(1-7). Hypertension 2014, 63, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.N.; Ali, Q.; Samuel, P.; Steckelings, U.M.; Hussain, T. Angiotensin II type 2 receptor and receptor mas are colocalized and functionally interdependent in obese zucker rat kidney. Hypertension 2017, 70, 831–838. [Google Scholar] [CrossRef]
- Duke, L.M.; Eppel, G.A.; Widdop, R.E.; Evans, R.G. Disparate roles of AT2 receptors in the renal cortical and medullary circula-tions of anesthetized rabbits. Hypertension 2003, 42, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Ali, Q.; Dhande, I.; Samuel, P.; Hussain, T. Angiotensin type 2 receptor null mice express reduced levels of renal angiotensin converting enzyme-2/angiotensin (1-7)/Mas receptor and exhibit greater high-fat diet-induced kidney injury. J. Renin-Angiotensin-Aldosterone Syst. 2016, 17. [Google Scholar] [CrossRef] [Green Version]
- Ali, Q.; Wu, Y.; Hussain, T. Chronic AT2 receptor activation increases renal ACE2 activity, attenuates AT1 receptor function and blood pressure in obese Zucker rats. Kidney Int. 2013, 84, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abassi, Z.; Skorecki, K.; Ben Hamo-Giladi, D.; Kruzel-Davila, E.; Heyman, S.N. Kinins and chymase: The forgotten components of the renin-angiotensin system and their implications in COVID-19 disease. Am. J. Physiol. Cell. Mol. Physiol. 2021, 320, L422–L429. [Google Scholar] [CrossRef] [PubMed]
- Brosnihan, K.B.; Li, P.; Ferrario, C.M. Angiotensin-(1-7) Dilates canine coronary arteries through kinins and nitric oxide. Hypertension 1996, 27, 523–528. [Google Scholar] [CrossRef]
- Dos Santos, R.A.S.; Passaglio, K.T.; Pesquero, J.B.; Bader, M.; Silva, E.A.C.S. Interactions between angiotensin-(1-7), kinins, and angiotensin II in kidney and blood vessels. Hypertension 2001, 38, 660–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, L.; Fortes, Z.B.; Nigro, D.; Tostes, R.C.; Santos, R.A.; Catelli De Carvalho, M.H. Potentiation of bradykinin by angioten-sin-(1-7) on arterioles of spontaneously hypertensive rats studied in vivo. Hypertension 2001, 37, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosnihan, K.B.; Li, P.; Tallant, E.A.; Ferrario, C.M. Angiotensin-(1-7): A novel vasodilator of the coronary circulation. Biol. Res. 1998, 31, 227–234. [Google Scholar] [PubMed]
- Liu, Y.F.; Zhang, Z.; Pan, X.L.; Xing, G.L.; Zhang, Y.; Liu, Z.S.; Tu, S.H. The chronic kidney disease and acute kidney injury in-volvement in COVID-19 pandemic: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0244779. [Google Scholar]
- Izzedine, H.; Jhaveri, K.D. Acute kidney injury in patients with COVID-19: An update on the pathophysiology Nephrol. Dial. Transpl. 2021, 36, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Chueh, T.-I.; Zheng, C.-M.; Hou, Y.-C.; Lu, K.-C. Novel evidence of acute kidney injury in COVID-19. J. Clin. Med. 2020, 9, 3547. [Google Scholar] [CrossRef] [PubMed]
- Koitka, A.; Cooper, M.E.; Thomas, M.C.; Tikellis, C. Angiotensin converting enzyme 2 in the kidney. Clin. Exp. Pharmacol. Physiol. 2008, 35, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Lores, E.; Wysocki, J.; Batlle, D. ACE2, the kidney and the emergence of COVID-19 two decades after ACE2 discovery. Clin. Sci. 2020, 134, 2791–2805. [Google Scholar] [CrossRef] [PubMed]
