
Renal Manifestations of Covid-19: Physiology and Pathophysiology

{kind=link}
{kind=link}
Abstract
:1. Introduction
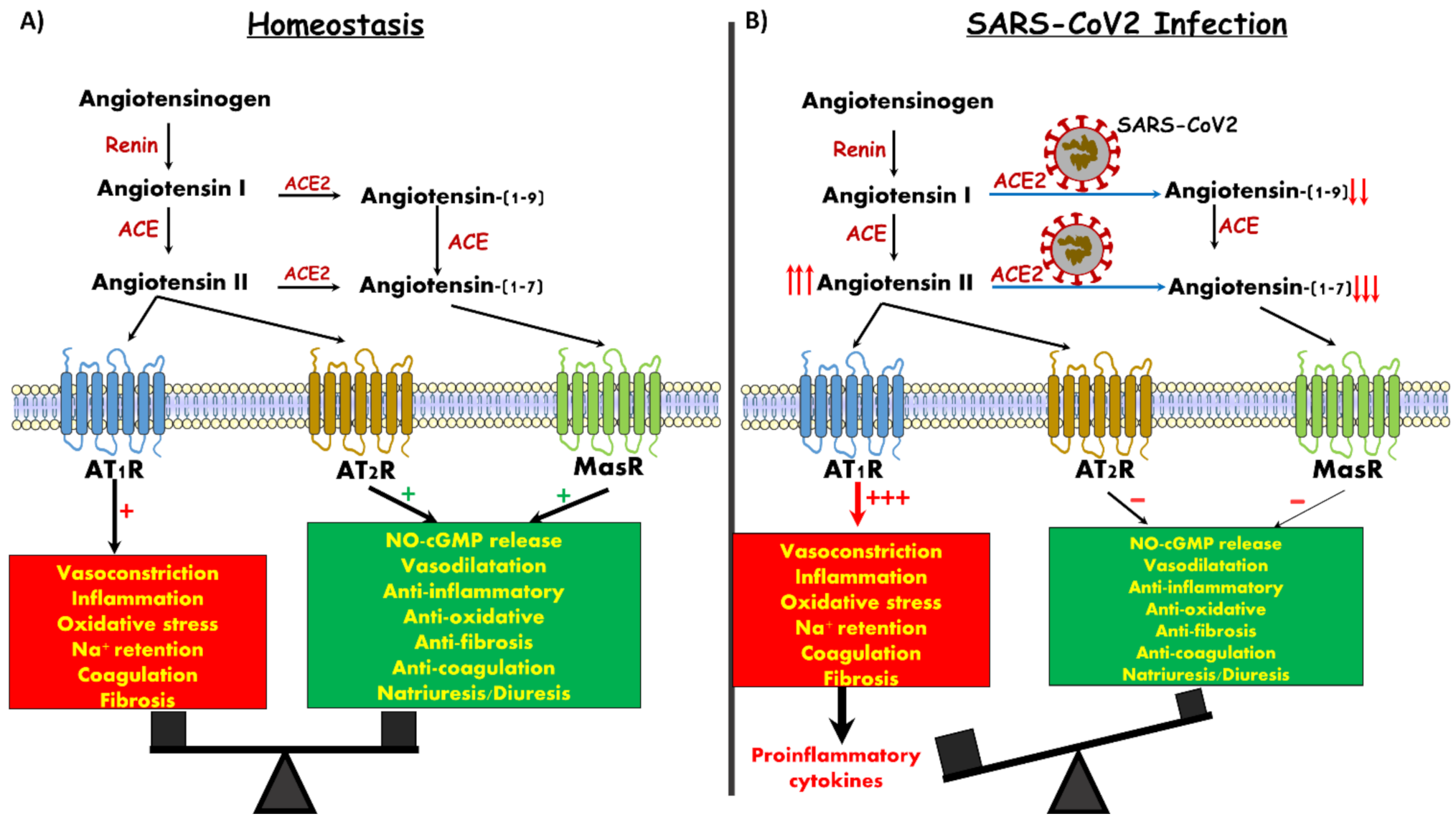
2. Angiotensin Converting Enzyme 2 and Kidney Function
3. Angiotensin Converting Enzyme 2 and Kidney Diseases
4. COVID-19
5. The Cardinal Clinical Manifestations
6. Laboratory Tests
7. Kidney Involvement
8. The Etiology of COVID-19-Induced AKI
9. Treatment
10. Renal Replacement Therapy
11. Kidney Transplanted Patients
12. Use of RAAS Blockers among COVID-19
- Continue ARBs or ACE inhibitors in patients with indications.
- Do not start ARBs or ACE inhibitors solely for COVID-19.
13. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.; Winaver, J.; Skorecki, K. Control of extracellular fluid volume and pathophysiology of edema formation. In The Kidney, 7th ed.; Brenner., B., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2003; pp. 777–855. [Google Scholar]
- Akhmerov, A.; Marban, E. COVID-19 and the Heart. Circ. Res. 2020, 126, 1443–1455. [Google Scholar] [CrossRef] [Green Version]
- Bader, M. ACE2, angiotensin-(1–7), and Mas: The other side of the coin. Pflügers Archiv. Eur. J. Physiol. 2013, 465, 79–85. [Google Scholar] [CrossRef]
- Clarke, N.E.; Turner, A.J. Angiotensin-Converting Enzyme 2: The First Decade. Int. J. Hypertens. 2012, 2012, 307315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.A.; Ferreira, A.J.; Simoes, E.S.A.C. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp. Physiol. 2008, 93, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [Green Version]
- Ingelfinger, J.R. ACE2: A new target for prevention of diabetic nephropathy? J. Am. Soc. Nephrol. 2006, 17, 2957–2959. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A Novel Angiotensin-Converting Enzyme–Related Carboxypeptidase (ACE2) Converts Angiotensin I to Angiotensin 1-9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yi, Y.; Luo, X.; Xiong, N.; Liu, Y.; Li, S.; Sun, R.; Wang, Y.; Hu, B.; Chen, W.; et al. Development and clinical application of a rapid IgM-IgG combined antibody test for SARS-CoV-2 infection diagnosis. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Abassi, Z.; Assady, S.; Khoury, E.E.; Heyman, S.N. Letter to the Editor: Angiotensin-converting enzyme 2: An ally or a Trojan horse? Implications to SARS-CoV-2-related cardiovascular complications. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1080–H1083. [Google Scholar] [CrossRef]
- Abassi, Z.A.; Skorecki, K.; Heyman, S.N.; Kinaneh, S.; Armaly, Z. Covid-19 infection and mortality: A physiologist’s perspective enlightening clinical features and plausible interventional strategies. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1020–L1022. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Wysocki, J.; William, J.; Soler, M.J.; Cokic, I.; Batlle, D. Glomerular Localization and Expression of Angiotensin-Converting Enzyme 2 and Angiotensin-Converting Enzyme: Implications for Albuminuria in Diabetes. J. Am. Soc. Nephrol. 2006, 17, 3067–3075. [Google Scholar] [CrossRef] [Green Version]
- Lely, A.; Hamming, I.; van Goor, H.; Navis, G. Renal ACE2 expression in human kidney disease. J. Pathol. 2004, 204, 587–593. [Google Scholar] [CrossRef] [PubMed]
- da Silveira, K.D.; Pompermayer Bosco, K.S.; Diniz, L.R.; Carmona, A.K.; Cassali, G.D.; Bruna-Romero, O.; de Sousa, L.P.; Teixeira, M.M.; Santos, R.A.; Simoes e Silva, A.C.; et al. ACE2-angiotensin-(1-7)-Mas axis in renal ischaemia/reperfusion injury in rats. Clin. Sci. (Lond.) 2010, 119, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappel, M.C.; Ferrario, C.M. ACE and ACE2: Their role to balance the expression of angiotensin II and angiotensin-(1-7). Kidney Int. 2006, 70, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampaio, W.O.; Nascimento, A.A.S.; Santos, R.A.S. Systemic and regional hemodynamic effects of angiotensin-(1–7) in rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1985–H1994. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Garvin, J.L.; Carretero, O.A. Vasodilator action of angiotensin-(1-7) on isolated rabbit afferent arterioles. Hypertension 2002, 39, 799–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara, L.S.; Bica, R.B.; Sena, S.L.; Correa, J.S.; Marques-Fernandes, M.F.; Lopes, A.G.; Caruso-Neves, C. Angiotensin-(1-7) reverts the stimulatory effect of angiotensin II on the proximal tubule Na(+)-ATPase activity via a A779-sensitive receptor. Regul. Pept. 2002, 103, 17–22. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Silva, A.C.S.e.; Magaldi, A.J.; Khosla, M.C.; Cesar, K.R.; Passaglio, K.T.; Baracho, N.C.V. Evidence for a Physiological Role of Angiotensin-(1-7) in the Control of Hydroelectrolyte Balance. Hypertension 1996, 27, 875–884. [Google Scholar] [CrossRef]
- Magaldi, A.J.; Cesar, K.R.; de Araújo, M.; Simões e Silva, A.C.; Santos, R.A.S. Angiotensin-(1–7) stimulates water transport in rat inner medullary collecting duct: Evidence for involvement of vasopressin V2 receptors. Pflügers Arch. 2003, 447, 223–230. [Google Scholar] [CrossRef]
- Vallon, V.; Heyne, N.; Richter, K.; Khosla, M.C.; Fechter, K. [7-D-ALA]-angiotensin 1-7 blocks renal actions of angiotensin 1-7 in the anesthetized rat. J. Cardiovasc. Pharm. 1998, 32, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Simoes e Silva, A.C.; Bello, A.P.; Baracho, N.C.; Khosla, M.C.; Santos, R.A. Diuresis and natriuresis produced by long term administration of a selective Angiotensin-(1-7) antagonist in normotensive and hypertensive rats. Regul. Pept. 1998, 74, 177–184. [Google Scholar] [CrossRef]
- Su, Z.; Zimpelmann, J.; Burns, K.D. Angiotensin-(1-7) inhibits angiotensin II-stimulated phosphorylation of MAP kinases in proximal tubular cells. Kidney Int. 2006, 69, 2212–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, C.M.; Varagic, J. The ANG-(1-7)/ACE2/mas axis in the regulation of nephron function. Am. J. Physiol Ren. Physiol. 2010, 298, F1297–F1305. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, S.V.B.; Ferreira, A.J.; Kitten, G.T.; da Silveira, K.D.; da Silva, D.A.; Santos, S.H.S.; Gava, E.; Castro, C.H.; Magalhaes, J.A.; da Mota, R.K.; et al. Genetic deletion of the angiotensin-(1-7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int. 2009, 75, 1184–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, S.V.; Simoes, E.S.A.C. Angiotensin converting enzyme 2, Angiotensin-(1-7), and receptor MAS axis in the kidney. Int. J. Hypertens. 2012, 2012, 414128. [Google Scholar] [CrossRef] [Green Version]
