
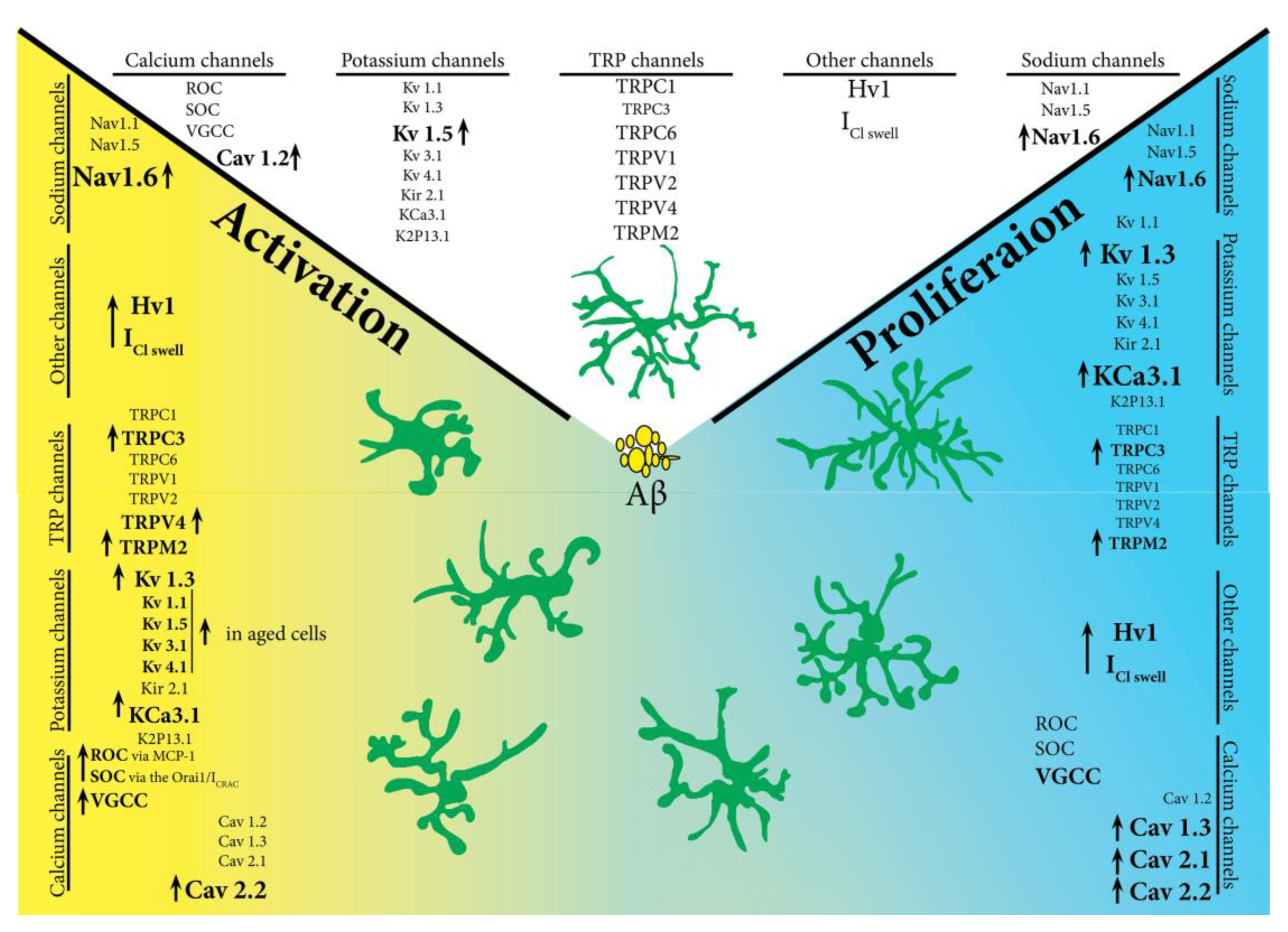
Roles of Microglial Ion Channel in Neurodegenerative Diseases


 , ,
, ,  and
and 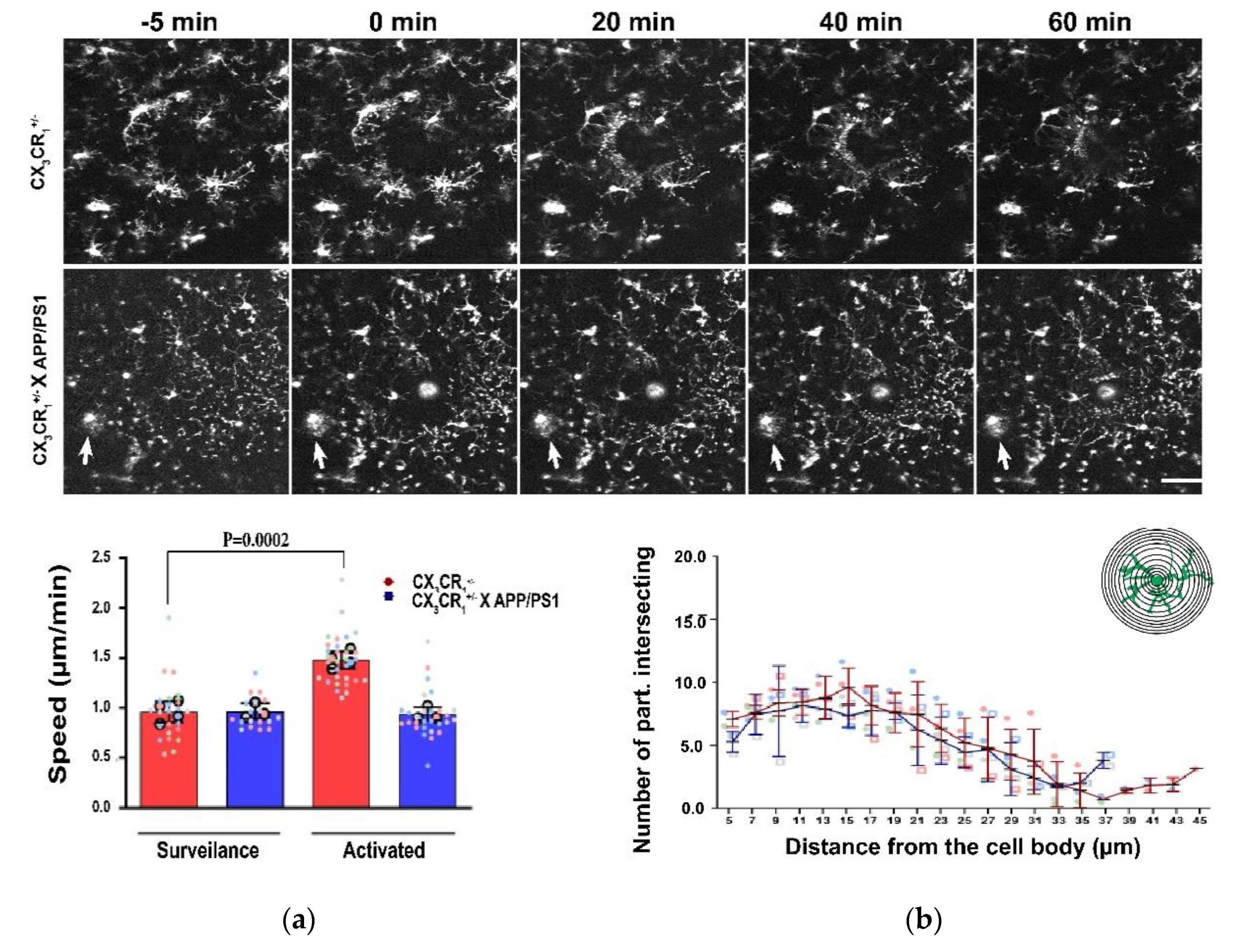{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Microglia Potassium Channels
3. Microglia Calcium Channels
4. Microglia TRP Channels
5. Microglia Voltage-Gated Sodium Channels
6. Other Microglial Channels
7. Ion Channels Modulation in Neurodegenerative Diseases. In Vivo Studies
8. The Future of Microglia Ion Channels in Pathology
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Baron, J.-C.; Yamauchi, H.; Fujioka, M.; Endres, M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 34, 2–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.Q.; Zhao, Y.-Y.; Cao, W.; Wei, Z.Z.; Gu, X.; Wei, L.; Yu, S.P. Long-term survival and regeneration of neuronal and vasculature cells inside the core region after ischemic stroke in adult mice. Brain Pathol. 2016, 27, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Cătălin, B.; Cupido, A.; Iancău, M.; Albu, C.V.; Kirchhoff, F. Microglia: First responders in the central nervous system. Romanian J. Morphol. Embryol. Rev. Roum. Morphol. Embryol. 2013, 54, 467–472. [Google Scholar]
- Black, J.A.; Liu, S.; Carrithers, M.; Carrithers, L.M.; Waxman, S.G. Exacerbation of experimental autoimmune encephalomyelitis after withdrawal of phenytoin and carbamazepine. Ann. Neurol. 2007, 62, 21–33. [Google Scholar] [CrossRef]
- Wu, W.K.K.; Li, G.R.; Wong, H.P.S.; Hui, M.K.C.; Tai, E.K.K.; Lam, E.K.Y.; Shin, V.Y.; Ni Ye, Y.; Li, P.; Yang, Y.H.; et al. Involvement of Kv1.1 and Nav1.5 in proliferation of gastric epithelial cells. J. Cell. Physiol. 2006, 207, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.K.; Li, G.R.; Wong, T.M.; Wang, J.Y.; Yu, L.; Cho, C.H. Involvement of voltage-gated K+ and Na+ channels in gastric epithelial cell migration. Mol. Cell. Biochem. 2007, 308, 219–226. [Google Scholar] [CrossRef]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colton, C.A.; Wilcock, D.M. Assessing activation states in microglia. CNS Neurol. Disord. Drug Targets 2010, 9, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.-X. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhou, X.-W.; Wang, J.-Z. The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 2016, 5, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Cunha, R.A.; Oliveira, C. Neuroinflammation, oxidative stress and the pathogenesis of alzheimers disease. Curr. Pharm. Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; Di Nunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 Alters immunoproteostasis in app mice, increasing plaque burden and worsening cognitive behavior. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.S.; Ma, J.; Jegathees, T.; Goldsbury, A.C. Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 2016, 27, 795–808. [Google Scholar] [CrossRef]
- Paasila, P.J.; Davies, D.S.; Kril, J.J.; Goldsbury, C.; Sutherland, G.T. The relationship between the morphological subtypes of microglia and Alzheimer’s disease neuropathology. Brain Pathol. 2019, 29, 726–740. [Google Scholar] [CrossRef] [Green Version]
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.-W. Amyloid-β protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 2011, 286, 3693–3706. [Google Scholar] [CrossRef] [Green Version]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Stern, Y. Epidemiology of alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [Green Version]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Wu, Y.-P.; Ling, E.-A. Induction of microglial and astrocytic response in the adult rat lumbar spinal cord following middle cerebral artery occlusion. Exp. Brain Res. 1998, 118, 235–242. [Google Scholar] [CrossRef]
- Kotecha, S.A.; Schlichter, L.C. A Kv1.5 to Kv1.3 Switch in Endogenous Hippocampal Microglia and a Role in Proliferation. J. Neurosci. 1999, 19, 10680–10693. [Google Scholar] [CrossRef] [Green Version]
- Pannasch, U.; Färber, K.; Nolte, C.; Blonski, M.; Chiu, S.Y.; Messing, A.; Kettenmann, H. The potassium channels Kv1.5 and Kv1.3 modulate distinct functions of microglia. Mol. Cell. Neurosci. 2006, 33, 401–411. [Google Scholar] [CrossRef]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef] [Green Version]
- LaLonde, J.; Saia, G.; Gill, G. Store-operated calcium entry promotes the degradation of the transcription factor sp4 in resting neurons. Sci. Signal. 2014, 7, ra51. [Google Scholar] [CrossRef] [Green Version]