- Lely, A.T.; Hamming, I.; van Goor, H.; Navis, G.J. Renal ACE2 expression in human kidney disease. J. Pathol. 2004, 204, 587–593. [Google Scholar] [CrossRef]
- Ye, M.; Wysocki, J.; William, J.; Soler, M.J.; Cokic, I.; Batlle, D. Glomerular localization and expression of angiotensin-converting enzyme 2 and angiotensin-converting enzyme: Implications for albuminuria in diabetes. J. Am. Soc. Nephrol. 2006, 17, 3067–3075. [Google Scholar] [CrossRef] [Green Version]
- Soler, M.J.; Ye, M.; Wysocki, J.; William, J.; Lloveras, J.; Batlle, D. Localization of ACE2 in the renal vasculature: Amplification by angiotensin II type 1 receptor blockade using telmisartan. Am. J. Physiol. Physiol. 2009, 296, F398–F405. [Google Scholar] [CrossRef] [Green Version]
- Nobes, M.S.; Harris, P.J.; Yamada, H.; Mendelsohn, F.A. Effects of angiotensin on renal cortical and papillary blood flows meas-ured by laser-Doppler flowmetry. Am. J. Physiol. Renal Physiol. 1991, 261, F998–F1006. [Google Scholar] [CrossRef] [PubMed]
- Ba̧dzyńska, B.; Grzelec-Mojzesowicz, M.; Dobrowolski, L.; Sadowski, J. Differential effect of angiotensin II on blood circulation in the renal medulla and cortex of anaesthetised rats. J. Physiol. 2002, 538, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, N.W.; Oliver, J.J.; Evans, R.G. Nitric oxide in responses of regional kidney blood flow to vasoactive agents in anesthe-tized rabbits. J. Cardiovasc. Pharmacol. 2002, 40, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Miyata, N.; Park, F.; Li, X.F.; Cowley, A.W., Jr. Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am. J. Physiol. Renal Physiol. 1999, 277, F437–F446. [Google Scholar] [CrossRef]
- Heyman, S.N.; Khamaisi, M.; Rosen, S.; Rosenberger, C. Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am. J. Nephrol. 2008, 28, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Chappell, M.C.; Pirro, N.T.; South, A.M.; Gwathmey, T.M. Concerns on the specificity of commercial elisas for the measurement of angiotensin (1-7) And angiotensin II in human plasma. Hypertension 2021, 77, e29–e31. [Google Scholar] [CrossRef]
- Banday, A.A.; Diaz, A.D.; Lokhandwala, M. Kidney dopamine D1-like receptors and angiotensin 1-7 interaction inhibits renal Na+ transporters. Am. J. Physiol. Physiol. 2019, 317, F949–F956. [Google Scholar] [CrossRef] [PubMed]
- Yousif, M.H.; Benter, I.F.; Diz, D.I.; Chappell, M.C. Angiotensin-(1-7)-dependent vasorelaxation of the renal artery exhibits unique angiotensin and bradykinin receptor selectivity. Peptides 2017, 90, 10–16. [Google Scholar] [CrossRef]
- Bürgelová, M.; Kramer, H.J.; Teplan, V.; Thumová, M.; Červenka, L. Effects of angiotensin-(1-7) blockade on renal function in rats with enhanced intrarenal Ang II activity. Kidney Int. 2005, 67, 1453–1461. [Google Scholar] [CrossRef] [Green Version]
- Handa, R.K.; Ferrario, C.M.; Strandhoy, J.W. Renal actions of angiotensin-(1-7): In vivo and in vitro studies. Am. J. Physiol. Physiol. 1996, 270, F141–F147. [Google Scholar] [CrossRef]
- Leyen, S.A.-V.; Romero, M.F.; Khosla, M.C.; Douglas, J.G. Modulation of phospholipase A2 activity and sodium transport by angiotensin-(1-7). Kidney Int. 1993, 44, 932–936. [Google Scholar] [CrossRef] [Green Version]