- Esteban, V.; Heringer-Walther, S.; Sterner-Kock, A.; de Bruin, R.; van den Engel, S.; Wang, Y.; Mezzano, S.; Egido, J.; Schultheiss, H.P.; Ruiz-Ortega, M.; et al. Angiotensin-(1-7) and the g protein-coupled receptor MAS are key players in renal inflammation. PLoS ONE 2009, 4, e5406. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.D. The emerging role of angiotensin-converting enzyme-2 in the kidney. Curr. Opin. Nephrol. Hypertens 2007, 16, 116–121. [Google Scholar] [CrossRef]
- Konoshita, T.; Wakahara, S.; Mizuno, S.; Motomura, M.; Aoyama, C.; Makino, Y.; Kawai, Y.; Kato, N.; Koni, I.; Miyamori, I.; et al. Tissue gene expression of renin-angiotensin system in human type 2 diabetic nephropathy. Diabetes Care 2006, 29, 848–852. [Google Scholar] [CrossRef] [Green Version]
- Mizuiri, S.; Hemmi, H.; Arita, M.; Ohashi, Y.; Tanaka, Y.; Miyagi, M.; Sakai, K.; Ishikawa, Y.; Shibuya, K.; Hase, H.; et al. Expression of ACE and ACE2 in individuals with diabetic kidney disease and healthy controls. Am. J. Kidney Dis. 2008, 51, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, J.; Ye, M.; Soler, M.J.; Gurley, S.B.; Xiao, H.D.; Bernstein, K.E.; Coffman, T.M.; Chen, S.; Batlle, D. ACE and ACE2 activity in diabetic mice. Diabetes 2006, 55, 2132–2139. [Google Scholar] [CrossRef] [Green Version]
- Tikellis, C.; Johnston, C.I.; Forbes, J.M.; Burns, W.C.; Burrell, L.M.; Risvanis, J.; Cooper, M.E. Characterization of renal angiotensin-converting enzyme 2 in diabetic nephropathy. Hypertension 2003, 41, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, M.J.; Wysocki, J.; Ye, M.; Lloveras, J.; Kanwar, Y.; Batlle, D. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007, 72, 614–623. [Google Scholar] [CrossRef] [Green Version]
- Oudit, G.Y.; Herzenberg, A.M.; Kassiri, Z.; Wong, D.; Reich, H.; Khokha, R.; Crackower, M.A.; Backx, P.H.; Penninger, J.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am. J. Pathol. 2006, 168, 1808–1820. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.W.; Oudit, G.Y.; Reich, H.; Kassiri, Z.; Zhou, J.; Liu, Q.C.; Backx, P.H.; Penninger, J.M.; Herzenberg, A.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 (Ace2) accelerates diabetic kidney injury. Am. J. Pathol. 2007, 171, 438–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benter, I.F.; Yousif, M.H.; Dhaunsi, G.S.; Kaur, J.; Chappell, M.C.; Diz, D.I. Angiotensin-(1-7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am. J. Nephrol. 2008, 28, 25–33. [Google Scholar] [CrossRef]
- Giani, J.F.; Mayer, M.A.; Munoz, M.C.; Silberman, E.A.; Hocht, C.; Taira, C.A.; Gironacci, M.M.; Turyn, D.; Dominici, F.P. Chronic infusion of angiotensin-(1-7) improves insulin resistance and hypertension induced by a high-fructose diet in rats. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E262–E271. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.Y.; Tanimoto, M.; Gohda, T.; Hagiwara, S.; Yamazaki, T.; Ohara, I.; Murakoshi, M.; Aoki, T.; Ishikawa, Y.; Lee, S.H.; et al. Attenuating effect of angiotensin-(1-7) on angiotensin II-mediated NAD(P)H oxidase activation in type 2 diabetic nephropathy of KK-A(y)/Ta mice. Am. J. Physiol. Ren. Physiol. 2011, 300, F1271–F1282. [Google Scholar] [CrossRef]
- Barroso, L.C.; Silveira, K.D.; Lima, C.X.; Borges, V.; Bader, M.; Rachid, M.; Santos, R.A.; Souza, D.G.; Simoes, E.S.A.C.; Teixeira, M.M. Renoprotective Effects of AVE0991, a Nonpeptide Mas Receptor Agonist, in Experimental Acute Renal Injury. Int. J. Hypertens. 2012, 2012, 808726. [Google Scholar] [CrossRef] [PubMed]
- Gurley, S.B.; Allred, A.; Le, T.H.; Griffiths, R.; Mao, L.; Philip, N.; Haystead, T.A.; Donoghue, M.; Breitbart, R.E.; Acton, S.L.; et al. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J. Clin. Invest. 2006, 116, 2218–2225. [Google Scholar] [CrossRef] [Green Version]
- Tikellis, C.; Cooper, M.E.; Bialkowski, K.; Johnston, C.I.; Burns, W.C.; Lew, R.A.; Smith, A.I.; Thomas, M.C. Developmental expression of ACE2 in the SHR kidney: A role in hypertension? Kidney Int. 2006, 70, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessup, J.A.; Gallagher, P.E.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Chappell, M.C.; Ferrario, C.M. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic Ren-2 rats. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2166–H2172. [Google Scholar] [CrossRef]
- Koka, V.; Huang, X.R.; Chung, A.C.; Wang, W.; Truong, L.D.; Lan, H.Y. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am. J. Pathol. 2008, 172, 1174–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakahara, S.; Konoshita, T.; Mizuno, S.; Motomura, M.; Aoyama, C.; Makino, Y.; Kato, N.; Koni, I.; Miyamori, I. Synergistic expression of angiotensin-converting enzyme (ACE) and ACE2 in human renal tissue and confounding effects of hypertension on the ACE to ACE2 ratio. Endocrinology 2007, 148, 2453–2457. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Guo, D.; Chen, C.B.; Wang, W.; Schuster, M.; Loibner, H.; Penninger, J.M.; Scholey, J.W.; Kassiri, Z.; Oudit, G.Y. Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension 2011, 57, 314–322. [Google Scholar] [CrossRef]