- Grissmer, S.; Nguyen, A.N.; Aiyar, J.; Hanson, D.C.; Mather, R.J.; A Gutman, G.; Karmilowicz, M.J.; Auperin, D.D.; Chandy, K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 1994, 45, 1227–1234. [Google Scholar]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.M.; Blomster, L.V.; Christophersen, P.; Wulff, H. Potassium channel expression and function in microglia: Plasticity and possible species variations. Channels 2017, 11, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattlen, C.; Deftu, A.-F.; Tonello, R.; Ling, Y.; Berta, T.; Ristoiu, V.; Suter, M.R. The inhibition of Kir2.1 potassium channels depolarizes spinal microglial cells, reduces their proliferation, and attenuates neuropathic pain. Glia 2020, 68, 2119–2135. [Google Scholar] [CrossRef]
- Lam, D.; Lively, S.; Schlichter, L.C. Responses of rat and mouse primary microglia to pro- and anti-inflammatory stimuli: Molecular profiles, K+ channels and migration. J. Neuroinflamm. 2017, 14, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Anton, R.; Ghenghea, M.; Ristoiu, V.; Gattlen, C.; Suter, M.-R.; Cojocaru, P.; Popa-Wagner, A.; Catalin, B.; Deftu, A.-F. Potassium channels Kv1.3 and Kir2.1 but Not Kv1.5 Contribute to BV2 cell line and primary microglial migration. Int. J. Mol. Sci. 2021, 22, 2081. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Grössinger, E.M.; Horiuchi, M.; Davis, K.W.; Jin, L.-W.; Maezawa, I.; Wulff, H. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017, 65, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Di Lucente, J.; Nguyen, H.M.; Wulff, H.; Jin, L.-W.; Maezawa, I. The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia 2018, 66, 1881–1895. [Google Scholar] [CrossRef]
- Blomster, L.V.; Strøbaek, D.; Hougaard, C.; Klein, J.; Pinborg, L.H.; Mikkelsen, J.D.; Christophersen, P. Quantification of the functional expression of the Ca(2+)-activated K+channel KCa3.1 on microglia from adult human neocortical tissue. Glia 2016, 64, 2065–2078. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, S.; Gearing, M.; Jin, L.-W.; Levey, A. Potassium channel Kv1.3 Is highly expressed by microglia in human alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Rus, H.; Pardo, C.A.; Hu, L.; Darrah, E.; Cudrici, C.; Niculescu, T.; Niculescu, F.; Mullen, K.M.; Allie, R.; Guo, L.; et al. The voltage-gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc. Natl. Acad. Sci. USA 2005, 102, 11094–11099. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-J.; Nguyen, H.M.; Maezawa, I.; Grössinger, E.M.; Garing, A.L.; Köhler, R.; Jin, L.-W.; Wulff, H. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J. Cereb. Blood Flow Metab. 2016, 36, 2146–2161. [Google Scholar] [CrossRef] [Green Version]
- Arnoux, I.; Hoshiko, M.; Mandavy, L.; Avignone, E.; Yamamoto, N.; Audinat, E. Adaptive phenotype of microglial cells during the normal postnatal development of the somatosensory “Barrel” cortex. Glia 2013, 61, 1582–1594. [Google Scholar] [CrossRef]
- Arnoux, I.; Hoshiko, M.; Diez, A.S.; Audinat, E. Paradoxical effects of minocycline in the developing mouse somatosensory cortex. Glia 2013, 62, 399–410. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Microglial K + channel expression in young adult and aged mice. Glia 2015, 63, 664–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maezawa, I.; Nguyen, H.M.; Di Lucente, J.; Jenkins, D.P.; Singh, V.; Hilt, S.; Kim, K.; Rangaraju, S.; I Levey, A.; Wulff, H.; et al. Kv1.3 inhibition as a potential microglia-targeted therapy for Alzheimer’s disease: Preclinical proof of concept. Brain 2017, 141, 596–612. [Google Scholar] [CrossRef] [Green Version]
- Lioudyno, M.I.; Broccio, M.; Sokolov, Y.; Rasool, S.; Wu, J.; Alkire, M.T.; Liu, V.; Kozak, J.A.; Dennison, P.R.; Glabe, C.G.; et al. Effect of Synthetic Aβ Peptide Oligomers and Fluorinated Solvents on Kv1.3 Channel Properties and Membrane Conductance. PLoS ONE 2012, 7, e35090. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Di Lucente, J.; Nguyen, H.M.; Singh, V.; Singh, L.; Chavez, M.; Bushong, T.; Wulff, H.; Maezawa, I. Repurposing the KCa3.1 inhibitor senicapoc for Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Dou, F.; Yu, Z. The potassium channel KCa3.1 represents a valid pharmacological target for microgliosis-induced neuronal impairment in a mouse model of Parkinson’s disease. J. Neuroinflamm. 2019, 16, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franciosi, S.; Ryu, J.K.; Choi, H.B.; Radov, L.; Kim, S.U.; McLarnon, J.G. Broad-spectrum effects of 4-aminopyridine to modulate amyloid beta1-42-Induced cell signaling and functional responses in human microglia. J. Neurosci. 2006, 26, 11652–11664. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Amyloid-β-induced reactive oxygen species production and priming are differentially regulated by ion channels in microglia. J. Cell. Physiol. 2011, 226, 3295–3302. [Google Scholar] [CrossRef]