- Maksimowski, N.; Williams, V.R.; Scholey, J.W. Kidney ACE2 expression: Implications for chronic kidney disease. PLoS ONE 2020, 15, e0241534. [Google Scholar] [CrossRef]
- Borges, C.C.; Penna-de-Carvalho, A.; Medeiros Junior, J.L.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Ovariectomy modify local renin-angiotensin-aldosterone system gene expressions in the heart of ApoE (-/-) mice. Life Sci. 2017, 191, 1–8. [Google Scholar] [CrossRef]
- Chanana, N.; Palmo, T.; Sharma, K.; Kumar, R.; Graham, B.B.; Pasha, Q. Sex-derived attributes contributing to SARS-CoV-2 mor-tality. Am. J Physiol. Endocrinol. Metab. 2020, 319, E562–E567. [Google Scholar] [CrossRef] [PubMed]
- Wangensteen, R.; Moreno, J.M.; Sainz, J. Rodríguez-Gómez, I.; Chamorro, V.; de Dios Luna, J.; Osuna, A.; Vargas, F. Gender dif-ference in the role of endothelium-derived relaxing factors modulating renal vascular reactivity. Eur. J. Pharmacol. 2004, 486, 281–288. [Google Scholar] [CrossRef]
- Wei, Q.; Wang, M.-H.; Dong, Z. Differential gender differences in ischemic and nephrotoxic acute renal failure. Am. J. Nephrol. 2005, 25, 491–499. [Google Scholar] [CrossRef]
- Hutchens, M.P.; Dunlap, J.; Hurn, P.D.; Jarnberg, P.O. Renal ischemia: Does sex matter? Anesth. Analg. 2008, 107, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Herzenberg, A.M.; Kassiri, Z.; Wong, D.; Reich, H.; Khokha, R.; Crackower, M.A.; Backx, P.H.; Penninger, J.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclero-sis. Am. J. Pathol. 2006, 168, 1808–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Guo, D.; Chen, C.B.; Wang, W.; Schuster, M.; Loibner, H.; Penninger, J.M.; Scholey, J.W.; Kassiri, Z.; Oudit, G.Y. Preven-tion of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension 2011, 57, 314–322. [Google Scholar] [CrossRef]
- Soler, M.J.; Wysocki, J.; Batlle, D. ACE2 alterations in kidney disease. Nephrol. Dial. Transplant. 2013, 28, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-Dos-Santos, A.J.; Da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nat. Cell Biol. 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Tikellis, C.; Cooper, M.E.; Bialkowski, K.; Johnston, C.I.; Burns, W.C.; Lew, R.A.; Smith, A.I.; Thomas, M.C. Developmental expres-sion of ACE2 in the SHR kidney: A role in hypertension? Kidney Int. 2006, 70, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Yang, F.; Huang, X.; Meng, J.; Chen, J.; Bader, M.; Penninger, J.M.; Fung, E.; Yu, X.; Lan, H. Dual deficiency of angiotensin-converting enzyme-2 and Mas receptor enhances angiotensin II-induced hypertension and hypertensive nephropathy. J. Cell. Mol. Med. 2020, 24, 13093–13103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, X.-R.; Chen, H.-Y.; Fung, E.; Liu, J.; Lan, H.-Y. Deletion of angiotensin-converting enzyme-2 promotes hypertensive nephropathy by targeting smad7 for ubiquitin degradation. Hypertension 2017, 70, 822–830. [Google Scholar] [CrossRef]
- Joyner, J.; Neves, L.A.; Granger, J.P.; Alexander, B.T.; Merrill, D.C.; Chappell, M.C.; Ferrario, C.M.; Davis, W.P.; Brosnihan, K.B. Temporal-spatial expression of ANG-(1-7) and angiotensin-converting enzyme 2 in the kidney of normal and hypertensive preg-nant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R169–R177. [Google Scholar] [CrossRef] [PubMed]