- Simoes e Silva, A.C.; Diniz, J.S.; Pereira, R.M.; Pinheiro, S.V.; Santos, R.A. Circulating renin Angiotensin system in childhood chronic renal failure: Marked increase of Angiotensin-(1-7) in end-stage renal disease. Pediatr. Res. 2006, 60, 734–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; Lau, S.K.; To, K.K.; Cheng, V.C.; Woo, P.C.; Yuen, K.Y. Middle East respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, T.T.; Shum, M.H.; Zhu, H.C.; Tong, Y.G.; Ni, X.B.; Liao, Y.S.; Wei, W.; Cheung, W.Y.; Li, W.J.; Li, L.F.; et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature 2020. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A. COVID-19 and Dialysis Units: What Do We Know Now and What Should We Do? Am. J. Kidney Dis. 2020. [Google Scholar] [CrossRef]
- Russell, C.D.; Millar, J.E.; Baillie, J.K. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet 2020, 395, 473–475. [Google Scholar] [CrossRef] [Green Version]
- Ronco, C.; Navalesi, P.; Vincent, J.L. Coronavirus epidemic: Preparing for extracorporeal organ support in intensive care. Lancet Respir Med. 2020, 8, 240–241. [Google Scholar] [CrossRef]
- Kutter, J.S.; Spronken, M.I.; Fraaij, P.L.; Fouchier, R.A.; Herfst, S. Transmission routes of respiratory viruses among humans. Curr. Opin. Virol. 2018, 28, 142–151. [Google Scholar] [CrossRef]
- Pica, N.; Bouvier, N.M. Environmental factors affecting the transmission of respiratory viruses. Curr. Opin. Virol. 2012, 2, 90–95. [Google Scholar] [CrossRef]
- Fernstrom, A.; Goldblatt, M. Aerobiology and its role in the transmission of infectious diseases. J. Pathog. 2013, 2013, 493960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowen, A.C.; Mubareka, S.; Steel, J.; Palese, P. Influenza virus transmission is dependent on relative humidity and temperature. PLoS Pathog. 2007, 3, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Harper, G.J. Airborne micro-organisms: Survival tests with four viruses. J. Hyg. (Lond.) 1961, 59, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.W. The effect of environmental parameters on the survival of airborne infectious agents. J. R. Soc. Interface 2009, 6 (Suppl. S6), S737–S746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Piccoli, G.B. Hospitals as health factories and the coronavirus epidemic. J. Nephrol. 2020, 33, 189–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Mahase, E. Coronavirus covid-19 has killed more people than SARS and MERS combined, despite lower case fatality rate. BMJ 2020, 368, m641. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Luo, R.; Wang, K.; Zhang, M.; Wang, Z.; Dong, L.; Li, J.; Yao, Y.; Ge, S.; Xu, G. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020, 97, 829–838. [Google Scholar] [CrossRef]
- Li, Z.; Wu, M.; Yao, J.; Guo, J.; Liao, X.; Song, S.; Li, J.; Duan, G.; Zhou, Y.; Wu, X.; et al. Caution on Kidney Dysfunctions of COVID-19 Patients. medRxiv 2002. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Xie, J.; Tong, Z.; Guan, X.; Du, B.; Qiu, H.; Slutsky, A.S. Critical care crisis and some recommendations during the COVID-19 epidemic in China. Intensive Care Med. 2020, 46, 837–840. [Google Scholar] [CrossRef] [Green Version]
- Young, B.E.; Ong, S.W.X.; Kalimuddin, S.; Low, J.G.; Tan, S.Y.; Loh, J.; Ng, O.T.; Marimuthu, K.; Ang, L.W.; Mak, T.M.; et al. Epidemiologic Features and Clinical Course of Patients Infected With SARS-CoV-2 in Singapore. JAMA 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cangemi, R.; Della Valle, P.; Calvieri, C.; Taliani, G.; Ferroni, P.; Falcone, M.; Carnevale, R.; Bartimoccia, S.; D’Angelo, A.; Violi, F. Low-grade endotoxemia and clotting activation in the early phase of pneumonia. Respirology 2016, 21, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Carnevale, R.; Calvieri, C.; Nocella, C.; Falcone, M.; Farcomeni, A.; Taliani, G.; Cangemi, R. Nox2 up-regulation is associated with an enhanced risk of atrial fibrillation in patients with pneumonia. Thorax 2015, 70, 961–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempfle, C.E. Use of D-dimer assays in the diagnosis of venous thrombosis. Semin. Thromb. Hemost. 2000, 26, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Falcone, M.; Russo, A.; Cangemi, R.; Farcomeni, A.; Calvieri, C.; Barilla, F.; Scarpellini, M.G.; Bertazzoni, G.; Palange, P.; Taliani, G.; et al. Lower mortality rate in elderly patients with community-onset pneumonia on treatment with aspirin. J. Am. Heart Assoc. 2015, 4, e001595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, X.; Chen, H.; Yan, S.; Li, D.; Li, Y.; Gong, Z. Coronavirus Disease 19 Infection Does Not Result in Acute Kidney Injury: An Analysis of 116 Hospitalized Patients from Wuhan, China. Am. J. Nephrol. 2020, 51, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; Liu, Y.; Liu, Y.; et al. Human Kidney is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. medRxiv 2003. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, J.S.; Ng, J.H.; Ross, D.W.; Sharma, P.; Shah, H.H.; Barnett, R.L.; Hazzan, A.D.; Fishbane, S.; Jhaveri, K.D.; Abate, M.; et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020. [Google Scholar] [CrossRef]
- Yao, X.; Li, T.; He, Z.; Ping, Y.; Liu, H.; Yu, S.; Mou, H.; Wang, L.; Zhang, H.; Fu, W.; et al. A pathological report of three COVID-19 cases by minimal invasive autopsies. Zhonghua Bing Li Xue Za Zhi Chin. J. Pathol. 2020, 49, E009. [Google Scholar] [CrossRef]