- Chen, Y.; Nguyen, H.M.; Maezawa, I.; Jin, L.; Wulff, H. Inhibition of the potassium channel Kv1.3 reduces infarction and inflammation in ischemic stroke. Ann. Clin. Transl. Neurol. 2017, 5, 147–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-J.; Raman, G.; Bodendiek, S.; E O’Donnell, M.; Wulff, H. The KCa3.1 Blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J. Cereb. Blood Flow Metab. 2011, 31, 2363–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valerie, N.C.K.; Dziegielewska, B.; Hosing, A.S.; Augustin, E.; Gray, L.S.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. Inhibition of T-type calcium channels disrupts Akt signaling and promotes apoptosis in glioblastoma cells. Biochem. Pharmacol. 2013, 85, 888–897. [Google Scholar] [CrossRef]
- Ertel, E.A.; Campbell, K.P.; Harpold, M.M.; Hofmann, F.; Mori, Y.; Perez-Reyes, E.; Schwartz, A.; Snutch, T.P.; Tanabe, T.; Birnbaumer, L.; et al. Nomenclature of voltage-gated calcium channels. Neuron 2000, 25, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Hashioka, S.; Klegeris, A.; McGeer, P.L. Inhibition of human astrocyte and microglia neurotoxicity by calcium channel blockers. Neuropharmacology 2012, 63, 685–691. [Google Scholar] [CrossRef]
- Nicoletti, N.F.; Erig, T.C.; Zanin, R.F.; Roxo, M.R.; Ferreira, N.P.; Gomez, M.V.; Morrone, F.B.; Campos, M.M. Pre-clinical evaluation of voltage-gated calcium channel blockers derived from the spider P. nigriventer in glioma progression. Toxicon 2017, 129, 58–67. [Google Scholar] [CrossRef]
- Silei, V.; Fabrizi, C.; Venturini, G.; Salmona, M.; Bugiani, O.; Tagliavini, F.; Lauro, G.M. Activation of microglial cells by PrP and β-amyloid fragments raises intracellular calcium through L-type voltage sensitive calcium channels. Brain Res. 1999, 818, 168–170. [Google Scholar] [CrossRef]
- Toescu, E.C.; Möller, T.; Kettenmann, H.; Verkhratsky, A. Long-term activation of capacitative Ca2+ entry in mouse microglial cells. Neuroscience 1998, 86, 925–935. [Google Scholar] [CrossRef]
- Espinosa-Parrilla, J.; Martínez-Moreno, M.; Gasull, X.; Mahy, N.; Rodríguez, M. The L-type voltage-gated calcium channel modulates microglial pro-inflammatory activity. Mol. Cell. Neurosci. 2015, 64, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Mai, A.; Valente, S.; Meade, S.; Carafa, V.; Tardugno, M.; Nebbioso, A.; Galmozzi, A.; Mitro, N.; De Fabiani, E.; Altucci, L.; et al. Study of 1,4-Dihydropyridine structural scaffold: Discovery of novel sirtuin activators and inhibitors. J. Med. Chem. 2009, 52, 5496–5504. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Deng, Y.; Qing, H. Calcium channel blockers and Alzheimer’s disease. Neural Regen. Res. 2012, 7, 137–140. [Google Scholar] [CrossRef]
- Tong, B.C.-K.; Wu, A.J.; Li, M.; Cheung, K.-H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Bachmeier, C.; Beaulieu-Abdelahad, D.; Mullan, M.; Paris, D. Selective dihydropyiridine compounds facilitate the clearance of β-amyloid across the blood–brain barrier. Eur. J. Pharmacol. 2011, 659, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Barhwal, K.; Hota, S.K.; Baitharu, I.; Prasad, D.; Singh, S.B.; Ilavazhagan, G. Isradipine antagonizes hypobaric hypoxia induced CA1 damage and memory impairment: Complementary roles of L-type calcium channel and NMDA receptors. Neurobiol. Dis. 2009, 34, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Copenhaver, P.F.; Anekonda, T.S.; Musashe, D.; Robinson, K.M.; Ramaker, J.M.; Swanson, T.L.; Wadsworth, T.L.; Kretzschmar, R.; Woltjer, R.L.; Quinn, J.F. A translational continuum of model systems for evaluating treatment strategies in Alzheimer’s disease: Isradipine as a candidate drug. Dis. Model. Mech. 2011, 4, 634–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchinetti, F.; Fasolato, C.; DelGiudice, E.; Burgo, A.; Furegato, S.; Fusco, M.; Basso, E.; Seraglia, R.; Darrigo, A.; León, A. Nimodipine selectively stimulates β-amyloid 1–42 secretion by a mechanism independent of calcium influx blockage. Neurobiol. Aging 2006, 27, 218–227. [Google Scholar] [CrossRef]
- Morich, F.J.; Bieber, F.; Lewis, J.M.; Kaiser, L.; Cutler, N.R.; Escobar, J.I.; Willmer, J.; Petersen, R.C.; Reisberg, B. Nimodipine in the Treatment of Probable Alzheimerʼs Disease. Clin. Drug Investig. 1996, 11, 185–195. [Google Scholar] [CrossRef]
- Wang, X.; Saegusa, H.; Huntula, S.; Tanabe, T. Blockade of microglial Cav1.2 Ca2+ channel exacerbates the symptoms in a Parkinson’s disease model. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mogi, M.; Iwai, M.; Chen, R.; Iwanami, J.; Ide, A.; Tsukuda, K.; Yoshii, T.; Horiuchi, M. Amlodipine treatment reduces stroke size in apolipoprotein E–Deficient Mice. Am. J. Hypertens. 2006, 19, 1144–1149. [Google Scholar] [CrossRef] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, C.; Sun, Y.; Huang, W.; Ye, K. Cognitive disorders associated with hospitalization of COVID-19: Results from an observational cohort study. Brain Behav. Immun. 2021, 91, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Frey, T.K.; Yang, J.J. Viral calciomics: Interplays between Ca2+ and virus. Cell Calcium 2009, 46, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cao, R.; Zhong, W. Host calcium channels and pumps in viral infections. Cells 2019, 9, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivier, M. Modulation of host cell intracellular Ca2+. Parasitol. Today 1996, 12, 145–150. [Google Scholar] [CrossRef]