- Wakahara, S.; Konoshita, T.; Mizuno, S.; Motomura, M.; Aoyama, C.; Makino, Y.; Kato, N.; Koni, I.; Miyamori, I. Synergistic ex-pression of angiotensin-converting enzyme (ACE) and ACE2 in human renal tissue and confounding effects of hypertension on the ACE to ACE2 ratio. Endocrinology 2007, 148, 2453–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velkoska, E.; Dean, R.G.; Burchill, L.; Levidiotis, V.; Burrell, L.M. Reduction in renal ACE2 expression in subtotal nephrectomy in rats is ameliorated with ACE inhibition. Clin. Sci. 2009, 118, 269–279. [Google Scholar] [CrossRef] [Green Version]
- DiLauro, M.; Zimpelmann, J.; Robertson, S.J.; Genest, D.; Burns, K.D. Effect of ACE2 and angiotensin-(1-7) in a mouse model of early chronic kidney disease. Am. J. Physiol. Physiol. 2010, 298, F1523–F1532. [Google Scholar] [CrossRef]
- Burrell, L.M.; Burchill, L.; Dean, R.G.; Griggs, K.; Patel, S.K.; Velkoska, E. Chronic kidney disease: Cardiac and renal angiotensin-converting enzyme (ACE) 2 expression in rats after subtotal nephrectomy and the effect of ACE inhibition. Exp. Physiol. 2012, 97, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.N.; Oudit, G.Y.; Penninger, J.M.; Scholey, J.W.; Herzenberg, A.M. Decreased glomerular and tubular expression of ACE2 in patients with type 2 diabetes and kidney disease. Kidney Int. 2008, 74, 1610–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuiri, S.; Hemmi, H.; Arita, M.; Ohashi, Y.; Tanaka, Y.; Miyagi, M.; Sakai, K.; Ishikawa, Y.; Shibuya, K.; Hase, H.; et al. Expres-sion of ACE and ACE2 in individuals with diabetic kidney disease and healthy controls. Am. J. Kidney Dis. 2008, 51, 613–623. [Google Scholar] [CrossRef]
- Wysocki, J.; Ye, M.; Soler, M.J.; Gurley, S.B.; Xiao, H.D.; Bernstein, K.E.; Coffman, T.M.; Chen, S.; Batlle, D. ACE and ACE2 activi-ty in diabetic mice. Diabetes 2006, 55, 2132–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikellis, C.; Bialkowski, K.; Pete, J.; Sheehy, K.; Su, Q.; Johnston, C.; Cooper, M.E.; Thomas, M.C. ACE2 deficiency modifies reno-protection afforded by ACE inhibition in experimental diabetes. Diabetes 2008, 57, 1018–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.; Xin, H.; Jiang, X.-Y.; Wang, Y.-X.; Zhang, Y.-S. Relationship between renal injury and the antagonistic roles of angiotensin-converting enzyme (ACE) and ACE2. Genet. Mol. Res. 2014, 13, 2333–2342. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Ghosh, A.; Lo, C.S.; Chenier, I.; Scholey, J.W.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S.D. Nrf2 deficiency up-regulates intrarenal angiotensin-converting enzyme-2 and angiotensin 1-7 receptor expression and attenuates hypertension and nephropathy in diabetic mice. Endocrinology 2018, 159, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Samuel, P.; Ali, Q.; Sabuhi, R.; Wu, Y.; Hussain, T. High Na-intake increases renal angiotensin II levels and reduces the expression of ACE2-AT2R-MasR axis in obese Zucker rats. Am. J. Physiol. Renal Physiol. 2012, 303, F412–F419. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Lai, F.M.-M.; Lai, K.-B.; Chow, K.-M.; Kwan, C.-H.B.; Li, K.-T.P.; Szeto, C.-C. Urinary mRNA expression of ACE and ACE2 in human type 2 diabetic nephropathy. Diabetologia 2008, 51, 1062–1067. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.W.; Oudit, G.Y.; Reich, H.; Kassiri, Z.; Zhou, J.; Liu, Q.C.; Backx, P.H.; Penninger, J.M.; Herzenberg, A.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 (Ace2) Accelerates diabetic kidney injury. Am. J. Pathol. 2007, 171, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Huang, X.R.; Chen, H.Y.; Penninger, J.M.; Lan, H.Y. Loss of angiotensin-converting enzyme 2 enhances TGF-β/Smad-mediated renal fibrosis and NF-κB-driven renal inflammation in a mouse model of obstructive nephropathy. Lab. Investig. 2012, 92, 650–661. [Google Scholar] [CrossRef]
- Khoury, E.E.; Knaney, Y.; Fokra, A.; Kinaneh, S.; Azzam, Z.; Heyman, S.N.; Abassi, Z. Pulmonary, cardiac and renal distribution of ACE2, furin, TMPRSS2 and ADAM17 in rats with heart failure: Potential implication for COVID-19 disease. J. Cell. Mol. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Serebrovska, Z.O.; Chong, E.Y.; Serebrovska, T.V.; Tumanovska, L.V.; Xi, L. Hypoxia, HIF-1α, and COVID-19: From pathogenic factors to potential therapeutic targets. Acta Pharmacol. Sin. 2020, 41, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Rosenberger, C.; Heyman, S.N.; Eckardt, K.U. HIF regulation in kidney disease. Clin. Exp. Pharmacol. Physiol. 2013, 40, 148–157. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, Y.; Zhao, M.; Liu, C.; Zhou, L.; Shen, S.; Liao, S.; Yang, K.; Li, Q.; Wan, H. Role of HIF-1α in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L631–L640. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Uttarwar, L.; Gao, B.; Charbonneau, M.; Shi, Y.; Chan, J.S.; Dubois, C.M.; Krepinsky, J.C. High glucose up-regulates AD-AM17 through HIF-1alpha in mesangial cells. J. Biol. Chem. 2015, 290, 21603–21614. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, I.M.; Verma, A.; Waikar, S.S. Circulating plasma angiotensin-converting enzyme 2 concentrations in patients with kidney disease. Eur. Heart J. 2020, 41, 3097–3098. [Google Scholar] [CrossRef]
- Harmer, D.; Gilbert, M.; Borman, R.; Clark, K.L. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002, 532, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Ingelfinger, J.R. Angiotensin-converting enzyme 2: Implications for blood pressure and kidney disease. Curr. Opin. Nephrol. Hypertens. 2009, 18, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kurzhagen, J.T.; Dellepiane, S.; Cantaluppi, V.; Rabb, H. AKI: An increasingly recognized risk factor for CKD development and progression. J. Nephrol. 2020, 33, 1171–1187. [Google Scholar] [CrossRef]
- Agata, J.; Ura, N.; Yoshida, H.; Shinshi, Y.; Sasaki, H.; Hyakkoku, M.; Taniguchi, S.; Shimamoto, K. Olmesartan is an angiotensin II receptor blocker with an inhibitory effect on angiotensin-converting enzyme. Hypertens. Res. 2006, 29, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.; Yagil, Y.; Bursztyn, M.; Barkalifa, R.; Scharf, S.; Yagil, C. ACE2 expression and activity are enhanced during pregnancy. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R1953–R1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, D.; Wysocki, J.; Soler, M.J.; Ranganath, K. Angiotensin-converting enzyme 2: Enhancing the degradation of angiotensin II as a potential therapy for diabetic nephropathy. Kidney Int. 2012, 81, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Nadim, M.K.; Forni, L.G.; Mehta, R.L.; Connor, M.J., Jr.; Liu, K.D.; Ostermann, M.; Rimmelé, T.; Zarbock, A.; Bell, S.; Bihorac, A.; et al. COVID-19-associated acute kidney injury: Consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nat. Rev. Nephrol. 2020, 16, 747–764. [Google Scholar] [PubMed]
