
Mitochondrial Syndromes Revisited

 ,
, 
 ,
,
Abstract
:1. Background
2. Clinical Pictures
2.1. Isolated Mitochondrial Ataxias
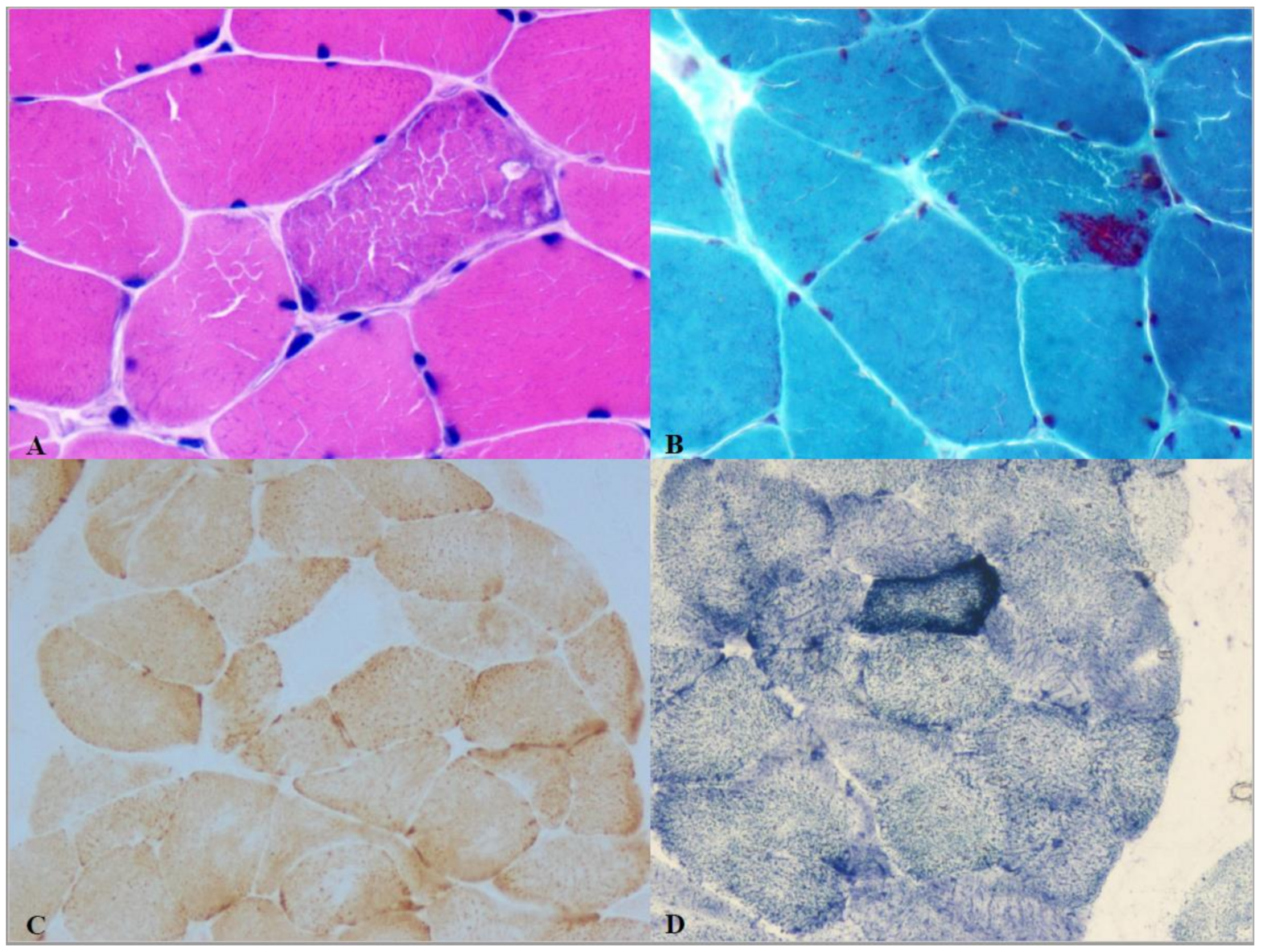
2.2. Isolated Mitochondrial Myopathies
2.3. Leigh Syndrome and NARP
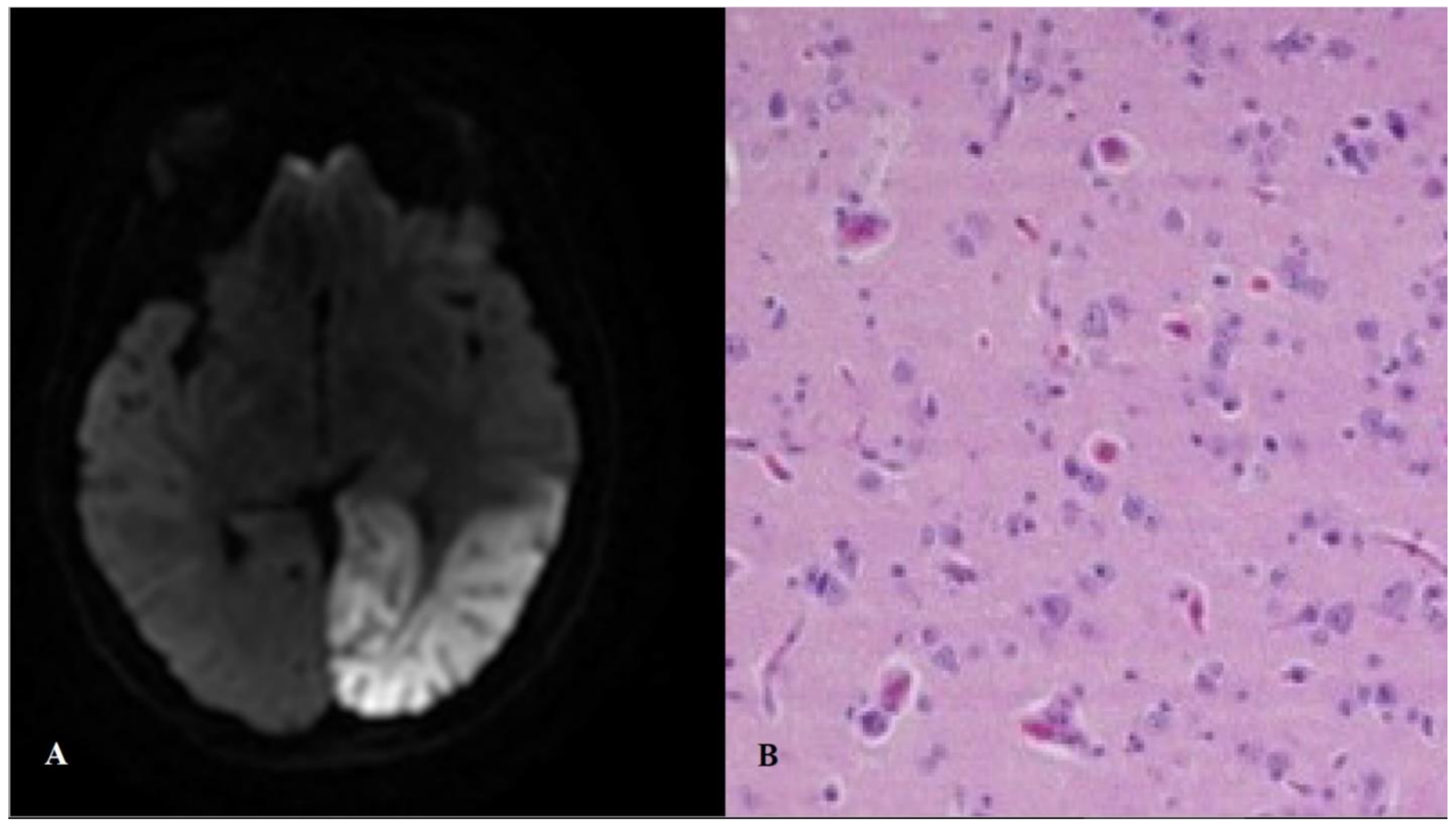
2.4. MELAS: Mitochondrial Encephalopathy with Lactic Acidosis and Stroke-Like Episodes
2.5. MERRF: Mitochondrial Encephalopathy with Ragged Red Fibers
2.6. Mitochondrial Cardiomyopathies
2.7. Mitochondrial Neuropathies
2.8. Mitochondrial Ocular Myopathies (I): PEO and PEO Plus
2.9. Mitochondrial Ocular Myopathies (II): Pearson Syndrome and “Kearns–Sayre Spectrum”
2.10. MNGIE: Mitochondrial Neurogastrointestinal Encephalomyopathy
2.11. Non-Syndromic Hearing Loss (NSHL)
2.12. Optic Neuropathies (I): LHON
2.13. Optic Neuropathies (II): Autosomal Dominant Optic Atrophy (ADOA)
2.14. Parkinsonisms
2.15. Pediatric Myocerebrohepatopathies, Including Alpers Syndrome
2.16. Tumors
2.17. Unspecified Mitochondrial Encephalomyopathies
3. Conclusions
Funding
Conflicts of Interest
References
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, C.; Wahbi, K.; Behin, A.; Bougouin, W.; Stojkovic, T.; Leonard-Louis, S.; Berber, N.; Lombès, A.; Duboc, D.; Jardel, C.; et al. Incidence and predictors of total mortality in 267 adults presenting with mitochondrial diseases. J. Inherit. Metab. Dis. 2020, 43, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondri-al disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ticci, C.; Sicca, F.; Ardissone, A.; Bertini, E.; Carelli, V.; Diodato, D.; Di Vito, L.; Filosto, M.; La Morgia, C.; Lamperti, C.; et al. Mitochondrial epilepsy: A cross-sectional nationwide Italian survey. Neurogenetics 2020, 21, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Filosto, M.; Mancuso, M. Mitochondrial diseases: A nosological update. Acta Neurol. Scand. 2007, 115, 211–221. [Google Scholar] [CrossRef]
- Luoma, P.T.; Luo, N.; Löscher, W.N.; Farr, C.L.; Horvath, R.; Wanschitz, J.; Kiechl, S.; Kaguni, L.S.; Suomalainen, A. Functional defects due to spacer-region mutations of human mitochondrial DNA polymerase in a family with an ataxia-myopathy syndrome. Hum. Mol. Genet. 2005, 14, 1907–1920. [Google Scholar] [CrossRef] [Green Version]
- Hakonen, A.H.; Heiskanen, S.; Juvonen, V.; Lappalainen, I.; Luoma, P.T.; Rantamäki, M.; Van Goethem, G.; Löfgren, A.; Hackman, P.; Paetau, A.; et al. Mitochondrial DNA Polymerase W748S Mutation: A Common Cause of Autosomal Recessive Ataxia with Ancient European Origin. Am. J. Hum. Genet. 2005, 77, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Schicks, J.; Synofzik, M.; Schulte, C.; Schöls, L. POLG, but notPEO1, is a frequent cause of cerebellar ataxia in Central Europe. Mov. Disord. 2010, 25, 2678–2682. [Google Scholar] [CrossRef]