- Danta, C.C. CNS Penetration ability: A critical factor for drugs in the treatment of SARS-CoV-2 brain infection. ACS Chem. Neurosci. 2020, 11, 2137–2144. [Google Scholar] [CrossRef]
- Straus, M.R.; Tang, T.; Lai, A.L.; Flegel, A.; Bidon, M.; Freed, J.H.; Daniel, S.; Whittaker, G.R. Ca2+ Ions promote fusion of middle east respiratory syndrome coronavirus with host cells and increase infectivity. J. Virol. 2020, 94, 00426-20. [Google Scholar] [CrossRef]
- Danta, C.C. Calcium channel blockers: A possible potential therapeutic strategy for the treatment of alzheimer’s dementia patients with SARS-CoV-2 infection. ACS Chem. Neurosci. 2020, 11, 2145–2148. [Google Scholar] [CrossRef]
- Madry, C.; Attwell, D. Receptors, ion channels, and signaling mechanisms underlying microglial dynamics. J. Biol. Chem. 2015, 290, 12443–12450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient receptor potential cation channels in disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef] [Green Version]
- Benemei, S.; Patacchini, R.; Trevisani, M.; Geppetti, P. TRP channels. Curr. Opin. Pharmacol. 2015, 22, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Eder, C. Regulation of microglial behavior by ion channel activity. J. Neurosci. Res. 2005, 81, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Ping, L. Calcium ion influx in microglial cells: Physiological and therapeutic significance. J. Neurosci. Res. 2014, 92, 409–423. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V. Store-operated calcium entry in neuroglia. Neurosci. Bull. 2013, 30, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Vennekens, R.; Menigoz, A.; Nilius, B. TRPs in the Brain. Rev. Physiol. Biochem. Pharmacol. 2012, 163, 27–64. [Google Scholar] [CrossRef]
- Ohana, L.; Newell, E.W.; Stanley, E.F.; Schlichter, L.C. The Ca2+ release-activated Ca2+ current (I(CRAC)) mediates store-operated Ca2+ entry in rat microglia. Channels 2009, 3, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Fusco, F.R.; Martorana, A.; Giampà, C.; De March, Z.; Vacca, F.; Tozzi, A.; Longone, P.; Piccirilli, S.; Paolucci, S.; Sancesario, G.; et al. Cellular localization of TRPC3 channel in rat brain: Preferential distribution to oligodendrocytes. Neurosci. Lett. 2004, 365, 137–142. [Google Scholar] [CrossRef]
- Putney, J.W. Capacitative calcium entry in the nervous system. Cell Calcium 2003, 34, 339–344. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, Y.; Kato, T.A.; Seki, Y.; Ohgidani, M.; Sagata, N.; Horikawa, H.; Yamauchi, Y.; Sato-Kasai, M.; Hayakawa, K.; Inoue, R.; et al. Brain-derived Neurotrophic Factor (BDNF) Induces Sustained Intracellular Ca2+ Elevation through the Up-regulation of Surface Transient Receptor Potential 3 (TRPC3) Channels in Rodent Microglia. J. Biol. Chem. 2014, 289, 18549–18555. [Google Scholar] [CrossRef] [Green Version]
- Amaral, M.D.; Chapleau, C.A.; Pozzo-Miller, L. Transient receptor potential channels as novel effectors of brain-derived neurotrophic factor signaling: Potential implications for Rett syndrome. Pharmacol. Ther. 2007, 113, 394–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holsinger, R.; Schnarr, J.; Henry, P.; Castelo, V.T.; Fahnestock, M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: Decreased levels in Alzheimer’s disease. Brain Res. Mol. Brain Res. 2000, 76, 347–354. [Google Scholar] [CrossRef]
- Michalski, B.; Fahnestock, M. Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer’s disease. Mol. Brain Res. 2003, 111, 148–154. [Google Scholar] [CrossRef]
- Lee, J.; Fukumoto, H.; Orne, J.; Klucken, J.; Raju, S.; Vanderburg, C.R.; Irizarry, M.C.; Hyman, B.T.; Ingelsson, M. Decreased levels of BDNF protein in Alzheimer temporal cortex are independent of BDNF polymorphisms. Exp. Neurol. 2005, 194, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-mediated modulation of capacitative calcium Entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Munakata, M.; Shirakawa, H.; Nagayasu, K.; Miyanohara, J.; Miyake, T.; Nakagawa, T.; Katsuki, H.; Kaneko, S. Transient receptor potential canonical 3 Inhibitor Pyr3 improves outcomes and attenuates astrogliosis after intracerebral hemorrhage in mice. Stroke 2013, 44, 1981–1987. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.R.; Kim, S.U.; Oh, U.; Jin, B.K. Transient Receptor Potential Vanilloid Subtype 1 Mediates Microglial Cell Death In Vivo and In Vitro via Ca2+-Mediated Mitochondrial Damage and Cytochrome c Release. J. Immunol. 2006, 177, 4322–4329. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Liao, P. Brain transient receptor potential channels and stroke. J. Neurosci. Res. 2015, 93, 1165–1183. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhang, L.; Westlund, K.N. Reactive Oxygen Species Mediate TNFR1 Increase after TRPV1 Activation in Mouse DRG Neurons. Mol. Pain 2009, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Park, E.S.; Kim, S.R.; Jin, B.K. Transient receptor potential vanilloid subtype 1 contributes to mesencephalic dopaminergic neuronal survival by inhibiting microglia-originated oxidative stress. Brain Res. Bull. 2012, 89, 92–96. [Google Scholar] [CrossRef]