- Shete, A. Urgent need for evaluating agonists of angiotensin-(1-7)/Mas receptor axis for treating patients with COVID-19. Int. J. Infect. Dis. 2020, 96, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.; Arnold, A.C. Angiotensin-(1-7): Translational avenues in cardiovascular control. Am. J. Hypertens. 2019, 32, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wysocki, J.; Souma, T.; Ye, M.; Ramirez, V.; Zhou, B.; Wilsbacher, L.D.; Quaggin, S.E.; Batlle, D.; Jin, J. Novel ACE2-Fc chimeric fusion provides long-lasting hypertension control and organ protection in mouse models of systemic renin angiotensin system activation. Kidney Int. 2018, 94, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lo, C.S.; Padda, R.; Abdo, S.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Angiotensin-(1-7) prevents systemic hypertension, attenuates oxidative stress and tubulointerstitial fibrosis, and normalizes renal angiotensin-converting en-zyme 2 and Mas receptor expression in diabetic mice. Clin. Sci. Lond. 2015, 128, 649–663. [Google Scholar] [CrossRef]
- Cassis, P.; Locatelli, M.; Corna, D.; Villa, S.; Rottoli, D.; Cerullo, D.; Abbate, M.; Remuzzi, G.; Benigni, A.; Zoja, C. Addition of cy-clic angiotensin-(1-7) to angiotensin-converting enzyme inhibitor therapy has a positive add-on effect in experimental diabetic nephropathy. Kidney Int. 2019, 96, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Oudit, G.Y.; Liu, G.C.; Zhong, J.; Basu, R.; Chow, F.L.; Zhou, J.; Loibner, H.; Janzek, E.; Schuster, M.; Penninger, J.M.; et al. Hu-man recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes 2010, 59, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Meems, L.M.; Andersen, I.A.; Sangaralingham, S.J.; McCormick, D.J.; Burnett, J.C.; Pan, S.; Harty, G.; Chen, Y.; Zheng, Y.; Harders, G.E.; et al. Design, synthesis, and actions of an innovative bispecific designer peptide. Hypertension 2019, 73, 900–909. [Google Scholar] [CrossRef]
- Silveira, K.D.; Barroso, L.C.; Vieira, A.T.; Cisalpino, D.; Lima, C.X.; Bader, M.; Arantes, R.M.E.; Dos Santos, R.A.S.; Simões, E.; Silva, A.C.; et al. Beneficial effects of the activation of the angiotensin-(1-7) Mas receptor in a murine model of adriamycin-induced nephropathy. PLoS ONE 2013, 8, e66082. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Han, W.; Zhang, M.; Fang, W.; Zhai, X.; Guan, S.; Qu, X. Long-term renal sympathetic denervation ameliorates renal fibrosis and delays the onset of hypertension in spontaneously hypertensive rats. Am. J. Transl. Res. 2018, 10, 4042–4053. [Google Scholar] [PubMed]
- Jang, I.-A.; Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Effects of resveratrol on the renin-angiotensin system in the aging kidney. Nutrients 2018, 10, 1741. [Google Scholar] [CrossRef] [Green Version]