- Kissling, S.; Rotman, S.; Gerber, C.; Halfon, M.; Lamoth, F.; Comte, D.; Lhopitallier, L.; Sadallah, S.; Fakhouri, F. Collapsing glomerulopathy in a COVID-19 patient. Kidney Int. 2020, 98, 228–231. [Google Scholar] [CrossRef]
- Larsen, C.P.; Bourne, T.D.; Wilson, J.D.; Saqqa, O.; Sharshir, M.A. Collapsing Glomerulopathy in a Patient With COVID-19. Kidney Int. Rep. 2020, 5, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Peleg, Y.; Kudose, S.; D’Agati, V.; Siddall, E.; Ahmad, S.; Kisselev, S.; Gharavi, A.; Canetta, P. Acute Kidney Injury Due to Collapsing Glomerulopathy Following COVID-19 Infection. Kidney Int. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Larsen, C.P.; Hernandez-Arroyo, C.F.; Mohamed, M.M.B.; Caza, T.; Sharshir, M.d.; Chughtai, A.; Xie, L.; Gimenez, J.M.; Sandow, T.A.; et al. AKI and Collapsing Glomerulopathy Associated with COVID-19 and APOL1 High-Risk Genotype. J. Am. Soc. Nephrol. 2020, 31, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Rolain, J.M.; Lagier, J.C.; Brouqui, P.; Raoult, D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int. J. Antimicrob. Agents 2020, 55, 105932. [Google Scholar] [CrossRef]
- Yan, Y.; Zou, Z.; Sun, Y.; Li, X.; Xu, K.F.; Wei, Y.; Jin, N.; Jiang, C. Anti-malaria drug chloroquine is highly effective in treating avian influenza A H5N1 virus infection in an animal model. Cell Res. 2013, 23, 300–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases? Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [Green Version]
- Boulware, D.R.; Pullen, M.F.; Bangdiwala, A.S.; Pastick, K.A.; Lofgren, S.M.; Okafor, E.C.; Skipper, C.P.; Nascene, A.A.; Nicol, M.R.; Abassi, M.; et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Cao, Z.; Han, M.; Wang, Z.; Chen, J.; Sun, W.; Wu, Y.; Xiao, W.; Liu, S.; Chen, E.; et al. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: Open label, randomised controlled trial. BMJ 2020, 369, m1849. [Google Scholar] [CrossRef]
- Chen, J.; Liu, D.; Liu, L.; Liu, P.; Xu, Q.; Xia, L.; Ling, Y.; Huang, D.; Song, S.; Zhang, D.; et al. [A pilot study of hydroxychloroquine in treatment of patients with moderate COVID-19]. Zhejiang Da Xue Xue Bao Yi Xue Ban 2020, 49, 215–219. [Google Scholar] [PubMed]
- Magagnoli, J.; Narendran, S.; Pereira, F.; Cummings, T.; Hardin, J.W.; Sutton, S.S.; Ambati, J. Outcomes of hydroxychloroquine usage in United States veterans hospitalized with Covid-19. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- America, I.D.S.o. Infectious Diseases Society of America Guidelines on the Treatment and Management of Patients with COVID-19. Available online: https://www.idsociety.org/practice-guideline/covid-19-guideline-treatment-and-management (accessed on 1 April 2020).
- Health, N.I.O. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/ (accessed on 1 April 2020).
- Geleris, J.; Sun, Y.; Platt, J.; Zucker, J.; Baldwin, M.; Hripcsak, G.; Labella, A.; Manson, D.; Kubin, C.; Barr, R.G.; et al. Observational Study of Hydroxychloroquine in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Pan, K.; Wu, B.; Li, X.; Chen, Z.; Xu, Q.; Lv, Q. Safety of hydroxychloroquine in COVID-19 and other diseases: A systematic review and meta-analysis of 53 randomized trials. Eur J. Clin. Pharm. 2020. [Google Scholar] [CrossRef]
- Misra, S.; Nath, M.; Hadda, V.; Vibha, D. Efficacy of various treatment modalities for nCOV-2019: A systematic review and meta-analysis. Eur J. Clin. Invest. 2020, e13383. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H.; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12, eabb5883. [Google Scholar] [CrossRef] [Green Version]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19 — Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Chu, C.M.; Poon, L.L.; Cheng, V.C.; Chan, K.S.; Hung, I.F.; Wong, M.M.; Chan, K.H.; Leung, W.S.; Tang, B.S.; Chan, V.L.; et al. Initial viral load and the outcomes of SARS. CMAJ 2004, 171, 1349–1352. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.S.; Lai, S.T.; Chu, C.M.; Tsui, E.; Tam, C.Y.; Wong, M.M.; Tse, M.W.; Que, T.L.; Peiris, J.S.; Sung, J.; et al. Treatment of severe acute respiratory syndrome with lopinavir/ritonavir: A multicentre retrospective matched cohort study. Hong Kong Med. J. 2003, 9, 399–406. [Google Scholar] [PubMed]
- Zhang, C.; Wu, Z.; Li, J.W.; Zhao, H.; Wang, G.Q. Cytokine release syndrome in severe COVID-19: Interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int. J. Antimicrob. Agents 2020, 55, 105954. [Google Scholar] [CrossRef] [PubMed]
- Veronese, N.; Demurtas, J.; Yang, L.; Tonelli, R.; Barbagallo, M.; Lopalco, P.; Lagolio, E.; Celotto, S.; Pizzol, D.; Zou, L.; et al. Use of Corticosteroids in Coronavirus Disease 2019 Pneumonia: A Systematic Review of the Literature. Front. Med. (Lausanne) 2020, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Arabi, Y.M.; Mandourah, Y.; Al-Hameed, F.; Sindi, A.A.; Almekhlafi, G.A.; Hussein, M.A.; Jose, J.; Pinto, R.; Al-Omari, A.; Kharaba, A.; et al. Corticosteroid Therapy for Critically Ill Patients with Middle East Respiratory Syndrome. Am. J. Respir. Crit. Care Med. 2018, 197, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- WHO. WHO Updates Clinical Care Guidance with Corticosteroid Recommendations. 20 April. Available online: https://www.who.int/news-room/feature-stories/detail/who-updates-clinical-care-guidance-with-corticosteroid-recommendations (accessed on 1 April 2020).