- Goffart, S.; Cooper, H.M.; Tyynismaa, H.; Wanrooij, S.; Suomalainen, A.; Spelbrink, J.N. Twinkle mutations associated with auto-somal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 2009, 18, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological dis-ease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef]
- Hikmat, O.; Naess, K.; Engvall, M.; Klingenberg, C.; Rasmussen, M.; Tallaksen, C.M.; Brodtkorb, E.; Ostergaard, E.; De Coo, I.F.M.; Pias-Peleteiro, L.; et al. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J. Inherit. Metab. Dis. 2020, 43, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Gerards, M.; Bosch, B.V.D.; Calis, C.; Schoonderwoerd, K.; Van Engelen, K.; Tijssen, M.; De Coo, R.; Van Der Kooi, A.; Smeets, H. Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion 2010, 10, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; McFarland, R.; Klopstock, T.; Hirano, M.; consortium on Trial Readiness in Mitochondrial Myopathies. International Workshop: Outcome measures and clinical trial readiness in primary mitochondrial myopathies in children and adults. Consensus recommendations. 16-18 November 2016, Rome, Italy. Neuromuscul. Disord. 2017, 27, 1126–1137. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Minetti, C.; Moggio, M.; Mongini, T.; Servidei, S.; Tonin, P.; et al. Nation-wide Italian Collaborative Network of Mitochondrial Diseases. Fatigue and exercise intolerance in mitochondrial diseases. Literature revision and experience of the Italian Network of mitochondrial diseases. Neuromuscul. Disord. 2012, 22, S226–S229. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Galioto, R.; Lapin, B.; Haas, R.; Hirano, M.; Koenig, M.K.; Saneto, R.P.; Zolkipli-Cunningham, Z.; Goldstein, A.; Karaa, A. Fatigue in primary genetic mitochondrial disease: No rest for the weary. Neuromuscul. Disord. 2019, 29, 895–902. [Google Scholar] [CrossRef]
- Montano, V.; Gruosso, F.; Carelli, V.; Comi, G.P.; Filosto, M.; Lamperti, C.; Mongini, T.; Musumeci, O.; Servidei, S.; Tonin, P.; et al. Primary mi-tochondrial myopathy: Clinical features and outcome measures in 118 cases from Italy. Neurol. Genet. 2020, 6, e519. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.-H.G.; Margeta, M. Educational Case: Mitochondrial Myopathy. Acad. Pathol. 