- Cao, G.; Della Penna, S.L.; Kouyoumdzian, N.M.; Choi, M.R.; Gorzalczany, S.; Fernández, B.E.; Toblli, J.E.; Rosón, M.I. Immuno-histochemical expression of intrarenal renin angiotensin system components in response to tempol in rats fed a high salt diet. World J. Nephrol. 2017, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Jessup, J.; Gallagher, P.E.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Smith, R.D.; Chappell, M.C. Effects of renin-angiotensin system blockade on renal angiotensin-(1-7) forming enzymes and receptors. Kidney Int. 2005, 68, 2189–2196. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-H.; Wang, Y.-H.; Wang, J.-J.; Liu, Y.-C.; Deng, W.; Qin, C.; Gao, J.-L.; Zhang, L.-Y. Role of angiotensin-converting enzyme (ACE and ACE2) imbalance on tourniquet-induced remote kidney injury in a mouse hindlimb ischemia- reperfusion model. Peptides 2012, 36, 60–70. [Google Scholar] [CrossRef]
- da Silveira, K.D.; Pompermayer Bosco, K.S.; Diniz, L.R.L.; Carmona, A.K.; Cassali, G.D.; Bruna-Romero, O.; de Sousa, L.P.; Teixeira, M.M.; Santos, R.A.S.; Simões e Silva, A.C.; et al. ACE2-angiotensin-(1-7)-Mas axis in renal ischaemia/reperfusion injury in rats. Clin. Sci. Lond. 2010, 119, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Malek, V.; Mulay, S.R.; Gaikwad, A.B. Angiotensin II type 2 receptor and angiotensin-converting enzyme 2 mediate ischemic renal injury in diabetic and non-diabetic rats. Life Sci. 2019, 235, 116796. [Google Scholar] [CrossRef] [PubMed]
- Barroso, L.C.; Silveira, K.D.; Lima, C.X.; Borges, V.; Bader, M.; Rachid, M.; Santos, R.A.S.; Souza, D.G.; Silva, A.C.S.E.; Teixeira, M.M. Renoprotective effects of AVE0991, a nonpeptide mas receptor agonist, in experimental acute renal injury. Int. J. Hypertens. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Fang, F.; Liu, G.C.; Scholey, J.W.; John, R.; Zhou, X.; Yang, S.; Reich, H.N.; Williams, V.; Hu, A.; Pan, J.; et al. Loss of ACE2 exacerbates murine renal ischemia-reperfusion injury. PLoS ONE 2013, 8, e71433. [Google Scholar] [CrossRef] [Green Version]
- Malek, M.; Nematbakhsh, M. The preventive effects of diminazene aceturate in renal ischemia/reperfusion injury in male and fe-male rats. Adv. Prev. Med. 2014, 2014, 740647. [Google Scholar]
- Safari, T.; Nematbakhsh, M.; Hilliard, L.M.; Evans, R.G.; Denton, K.M. Sex differences in the renal vascular response to angioten-sin II involves the Mas receptor. Acta Physiol. Oxf. 2012, 206, 150–156. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, T.C.S.; Lanza, K.; Filha, R.D.S.; Campos, L.M.D.C.C.; Fonseca, E.G.; Chagas, M.W.; Rocha, N.P.; De Sá, M.A.; Vieira, M.A.R.; Caliari, M.V.; et al. ACE2 activator diminazene aceturate exerts renoprotective effects in gentamicin-induced acute renal injury in rats. Clin. Sci. 2020, 134, 3093–3106. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, I.J.; Bae, E.H.; Ma, S.K.; Lee, J.; Kim, S.W. Angiotensin-(1-7) attenuates kidney injury due to obstructive nephropa-thy in rats. PLoS ONE 2015, 10, e0142664. [Google Scholar]