- Mair-Jenkins, J.; Saavedra-Campos, M.; Baillie, J.K.; Cleary, P.; Khaw, F.M.; Lim, W.S.; Makki, S.; Rooney, K.D.; Nguyen-Van-Tam, J.S.; Beck, C.R. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: A systematic review and exploratory meta-analysis. J. Infect. Dis. 2015, 211, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Zhou, J.; Huang, Y.; Liu, X.; Xu, Y.; Chen, S.; Liu, D.; Lin, Z.; Liu, X.; Li, Y. The efficacy of convalescent plasma for the treatment of severe influenza. medRxiv 2002. [Google Scholar] [CrossRef]
- Naicker, S.; Yang, C.W.; Hwang, S.J.; Liu, B.C.; Chen, J.H.; Jha, V. The Novel Coronavirus 2019 epidemic and kidneys. Kidney Int. 2020, 97, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.H.; Tsang, W.K.; Tang, C.S.; Lam, M.F.; Lai, F.M.; To, K.F.; Fung, K.S.; Tang, H.L.; Yan, W.W.; Chan, H.W.; et al. Acute renal impairment in coronavirus-associated severe acute respiratory syndrome. Kidney Int. 2005, 67, 698–705. [Google Scholar] [CrossRef] [Green Version]
- Arabi, Y.M.; Arifi, A.A.; Balkhy, H.H.; Najm, H.; Aldawood, A.S.; Ghabashi, A.; Hawa, H.; Alothman, A.; Khaldi, A.; Al Raiy, B. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann. Intern. Med. 2014, 160, 389–397. [Google Scholar] [CrossRef]
- Ghani, R.A.; Zainudin, S.; Ctkong, N.; Rahman, A.F.; Wafa, S.R.; Mohamad, M.; Manaf, M.R.; Ismail, R. Serum IL-6 and IL-1-ra with sequential organ failure assessment scores in septic patients receiving high-volume haemofiltration and continuous venovenous haemofiltration. Nephrol. (Carlton) 2006, 11, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Ankawi, G.; Xie, Y.; Yang, B.; Xie, Y.; Xie, P.; Ronco, C. What Have We Learned about the Use of Cytosorb Adsorption Columns? Blood Purif. 2019, 48, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Poli, E.C.; Rimmele, T.; Schneider, A.G. Hemoadsorption with CytoSorb((R)). Intensive Care Med. 2019, 45, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Ankawi, G.; Fan, W.; Pomare Montin, D.; Lorenzin, A.; Neri, M.; Caprara, C.; de Cal, M.; Ronco, C. A New Series of Sorbent Devices for Multiple Clinical Purposes: Current Evidence and Future Directions. Blood Purif. 2019, 47, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, S.R.; Su, W.; Liu, J.Y. Removal of humoral mediators and the effect on the survival of septic patients by hemoperfusion with neutral microporous resin column. Ther. Apher Dial. 2010, 14, 596–602. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, S.R.; Yang, Z.L.; Liu, J.Y. Effect on extrapulmonary sepsis-induced acute lung injury by hemoperfusion with neutral microporous resin column. Ther. Apher Dial. 2013, 17, 454–461. [Google Scholar] [CrossRef]
- Ronco, C.; Reis, T.; De Rosa, S. Coronavirus Epidemic and Extracorporeal Therapies in Intensive Care: Si vis pacem para bellum. Blood Purif. 2020, 49, 255–258. [Google Scholar] [CrossRef]
- van der Meer, J.W. The effects of recombinant interleukin-1 and recombinant tumor necrosis factor on non-specific resistance to infection. Biotherapy 1988, 1, 19–25. [Google Scholar] [CrossRef]
- Echtenacher, B.; Falk, W.; Mannel, D.N.; Krammer, P.H. Requirement of endogenous tumor necrosis factor/cachectin for recovery from experimental peritonitis. J. Immunol. 1990, 145, 3762–3766. [Google Scholar]