2019, 6, 2374289519888732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jou, C.; Ortigoza-Escobar, J.D.; O’Callaghan, M.M.; Nascimento, A.; Darling, A.; Pias-Peleteiro, L.; Perez-Dueñas, B.; Pineda, M.; Codina, A.; Arjona, C.; et al. Muscle Involve-ment in a Large Cohort of Pediatric Patients with Genetic Diagnosis of Mitochondrial Disease. J. Clin. Med. 2019, 8, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, C.A.P.F.; Teixeira, S.R.; Martin-Saavedra, J.S.; Guimarães Gonçalves, F.; Lo Russo, F.; Muraresku, C.; McCormick, E.M.; Falk, M.J.; Zolkipli-Cunningham, Z.; Ganetzky, R.; et al. Pediatric Leigh Syndrome: Neuroimaging Fea-tures and Genetic Correlations. Ann. Neurol. 2020, 88, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-M.; Na, J.-H.; Park, S.; Lee, Y.-M. Clinical Characteristics of Early-Onset and Late-Onset Leigh Syndrome. Front. Neurol. 2020, 11, 267. [Google Scholar] [CrossRef]
- Martikainen, M.H.; Ng, Y.S.; Gorman, G.S.; Alston, C.L.; Blakely, E.L.; Schaefer, A.M.; Chinnery, P.F.; Burn, D.J.; Taylor, R.W.; McFarland, R.; et al. Clinical, Genetic, and Radiological Features of Extrapyramidal Movement Disorders in Mitochondrial Dis-ease. JAMA Neurol. 2016, 73, 668–674. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Wu, Y.; Zhou, J.; Meng, H.; Zhang, W.; Guo, J. A meta-analysis and systematic review of Leigh syndrome: Clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine 2020, 99, e18634. [Google Scholar] [CrossRef] [PubMed]
- D’Aurelio, M.; Vives-Bauza, C.; Davidson, M.; Manfredi, G. Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Hum. Mol. Genet. 2009, 19, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debray, F.-G.; Lambert, M.; Lortie, A.; Vanasse, M.; Mitchell, G.A. Long-term outcome of Leigh syndrome caused by the NARP-T8993C mtDNA mutation. Am. J. Med. Genet. Part A 2007, 143, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Horvath, R.; Klopstock, T.; Mancuso, M.; Martikainen, M.H.; McFarland, R.; Nesbitt, V.; Pitceathly, R.D.S.; et al. Consensus-based statements for the management of mitochondrial stroke-like episodes. Wellcome Open Res. 2019, 4, 201. [Google Scholar] [CrossRef] [Green Version]
- Boggan, R.M.; Lim, A.; Taylor, R.W.; McFarland, R.; Pickett, S.J. Resolving complexity in mitochondrial disease: Towards preci-sion medicine. Mol. Genet. Metab. 2019, 128, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Y.; Lin, J.; Sun, C.; Chen, N.; Xu, W.; Hu, B.; Liu, X.; Geng, D.; Yang, L. Altered spontaneous brain activity at attack and remission stages in patients with mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS): Beyond stroke-like lesions. Mitochondrion 2020, 54, 49–56. [Google Scholar] [CrossRef]