- Gupta, A.; Rhodes, G.J.; Berg, D.T.; Gerlitz, B.; Molitoris, B.A.; Grinnell, B.W. Activated protein C ameliorates LPS-induced acute kidney injury and downregulates renal INOS and angiotensin 2. Am. J. Physiol. Physiol. 2007, 293, F245–F254. [Google Scholar] [CrossRef] [Green Version]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin–angiotensin–aldosterone system inhibitors in patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Esteban, V.; Heringer-Walther, S.; Sterner-Kock, A.; de Bruin, R.; van den Engel, S.; Wang, Y.; Mezzano, S.; Egido, J.; Schultheiss, H.P.; Ruiz-Ortega, M.; et al. Angiotensin-(1-7) and the g protein-coupled receptor MAS are key players in renal inflammation. PLoS ONE 2009, 4, e5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, L.M.; Gayed, D.; Griggs, K.; Patel, S.K.; Velkoska, E. Adverse cardiac effects of exogenous angiotensin 1-7 in rats with sub-total nephrectomy are prevented by ACE inhibition. PLoS ONE 2017, 12, e0171975. [Google Scholar] [CrossRef]
- Bi, J.; Contag, S.A.; Carey, L.C.; Tang, L.; Valego, N.K.; Chappell, M.C.; Rose, J.C. Antenatal betamethasone exposure alters renal responses to angiotensin-(1-7) in uninephrectomized adult male sheep. J. Renin Angiotensin Aldosterone Syst. 2012, 14, 290–298. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, D.L.; Zimpelmann, J.; Xiao, F.; Gutsol, A.; Touyz, R.; Burns, K.D. The effect of angiotensin-(1-7) In mouse unilateral ureteral obstruction. Am. J. Pathol. 2015, 185, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; He, M.; Zhou, L.; Yao, T.; Huang, Y.; Lu, L.-M. Chronic angiotensin (17) injection accelerates STZ-induced diabetic renal injury1. Acta Pharmacol. Sin. 2008, 29, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, M.A.; Baban, B.; Tipton, A.J.; O’Connor, P.M.; Sullivan, J.C. Chronic ANG II infusion induces sex-specific increases in renal T cells in Sprague-Dawley rats. Am. J. Physiol. Physiol. 2015, 308, F706–F712. [Google Scholar] [CrossRef] [Green Version]
- Darawshi, S.; Yassin, H.; Gorelik, Y.; Faor, C.; Szalet, A.; Heyman, S.N.; Khamaisi, M. Biomarker evidence for distal tubular dam-age but cortical sparing in hospitalized diabetic patients with acute kidney injury (AKI) while on SGLT2 inhibitors. Ren. Fail. 2020, 42, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.C.; Velkoska, E.; Dean, R.; Burrell, L.M.; Thomas, M.C. Angiotensin II mediates epithelial-to-mesenchymal transfor-mation in tubular cells by ANG 1-7/MAS-1-dependent pathways. Am. J. Physiol. Renal. Physiol. 2010, 299, F585–F593. [Google Scholar] [CrossRef] [PubMed]
- Safari, T.; Shahraki, M.R.; Miri, S.; Bakhshani, N.M.; Niazi, A.A.; Komeili, G.R.; Bagheri, H. The effect of angiotensin 1-7 and losartan on renal ischemic/reperfusion injury in male rats. Res. Pharm. Sci. 2019, 14, 441–447. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heyman, S.N.; Walther, T.; Abassi, Z. Angiotensin-(1-7)—A Potential Remedy for AKI: Insights Derived from the COVID-19 Pandemic. J. Clin. Med. 2021, 10, 1200. https://doi.org/10.3390/jcm10061200
Heyman SN, Walther T, Abassi Z. Angiotensin-(1-7)—A Potential Remedy for AKI: Insights Derived from the COVID-19 Pandemic. Journal of Clinical Medicine. 2021; 10(6):1200. https://doi.org/10.3390/jcm10061200
Chicago/Turabian StyleHeyman, Samuel N., Thomas Walther, and Zaid Abassi. 2021. "Angiotensin-(1-7)—A Potential Remedy for AKI: Insights Derived from the COVID-19 Pandemic" Journal of Clinical Medicine 10, no. 6: 1200. https://doi.org/10.3390/jcm10061200
APA StyleHeyman, S. N., Walther, T., & Abassi, Z. (2021). Angiotensin-(1-7)—A Potential Remedy for AKI: Insights Derived from the COVID-19 Pandemic. Journal of Clinical Medicine, 10(6), 1200. https://doi.org/10.3390/jcm10061200