- Miyake, T.; Shirakawa, H.; Nakagawa, T.; Kaneko, S. Activation of mitochondrial transient receptor potential vanilloid 1 channel contributes to microglial migration. Glia 2015, 63, 1870–1882. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.; Eldeeb, K.; Millns, P.J.; Bennett, A.J.; Alexander, S.P.H.; A Kendall, D. Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. Br. J. Pharmacol. 2014, 171, 2426–2439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, M.; Shirakawa, H.; Iida, S.; Sakimoto, S.; Matsutani, I.; Miyake, T.; Kageyama, K.; Nakagawa, T.; Shibasaki, K.; Kaneko, S. Stimulation of transient receptor potential vanilloid 4 channel suppresses abnormal activation of microglia induced by lipopolysaccharide. Glia 2012, 60, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Du, F.; Liu, Y.; Li, L.; Cai, J.; Zhang, G.-F.; Xu, X.-F.; Lin, T.; Cheng, H.-R.; Liu, X.-D.; et al. Glial cell-expressed mechanosensitive channel TRPV4 mediates infrasound-induced neuronal impairment. Acta Neuropathol. 2013, 126, 725–739. [Google Scholar] [CrossRef]
- Flockerzi, V.; Nilius, B. TRPs: Truly Remarkable Proteins. Handb. Exp. Pharmacol. 2014, 222, 1–12. [Google Scholar] [CrossRef]
- Haraguchi, K.; Kawamoto, A.; Isami, K.; Maeda, S.; Kusano, A.; Asakura, K.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; Kaneko, S. trpm2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J. Neurosci. 2012, 32, 3931–3941. [Google Scholar] [CrossRef] [Green Version]
- Miyake, T.; Shirakawa, H.; Kusano, A.; Sakimoto, S.; Konno, M.; Nakagawa, T.; Mori, Y.; Kaneko, S. TRPM2 contributes to LPS/IFNγ-induced production of nitric oxide via the p38/JNK pathway in microglia. Biochem. Biophys. Res. Commun. 2014, 444, 212–217. [Google Scholar] [CrossRef]
- Mortadza, S.A.S.; Sim, J.A.; Neubrand, V.E.; Jiang, L.-H. A critical role of TRPM2 channel in Aβ(42)-induced microglial activation and generation of tumor necrosis factor-α. Glia 2018, 66, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.-F.; LaVine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The Transient Receptor Potential Melastatin 2 (TRPM2) Channel Contributes to β-Amyloid Oligomer-Related Neurotoxicity and Memory Impairment. J. Neurosci. 2015, 35, 15157–15169. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Penner, R.; Fleig, A. Lipopolysaccharide-induced down-regulation of Ca2+release-activated Ca2+currents (ICRAC) but not Ca2+-activated TRPM4-like currents (ICAN) in cultured mouse microglial cells. J. Physiol. 2008, 586, 427–439. [Google Scholar] [CrossRef]
- Siddiqui, T.; Lively, S.; Ferreira, R.; Wong, R.; Schlichter, L.C. Expression and contributions of TRPM7 and KCa2.3/SK3 CHANNELS to the INCREASED migration and invasion of microglia in Anti-inflammatory Activation States. PLoS ONE 2014, 9, e106087. [Google Scholar] [CrossRef]
- Payandeh, J.; El-Din, T.M.G.; Scheuer, T.; Zheng, N.; Catterall, W.A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012, 486, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Brackenbury, W.J.; Isom, L.L. Voltage-gated Na+channels: Potential for β subunits as therapeutic targets. Expert Opin. Ther. Targets 2008, 12, 1191–1203. [Google Scholar] [CrossRef] [Green Version]
- Black, J.; Westenbroek, R.; Catterall, W.; Waxman, S. Type II brain sodium channel expression in non-neuronal cells: Embryonic rat osteoblasts. Mol. Brain Res. 1995, 34, 89–98. [Google Scholar] [CrossRef]
- Black, J.A.; Waxman, S.G. Noncanonical Roles of Voltage-Gated Sodium Channels. Neuron 2013, 80, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, L.W.; Black, J.A.; Waxman, S.G. Sodium channels in astroglia and microglia. Glia 2016, 64, 1628–1645. [Google Scholar] [CrossRef] [Green Version]
- DeCoursey, T.; Chandy, K.; Gupta, S.; Cahalan, M. Voltage-dependent ion channels in T-lymphocytes. J. Neuroimmunol. 1985, 10, 71–95. [Google Scholar] [CrossRef]
- E DeCoursey, T.; Chandy, K.G.; Gupta, S.; Cahalan, M.D. Mitogen induction of ion channels in murine T lymphocytes. J. Gen. Physiol. 1987, 89, 405–420. [Google Scholar] [CrossRef] [Green Version]