- Quadir, A.; Pontifex, C.S.; Robertson, H.L.; Labos, C.; Pfeffer, G. Systematic review and meta-analysis of cardiac involvement in mitochondrial myopathy. Neurol. Genet. 2019, 5, e339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fayssoil, A.; Laforêt, P.; Bougouin, W.; Jardel, C.; Lombès, A.; Bécane, H.M.; Berber, N.; Stojkovic, T.; Béhin, A.; Eymard, B.; et al. Prediction of long-term prognosis by heteroplasmy levels of the m.3243A>G mutation in patients with the mito-chondrial encephalomyopathy, lactic acidosis and stroke-like episodes syndrome. Eur. J. Neurol. 2017, 24, 255–261. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, A.; Minetti, C.; Moggio, M.; Mongini, T.; et al. The m.3243A>G mitochondrial DNA mutation and related phenotypes. A matter of gender? J. Neurol. 2014, 261, 504–510. [Google Scholar] [CrossRef]
- Sanger, T.D.; Chen, D.; Fehlings, D.L.; Hallett, M.; Lang, A.E.; Mink, J.W.; Singer, H.S.; Alter, K.; Ben-Pazi, H.; Ms, E.E.B.; et al. Definition and classification of hyperkinetic movements in childhood. Mov. Disord. 2010, 25, 1538–1549. [Google Scholar] [CrossRef]
- Shahwan, A.; Farrell, M.; Delanty, N. Progressive myoclonic epilepsies: A review of genetic and therapeutic aspects. Lancet Neurol. 2005, 4, 239–248. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Minetti, C.; Moggio, M.; Mongini, T.; Servidei, S.; et al. Phenotypic heterogeneity of the 8344A>G mtDNA “MERRF” mutation. Neurology 2013, 80, 2049–2054. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Catteruccia, M.; Pegoraro, E.; Carelli, V.; Valentino, M.L.; Comi, G.P.; Minetti, C.; et al. Myoclonus in mitochondrial dis-orders. Mov. Disord. 2014, 29, 722–728. [Google Scholar] [CrossRef]
- Giannoni, A.; Aimo, A.; Mancuso, M.; Piepoli, M.F.; Orsucci, D.; Aquaro, G.D.; Barison, A.; De Marchi, D.; Taddei, C.; Cameli, M.; et al. Autonomic, functional, skeletal muscle, and cardiac abnormalities are associ-ated with increased ergoreflex sensitivity in mitochondrial disease. Eur. J. Heart Fail. 2017, 19, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Bougouin, W.; Béhin, A.; Stojkovic, T.; Bécane, H.M.; Jardel, C.; Berber, N.; Mochel, F.; Lombès, A.; Eymard, B.; et al. Long-term cardiac prognosis and risk stratification in 260 adults presenting with mitochondrial diseases. Eur. Heart J. 2015, 36, 2886–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galetta, F.; Franzoni, F.; Mancuso, M.; Orsucci, D.; Tocchini, L.; Papi, R.; Speziale, G.; Gaudio, C.; Siciliano, G.; Santoro, G. Cardiac