- Ronco, C.; Bonello, M.; Bordoni, V.; Ricci, Z.; D’Intini, V.; Bellomo, R.; Levin, N.W. Extracorporeal therapies in non-renal disease: Treatment of sepsis and the peak concentration hypothesis. Blood Purif. 2004, 22, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Tetta, C.; Mariano, F.; Wratten, M.L.; Bonello, M.; Bordoni, V.; Cardona, X.; Inguaggiato, P.; Pilotto, L.; d’Intini, V.; et al. Interpreting the mechanisms of continuous renal replacement therapy in sepsis: The peak concentration hypothesis. Artif Organs 2003, 27, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Rimmele, T.; Kellum, J.A. Clinical review: Blood purification for sepsis. Crit. Care 2011, 15, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honore, P.M.; Hoste, E.; Molnar, Z.; Jacobs, R.; Joannes-Boyau, O.; Malbrain, M.; Forni, L.G. Cytokine removal in human septic shock: Where are we and where are we going? Ann. Intensive Care 2019, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Cook, J.D.; Lee, J.E. The secret life of viral entry glycoproteins: Moonlighting in immune evasion. PLoS Pathog. 2013, 9, e1003258. [Google Scholar] [CrossRef]
- Volchkova, V.A.; Dolnik, O.; Martinez, M.J.; Reynard, O.; Volchkov, V.E. Genomic RNA editing and its impact on Ebola virus adaptation during serial passages in cell culture and infection of guinea pigs. J. Infect. Dis. 2011, 204 (Suppl. S3), S941–S946. [Google Scholar] [CrossRef]
- Wilson, J.A.; Hevey, M.; Bakken, R.; Guest, S.; Bray, M.; Schmaljohn, A.L.; Hart, M.K. Epitopes involved in antibody-mediated protection from Ebola virus. Science 2000, 287, 1664–1666. [Google Scholar] [CrossRef]
- Mohan, G.S.; Li, W.; Ye, L.; Compans, R.W.; Yang, C. Antigenic subversion: A novel mechanism of host immune evasion by Ebola virus. PLoS Pathog. 2012, 8, e1003065. [Google Scholar] [CrossRef] [Green Version]
- Escudero-Perez, B.; Volchkova, V.A.; Dolnik, O.; Lawrence, P.; Volchkov, V.E. Shed GP of Ebola virus triggers immune activation and increased vascular permeability. PLoS Pathog. 2014, 10, e1004509. [Google Scholar] [CrossRef]
- Kim, S.Y.; Park, S.J.; Cho, S.Y.; Cha, R.H.; Jee, H.G.; Kim, G.; Shin, H.S.; Kim, Y.; Jung, Y.M.; Yang, J.S.; et al. Viral RNA in Blood as Indicator of Severe Outcome in Middle East Respiratory Syndrome Coronavirus Infection. Emerg. Infect. Dis. 2016, 22, 1813–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knust, B.; Schafer, I.J.; Wamala, J.; Nyakarahuka, L.; Okot, C.; Shoemaker, T.; Dodd, K.; Gibbons, A.; Balinandi, S.; Tumusiime, A.; et al. Multidistrict Outbreak of Marburg Virus Disease-Uganda, 2012. J. Infect. Dis. 2015, 212 (Suppl. S2), S119–S128. [Google Scholar] [CrossRef]
- McElroy, A.K.; Erickson, B.R.; Flietstra, T.D.; Rollin, P.E.; Nichol, S.T.; Towner, J.S.; Spiropoulou, C.F. Ebola hemorrhagic Fever: Novel biomarker correlates of clinical outcome. J. Infect. Dis. 2014, 210, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElroy, A.K.; Erickson, B.R.; Flietstra, T.D.; Rollin, P.E.; Nichol, S.T.; Towner, J.S.; Spiropoulou, C.F. Biomarker correlates of survival in pediatric patients with Ebola virus disease. Emerg. Infect. Dis. 2014, 20, 1683–1690. [Google Scholar] [CrossRef]
- Ewers, E.C.; Pratt, W.D.; Twenhafel, N.A.; Shamblin, J.; Donnelly, G.; Esham, H.; Wlazlowski, C.; Johnson, J.C.; Botto, M.; Hensley, L.E.; et al. Natural History of Aerosol Exposure with Marburg Virus in Rhesus Macaques. Viruses 2016, 8, 87. [Google Scholar] [CrossRef] [Green Version]
- Ajelli, M.; Merler, S. Transmission potential and design of adequate control measures for Marburg hemorrhagic fever. PLoS ONE 2012, 7, e50948. [Google Scholar] [CrossRef] [PubMed]
- ERA-EDTA. ERA-EDTA Working Group Descartes for Patients Living with a Kidney Transplant. Available online: https://www.era-edta.org/en/covid-19-news-and-information/#toggle-id-9 (accessed on 1 April 2020).
- Frequently asked questions from transplant candidates and recipients April 6, 2020. Am. Soc. Transplant. 2020.