- Fraser, S.P.; Diss, J.K.; Lloyd, L.J.; Pani, F.; Chioni, A.-M.; George, A.J.; Djamgoz, M.B. T-lymphocyte invasiveness: Control by voltage-gated Na+channel activity. FEBS Lett. 2004, 569, 191–194. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.-F.; Chen, Y.-Z.; Nishimura, Y.; Nishi, K. An amiloride-sensitive and voltage-dependent Na+ channel in an HLA-DR-restricted human T cell clone. J. Immunol. 2000, 165, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Lo, W.-L.; Donermeyer, D.L.; Allen, P.M. A voltage-gated sodium channel is essential for the positive selection of CD4+ T cells. Nat. Immunol. 2012, 13, 880–887. [Google Scholar] [CrossRef] [Green Version]
- A Black, J.; Newcombe, J.; Waxman, S.G. Nav1.5 sodium channels in macrophages in multiple sclerosis lesions. Mult. Scler. J. 2012, 19, 532–542. [Google Scholar] [CrossRef]
- Carrithers, L.M.; Hulseberg, P.; Sandor, M.; Carrithers, M.D. The human macrophage sodium channel NaV1.5 regulates mycobacteria processing through organelle polarization and localized calcium oscillations. FEMS Immunol. Med Microbiol. 2011, 63, 319–327. [Google Scholar] [CrossRef]
- Chatelier, A.; Mercier, A.; Tremblier, B.; Thériault, O.; Moubarak, M.; Benamer, N.; Corbi, P.; Bois, P.; Chahine, M.; Faivre, J.F. A distinct de novo expression of Nav1.5 sodium channels in human atrial fibroblasts differentiated into myofibroblasts. J. Physiol. 2012, 590, 4307–4319. [Google Scholar] [CrossRef] [PubMed]
- Bevan, S.; Chiu, S.Y.; Gray, P.T.; Ritchie, J.M. The presence of voltage-gated sodium, potassium and chloride channels in rat cultured astrocytes. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1985, 225, 299–313. [Google Scholar] [CrossRef]
- Chiu, S.Y.; Schrager, P.; Ritchie, J.M. Neuronal-type Na+ and K+ channels in rabbit cultured Schwann cells. Nat. Cell Biol. 1984, 311, 156–157. [Google Scholar] [CrossRef]
- Chen, P.-H.; Cai, W.-Q.; Wang, L.-Y.; Deng, Q.-Y. A morphological and electrophysiological study on the postnatal development of oligodendrocyte precursor cells in the rat brain. Brain Res. 2008, 1243, 27–37. [Google Scholar] [CrossRef]
- Káradóttir, R.; Hamilton, N.B.; Bakiri, Y.; Attwell, D. Spiking and nonspiking classes of oligodendrocyte precursor glia in CNS white matter. Nat. Neurosci. 2008, 11, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, A.-K.; Estacion, M.; Ahn, H.; Liu, S.; Stamboulian-Platel, S.; Waxman, S.G.; Black, J.A. Contribution of sodium channels to lamellipodial protrusion and Rac1 and ERK1/2 activation in ATP-stimulated microglia. Glia 2014, 62, 2080–2095. [Google Scholar] [CrossRef]
- Nicholson, E.; Randall, A. NaV1.5 sodium channels in a human microglial cell line. J. Neuroimmunol. 2009, 215, 25–30. [Google Scholar] [CrossRef]
- Black, J.A.; Liu, S.; Waxman, S.G. Sodium channel activity modulates multiple functions in microglia. Glia 2009, 57, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Black, J.A.; Waxman, S.G. Sodium channels and microglial function. Exp. Neurol. 2012, 234, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Craner, M.J.; Damarjian, T.G.; Liu, S.; Hains, B.C.; Lo, A.C.; Black, J.A.; Newcombe, J.; Cuzner, M.L.; Waxman, S.G. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia 2004, 49, 220–229. [Google Scholar] [CrossRef]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial Physiology: Unique Stimuli, Specialized Responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, J.D.; Schwender, S.; Imrich, H.; Dorries, R.; Butcher, G.W.; Ter Meulen, V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc. Natl. Acad. Sci. USA 1991, 88, 7438–7442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morsali, D.; Bechtold, D.; Lee, W.; Chauhdry, S.; Palchaudhuri, U.; Hassoon, P.; Snell, D.M.; Malpass, K.; Piers, T.; Pocock, J.; et al. Safinamide and flecainide protect axons and reduce microglial activation in models of multiple sclerosis. Brain 2013, 136, 1067–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohrman, D.C.; Smith, M.R.; Goldin, A.L.; Harris, J.; Meisler, M.H. A Missense Mutation in the Sodium Channel Scn8a Is Responsible for Cerebellar Ataxia in the Mouse Mutantjolting. J. Neurosci. 1996, 16, 5993–5999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisi, S.; Disanza, A.; Malinverno, C.; Frittoli, E.; Palamidessi, A.; Scita, G. Membrane and actin dynamics interplay at lamellipodia leading edge. Curr. Opin. Cell Biol. 2013, 25, 565–573. [Google Scholar] [CrossRef]
- A Siddiqui, T.; Lively, S.; Vincent, C.; Schlichter, L.C. Regulation of podosome formation, microglial migration and invasion by Ca2+-signaling molecules expressed in podosomes. J. Neuroinflamm. 2012, 9, 250. [Google Scholar] [CrossRef] [Green Version]
- Annunziato, L.; Pignataro, G.; Di Renzo, G.F. Pharmacology of Brain Na+/Ca2+ Exchanger: From Molecular Biology to Therapeutic Perspectives. Pharmacol. Rev. 2004, 56, 633–654. [Google Scholar] [CrossRef] [Green Version]