in-volvement in chronic progressive external ophthalmoplegia. J. Neurol. Sci. 2014, 345, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Friederich, M.W.; Timal, S.; Powell, C.A.; Dallabona, C.; Kurolap, A.; Palacios-Zambrano, S.; Bratkovic, D.; Derks, T.G.J.; Bick, D.; Bou-man, K.; et al. Pathogenic variants in glutam-yl-tRNAGln amidotransferase subunits cause a lethal mitochondrial cardiomyopathy disorder. Nat. Commun. 2018, 9, 4065. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, A.; Olivotto, I.; Favilli, S.; Spaziani, G.; Passantino, S.; Procopio, E.; Morrone, A.; Donati, M.A. Impact of cardiovascular involvement on the clinical course of paediatric mitochondrial disorders. Orphanet J. Rare Dis. 2020, 15, 1–12. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Federico, A.; Minetti, C.; Moggio, M.; Mongini, T.; et al. “Mitochondrial neuropathies”: A survey from the large cohort of the Italian Network. Neuromuscul. Disord. 2016, 26, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W.I. Mitofusin 2 Dysfunction and Disease in Mice and Men. Front. Physiol. 2020, 11, 782. [Google Scholar] [CrossRef] [PubMed]
- McClelland, C.; Manousakis, G.; Lee, M.S. Progressive External Ophthalmoplegia. Curr. Neurol. Neurosci. Rep. 2016, 16, 53. [Google Scholar] [CrossRef]
- Rodríguez-López, C.; García-Cárdaba, L.M.; Blázquez, A.; Serrano-Lorenzo, P.; Gutiérrez-Gutiérrez, G.; San Millán-Tejado, B.; Muelas, N.; Hernández-Laín, A.; Vílchez, J.J.; Gutiérrez-Rivas, E.; et al. Clinical, patho-logical and genetic spectrum in 89 cases of mitochondrial progressive external ophthalmoplegia. J. Med. Genet. 2020, 57, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Federico, A.; Minetti, C.; Moggio, M.; Mongini, T.; Santorelli, F.M.; et al. Revisiting mitochondrial ocular myopathies: A study from the Italian Network. J. Neurol. 2017, 264, 1777–1784. [Google Scholar] [CrossRef]
- Smits, B.W.; Fermont, J.; Delnooz, C.C.; Kalkman, J.S.; Bleijenberg, G.; van Engelen, B.G. Disease impact in chronic progressive ex-ternal ophthalmoplegia: More than meets the eye. Neuromuscul. Disord. 2011, 21, 272–278. [Google Scholar] [CrossRef] [Green Version]
- Smits, B.W.; Heijdra, Y.F.; Cuppen, F.W.; van Engelen, B.G. Nature and frequency of respiratory involvement in chronic progres-sive external ophthalmoplegia. J. Neurol. 2011, 258, 2020–2025. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, M.A.; Federico, A.; Minetti, C.; Moggio, M.; et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J. Neurol. 2015, 262, 1301–1309. [Google Scholar] [CrossRef]