- Chen, S.; Yin, Q.; Shi, H.; Du, D.; Chang, S.; Ni, L.; Qiu, H.; Chen, Z.; Zhang, J.; Zhang, W. A familial cluster, including a kidney transplant recipient, of Coronavirus Disease 2019 (COVID-19) in Wuhan, China. Am. J. Transplant. 2020, 20, 1869–1874. [Google Scholar] [CrossRef] [PubMed]
- Guillen, E.; Pineiro, G.J.; Revuelta, I.; Rodriguez, D.; Bodro, M.; Moreno, A.; Campistol, J.M.; Diekmann, F.; Ventura-Aguiar, P. Case report of COVID-19 in a kidney transplant recipient: Does immunosuppression alter the clinical presentation? Am. J. Transplant. 2020, 20, 1875–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfini, I.; Delsante, M.; Fiaccadori, E.; Zaza, G.; Manenti, L.; Degli Antoni, A.; Peruzzi, L.; Riella, L.V.; Cravedi, P.; Maggiore, U. COVID-19 in kidney transplant recipients. Am. J. Transplant. 2020, 20, 1941–1943. [Google Scholar] [CrossRef]
- Zhu, L.; Xu, X.; Ma, K.; Yang, J.; Guan, H.; Chen, S.; Chen, Z.; Chen, G. Successful recovery of COVID-19 pneumonia in a renal transplant recipient with long-term immunosuppression. Am. J. Transplant. 2020, 20, 1859–1863. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Torres, J.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Akalin, E.; Azzi, Y.; Bartash, R.; Seethamraju, H.; Parides, M.; Hemmige, V.; Ross, M.; Forest, S.; Goldstein, Y.D.; Ajaimy, M.; et al. Covid-19 and Kidney Transplantation. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Alberici, F.; Delbarba, E.; Manenti, C.; Econimo, L.; Valerio, F.; Pola, A.; Maffei, C.; Possenti, S.; Zambetti, N.; Moscato, M.; et al. A single center observational study of the clinical characteristics and short-term outcome of 20 kidney transplant patients admitted for SARS-CoV2 pneumonia. Kidney Int. 2020, 97, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, S.; Michelli, A.; Zuolo, G.; Candido, R.; Fabris, B. Update on RAAS Modulation for the Treatment of Diabetic Cardiovascular Disease. J. Diabetes Res. 2016, 2016, 8917578. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Chen, J.; Wang, X.; Zhang, F.; Liu, Y. Age- and gender-related difference of ACE2 expression in rat lung. Life Sci. 2006, 78, 2166–2171. [Google Scholar] [CrossRef]
- Gallagher, P.E.; Ferrario, C.M.; Tallant, E.A. MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am. J. Physiol. Cell Physiol. 2008, 295, C1169–C1174. [Google Scholar] [CrossRef] [Green Version]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- MaassenVanDenBrink, A.; de Vries, T.; Danser, A.H.J. Headache medication and the COVID-19 pandemic. J. Headache Pain 2020, 21, 38. [Google Scholar] [CrossRef] [Green Version]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Xie, Z.; Li, T.; Zhang, S.; Lai, C.; Zhu, P.; Wang, K.; Han, L.; Duan, Y.; Zhao, Z.; et al. Angiotensin-converting enzyme 2 inhibits lung injury induced by respiratory syncytial virus. Sci. Rep. 2016, 6, 19840. [Google Scholar] [CrossRef]
- Zou, Z.; Yan, Y.; Shu, Y.; Gao, R.; Sun, Y.; Li, X.; Ju, X.; Liang, Z.; Liu, Q.; Zhao, Y.; et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat. Commun. 2014, 5, 3594. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Campbell, D.J.; Zeitz, C.J.; Esler, M.D.; Horowitz, J.D. Evidence against a major role for angiotensin converting enzyme-related carboxypeptidase (ACE2) in angiotensin peptide metabolism in the human coronary circulation. J. Hypertens. 2004, 22, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Luque, M.; Martin, P.; Martell, N.; Fernandez, C.; Brosnihan, K.B.; Ferrario, C.M. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J. Hypertens. 1996, 14, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Ramchand, J.; Patel, S.K.; Srivastava, P.M.; Farouque, O.; Burrell, L.M. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS ONE 2018, 13, e0198144. [Google Scholar] [CrossRef]
- Ramchand, J.; Patel, S.K.; Kearney, L.G.; Matalanis, G.; Farouque, O.; Srivastava, P.M.; Burrell, L.M. Plasma ACE2 Activity Predicts Mortality in Aortic Stenosis and Is Associated With Severe Myocardial Fibrosis. JACC Cardiovasc. Imaging 2020, 13, 655–664. [Google Scholar] [CrossRef]
- Walters, T.E.; Kalman, J.M.; Patel, S.K.; Mearns, M.; Velkoska, E.; Burrell, L.M. Angiotensin converting enzyme 2 activity and human atrial fibrillation: Increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Europace 2017, 19, 1280–1287. [Google Scholar] [CrossRef]
- Epelman, S.; Shrestha, K.; Troughton, R.W.; Francis, G.S.; Sen, S.; Klein, A.L.; Tang, W.H. Soluble angiotensin-converting enzyme 2 in human heart failure: Relation with myocardial function and clinical outcomes. J. Card. Fail. 2009, 15, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Reddy, R.; Asante, I.; Liu, S.; Parikh, P.; Liebler, J.; Borok, Z.; Rodgers, K.; Baydur, A.; Louie, S.G. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS ONE 2019, 14, e0213096. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armaly, Z.; Kinaneh, S.; Skorecki, K. Renal Manifestations of Covid-19: Physiology and Pathophysiology. J. Clin. Med. 2021, 10, 1216. https://doi.org/10.3390/jcm10061216
Armaly Z, Kinaneh S, Skorecki K. Renal Manifestations of Covid-19: Physiology and Pathophysiology. Journal of Clinical Medicine. 2021; 10(6):1216. https://doi.org/10.3390/jcm10061216
Chicago/Turabian StyleArmaly, Zaher, Safa Kinaneh, and Karl Skorecki. 2021. "Renal Manifestations of Covid-19: Physiology and Pathophysiology" Journal of Clinical Medicine 10, no. 6: 1216. https://doi.org/10.3390/jcm10061216
APA StyleArmaly, Z., Kinaneh, S., & Skorecki, K. (2021). Renal Manifestations of Covid-19: Physiology and Pathophysiology. Journal of Clinical Medicine, 10(6), 1216. https://doi.org/10.3390/jcm10061216