- Tong, X.-P.; Li, X.-Y.; Zhou, B.; Shen, W.; Zhang, Z.-J.; Xu, T.-L.; Duan, S. Ca(2+) signaling evoked by activation of Na(+) channels and Na(+)/Ca(2+) exchangers is required for GABA-induced NG2 cell migration. J. Cell Biol. 2009, 186, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Pappalardo, L.W.; Samad, O.A.; Black, J.A.; Waxman, S.G. Voltage-gated sodium channel Nav1.5 contributes to astrogliosis in anin vitromodel of glial injury via reverse Na+/Ca2+exchange. Glia 2014, 62, 1162–1175. [Google Scholar] [CrossRef] [Green Version]
- Wiegert, J.S.; Bading, H. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: A link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 2011, 49, 296–305. [Google Scholar] [CrossRef]
- Hossain, M.M.; Weig, B.; Reuhl, K.; Gearing, M.; Wu, L.-J.; Richardson, J.R. The anti-parkinsonian drug zonisamide reduces neuroinflammation: Role of microglial Nav 1.6. Exp. Neurol. 2018, 308, 111–119. [Google Scholar] [CrossRef]
- Wu, L.-J.; Wu, G.; Sharif, M.R.A.; Baker, A.; Jia, Y.; Fahey, F.H.; Luo, H.R.; Feener, E.P.; Clapham, D.E. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat. Neurosci. 2012, 15, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.-J. Voltage-Gated Proton Channel HV1 in Microglia. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2014, 20, 599–609. [Google Scholar] [CrossRef]
- Yu, Y.; Luo, X.; Li, C.; Ding, F.; Wang, M.; Xie, M.; Yu, Z.; Ransom, B.R.; Wang, W. Microglial Hv1 proton channels promote white matter injuries after chronic hypoperfusion in mice. J. Neurochem. 2019, 152, 350–367. [Google Scholar] [CrossRef]
- Tian, D.-S.; Li, C.-Y.; Qin, C.; Murugan, M.; Wu, L.-J.; Liu, J.-L. Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J. Neurochem. 2016, 139, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yu, Z.; Xie, M.; Wang, W.; Luo, X. Hv1 proton channel facilitates production of ROS and pro-inflammatory cytokines in microglia and enhances oligodendrocyte progenitor cells damage from oxygen-glucose deprivation in vitro. Biochem. Biophys. Res. Commun. 2018, 498, 1–8. [Google Scholar] [CrossRef] [PubMed]
- De Simoni, A.; Allen, N.J.; Attwell, D. Charge compensation for NADPH oxidase activity in microglia in rat brain slices does not involve a proton current. Eur. J. Neurosci. 2008, 28, 1146–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlichter, L.C.; Mertens, T.; Liu, B. Swelling activated Cl- channels in microglia: Biophysics, pharmacology and role in glutamate release. Channels 2011, 5, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, T.; Kang, J.; Jaiswal, J.K.; Simon, S.M.; Lin, J.H.-C.; Yu, Y.; Li, Y.; Yang, J.; Dienel, G.; Zielke, H.R.; et al. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 16466–16471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlichter, L.; Sakellaropoulos, G.; Ballyk, B.; Pennefather, P.; Phipps, D. Properties of K+ and Cl− channels and their involvement in proliferation of rat microglial cells. Glia 1996, 17, 225–236. [Google Scholar] [CrossRef]
- Ducharme, G.; Newell, E.W.; Pinto, C.; Schlichter, L.C. Small-conductance Cl- channels contribute to volume regulation and phagocytosis in microglia. Eur. J. Neurosci. 2007, 26, 2119–2130. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G.; Möbius, H.-J.; Stöffler, A.; Quack, G. Neuroprotective and symptomatological action of memantine relevant for alzheimer’s disease—A unified glutamatergic hypothesis on the mechanism of action. Neurotox. Res. 2000, 2, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, N.; Gauthier, S.; Boneva, N.; Lemming, O.M. A randomized, double-blind, placebo-controlled trial of memantine in a behaviorally enriched sample of patients with moderate-to-severe Alzheimer’s disease. Int. Psychogeriatr. 2013, 25, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.; Gimeno-Bayon, J.; Espinosa-Parrilla, J.; Carrasco, J.; Batlle, M.; Pugliese, M.; Mahy, N.; Rodríguez, M. ATP-dependent potassium channel blockade strengthens microglial neuroprotection after hypoxia–ischemia in rats. Exp. Neurol. 2012, 235, 282–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Maniskas, M.; Roberts, J.M.; Aron, I.; Fraser, J.F.; Bix, G.J. Stroke neuroprotection revisited: Intra-arterial verapamil is profoundly neuroprotective in experimental acute ischemic stroke. J. Cereb. Blood Flow Metab. 2015, 36, 721–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, S.; Lu, K.; Wang, F.; Deng, J.; Xu, Z.; Wang, X.; Zhou, Q.; Le, W.; Zhao, Y. Verapamil Ameliorates Motor Neuron Degeneration and Improves Lifespan in the SOD1G93A Mouse Model of ALS by Enhancing Autophagic Flux. Aging Dis. 2019, 10, 1159–1173. [Google Scholar] [CrossRef] [Green Version]