- Wild, K.T.; Goldstein, A.C.; Muraresku, C.; Ganetzky, R.D. Broadening the phenotypic spectrum of Pearson syndrome: Five new cases and a review of the literature. Am. J. Med. Genet. Part A 2019, 182, 365–373. [Google Scholar] [CrossRef]
- Broomfield, A.; Sweeney, M.G.; Woodward, C.E.; Fratter, C.; Morris, A.M.; Leonard, J.V.; Abulhoul, L.; Grunewald, S.; Clayton, P.T.; Hanna, M.G.; et al. Paediatric single mitochondrial DNA deletion disorders: An overlapping spectrum of disease. J. Inherit. Metab. Dis. 2015, 38, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Pitceathly, R.D.; Rahman, S.; Hanna, M.G. Single deletions in mitochondrial DNA—Molecular mechanisms and disease pheno-types in clinical practice. Neuromuscul. Disord. 2012, 22, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Filosto, M.; Cotti Piccinelli, S.; Caria, F.; Gallo Cassarino, S.; Baldelli, E.; Galvagni, A.; Volonghi, I.; Scarpelli, M.; Padovani, A. Mito-chondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1). J. Clin. Med. 2018, 7, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, M.; Carelli, V.; De Giorgio, R.; Pironi, L.; Accarino, A.; Cenacchi, G.; D’Alessandro, R.; Filosto, M.; Martí, R.; Nonino, F.; et al. Mitochondrial neurogastrointestinal enceph-alomyopathy (MNGIE): Position paper on diagnosis, prognosis, and treatment by the MNGIE International Network. J. Inherit. Metab. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Ienco, E.C.; Siciliano, G.; Mancuso, M. Mitochondrial disorders and drugs: What every physician should know. Drugs Context 2019, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Marotta, R.; Chin, J.; Chiotis, M.; Shuey, N.; Collins, S.J. Long-term screening for primary mitochondrial DNA variants associated with Leber hereditary optic neuropathy: Incidence, penetrance and clinical features. Mitochondrion 2020, 54, 128–132. [Google Scholar] [CrossRef]
- Newman, N.J.; Carelli, V.; Taiel, M.; Yu-Wai-Man, P. Visual Outcomes in Leber Hereditary Optic Neuropathy Patients With the m.11778G>A (MTND4) Mitochondrial DNA Mutation. J. Neuro Ophthalmol. 2020, 40, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-T.; Shen, M.-X.; Chen, C.; Huang, S.-H.; Zhuang, X.-R.; Ma, Q.-K.; Chen, Q.; Lu, F.; Yuan, Y.-M. Foveal pit morphological changes in asymptomatic carriers of the G11778A mutation with Leber’s hereditary optic neuropathy. Int. J. Ophthalmol. 2020, 13, 766–772. [Google Scholar] [CrossRef]