- Nicita, F.; Spalice, A.; Papetti, L.; Nikanorova, M.; Iannetti, P.; Parisi, P. Efficacy of verapamil as an adjunctive treatment in children with drug-resistant epilepsy: A pilot study. Seizure 2014, 23, 36–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, J.; Frech, R.; Walters, S.; Patel, V.; Frigerio, R.; Maraganore, D.M. Low dose verapamil as an adjunct therapy for medically refractory epilepsy—An open label pilot study. Epilepsy Res. 2016, 126, 197–200. [Google Scholar] [CrossRef]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Christian, K.M.; Song, H.; Ming, G.-L. Modeling psychiatric disorders with patient-derived iPSCs. Curr. Opin. Neurobiol. 2016, 36, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Bechtold, D.A.; Smith, K.J. Sodium-mediated axonal degeneration in inflammatory demyelinating disease. J. Neurol. Sci. 2005, 233, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta 2011, 1812, 1584–1590. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Lu, K.; Li, Z.; Zhao, Y.; Wang, Y.; Hu, B.; Xu, P.; Shi, X.; Zhou, B.; Pennington, M.; et al. Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro Oncol. 2014, 16, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.-M.; Wang, F.; Qi, D.; Liu, W.-W.; Gu, C.; Mao, C.-J.; Yang, Y.-P.; Zhao, Z.; Hu, L.-F.; Liu, C.-F. A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Front. Aging Neurosci. 2018, 10, 378. [Google Scholar] [CrossRef] [PubMed]
- Berglund, R.; Guerreiro-Cacais, A.O.; Adzemovic, M.Z.; Zeitelhofer, M.; Lund, H.; Ewing, E.; Ruhrmann, S.; Nutma, E.; Parsa, R.; Thessen-Hedreul, M.; et al. Microglial autophagy–associated phagocytosis is essential for recovery from neuroinflammation. Sci. Immunol. 2020, 5, eabb5077. [Google Scholar] [CrossRef] [PubMed]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Prevarskaya, N. Ion channels in the regulation of apoptosis. Biochim. Biophys. Acta 2015, 1848, 2532–2546. [Google Scholar] [CrossRef] [Green Version]
- A Razik, M.; A Cidlowski, J. Molecular interplay between ion channels and the regulation of apoptosis. Biol. Res. 2002, 35, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Abeele, F.V.; Prevarskaya, N. Targeting apoptosis by the remodelling of calcium-transporting proteins in cancerogenesis. FEBS J. 2013, 280, 5500–5510. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Hartnett, K.A.; Nerbonne, J.M.; Levitan, E.S.; Aizenman, E. Mediation of Neuronal Apoptosis by Kv2.1-Encoded Potassium Channels. J. Neurosci. 2003, 23, 4798–4802. [Google Scholar] [CrossRef]
- McFerrin, M.B.; Turner, K.L.; Cuddapah, V.A.; Sontheimer, H. Differential role of IK and BK potassium channels as mediators of intrinsic and extrinsic apoptotic cell death. Am. J. Physiol. Physiol. 2012, 303, C1070–C1078. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.K.; Ryu, H.J.; Kim, J.-E.; Jo, S.-M.; Choi, H.-C.; Song, H.-K.; Kang, T.-C. The roles of P2X7 receptor in regional-specific microglial responses in the rat brain following status epilepticus. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2011, 33, 515–525. [Google Scholar] [CrossRef]
- Huang, C.; Chi, X.-S.; Li, R.; Hu, X.; Xu, H.-X.; Li, J.-M.; Zhou, D. Inhibition of P2X7 Receptor Ameliorates Nuclear Factor-Kappa B Mediated Neuroinflammation Induced by Status Epilepticus in Rat Hippocampus. J. Mol. Neurosci. 2017, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Guo, Z.; Liu, X.; Ouyang, Q.; He, C.; Burnstock, G.; Yuan, H.; Xiang, Z. Block of P2X7 receptors could partly reverse the delayed neuronal death in area CA1 of the hippocampus after transient global cerebral ischemia. Purinergic Signal. 2013, 9, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhao, Z.; Ji, R.; Zhu, J.; Sui, Q.-Q.; Knight, G.E.; Burnstock, G.; He, C.; Yuan, H.; Xiang, Z. Inhibition of P2X7 receptors improves outcomes after traumatic brain injury in rats. Purinergic Signal. 2017, 13, 529–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cojocaru, A.; Burada, E.; Bălșeanu, A.-T.; Deftu, A.-F.; Cătălin, B.; Popa-Wagner, A.; Osiac, E. Roles of Microglial Ion Channel in Neurodegenerative Diseases. J. Clin. Med. 2021, 10, 1239. https://doi.org/10.3390/jcm10061239
Cojocaru A, Burada E, Bălșeanu A-T, Deftu A-F, Cătălin B, Popa-Wagner A, Osiac E. Roles of Microglial Ion Channel in Neurodegenerative Diseases. Journal of Clinical Medicine. 2021; 10(6):1239. https://doi.org/10.3390/jcm10061239
Chicago/Turabian StyleCojocaru, Alexandru, Emilia Burada, Adrian-Tudor Bălșeanu, Alexandru-Florian Deftu, Bogdan Cătălin, Aurel Popa-Wagner, and Eugen Osiac. 2021. "Roles of Microglial Ion Channel in Neurodegenerative Diseases" Journal of Clinical Medicine 10, no. 6: 1239. https://doi.org/10.3390/jcm10061239
APA StyleCojocaru, A., Burada, E., Bălșeanu, A. -T., Deftu, A. -F., Cătălin, B., Popa-Wagner, A., & Osiac, E. (2021). Roles of Microglial Ion Channel in Neurodegenerative Diseases. Journal of Clinical Medicine, 10(6), 1239. https://doi.org/10.3390/jcm10061239