- Kirkman, M.A.; Yu-Wai-Man, P.; Korsten, A.; Leonhardt, M.; Dimitriadis, K.; De Coo, I.F.; Klopstock, T.; Chinnery, P.F. Gene–environment interactions in Leber hereditary optic neuropathy. Brain 2009, 132, 2317–2326. [Google Scholar] [CrossRef]
- Orsucci, D.; Caldarazzo Ienco, E.; Mancuso, M.; Siciliano, G. POLG1-related and other “mitochondrial Parkinsonisms”: An over-view. J. Mol. Neurosci. 2011, 44, 17–24. [Google Scholar] [CrossRef]
- Kiferle, L.; Orsucci, D.; Mancuso, M.; Gerfo, A.L.; Petrozzi, L.; Siciliano, G.; Ceravolo, R.; Bonuccelli, U. Twinkle mutation in an Italian family with external progressive ophthalmoplegia and parkinsonism: A case report and an update on the state of art. Neurosci. Lett. 2013, 556, 1–4. [Google Scholar] [CrossRef]
- Horvath, R.; Hudson, G.; Ferrari, G.; Fütterer, N.; Ahola, S.; Lamantea, E.; Prokisch, H.; Lochmüller, H.; McFarland, R.; Ramesh, V.; et al. Phenotypic spectrum associated with mutations of the mitochondrial poly-merase gamma gene. Brain 2006, 129, 1674–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saneto, R.P.; Cohen, B.H.; Copeland, W.C.; Naviaux, R.K. Alpers-Huttenlocher Syndrome. Pediatr. Neurol. 2013, 48, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Hakonen, A.H.; Isohanni, P.; Paetau, A.; Herva, R.; Suomalainen, A.; Lönnqvist, T. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain 2007, 130, 3032–3040. [Google Scholar] [CrossRef]
- Musumeci, O.; Barca, E.; Lamperti, C.; Servidei, S.; Comi, G.P.; Moggio, M.; Mongini, T.; Siciliano, G.; Filosto, M.; Pegoraro, E.; et al. Lipomatosis Incidence and Characteristics in an Italian Cohort of Mitochondrial Patients. Front. Neurol. 2019, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Kantorovich, V.; King, K.S.; Pacak, K. SDH-related pheochromocytoma and paraganglioma. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barca, E.; Long, Y.; Cooley, V.; Schoenaker, R.; Emmanuele, V.; DiMauro, S.; Cohen, B.H.; Karaa, A.; Vladutiu, G.D. Mitochondrial diseases in North America: An analysis of the NAMDC Registry. Neurol. Genet. 2020, 6, e402. [Google Scholar] [CrossRef] [Green Version]
- Ng, Y.S.; Martikainen, M.H.; Gorman, G.S.; Blain, A.; Bugiardini, E.; Bunting, A.; Schaefer, A.M.; Alston, C.L.; Blakely, E.L.; Sharma, S.; et al. Pathogenic variants in MT-ATP6: A United Kingdom–based mitochondrial disease cohort study. Ann. Neurol. 2019, 86, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Quinzii, C.M.; Lopez, L.C.; Naini, A.; DiMauro, S.; Hirano, M. Human CoQ10 deficiencies. Biofactors 2008, 32, 113–118. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the oxphos system. FEBS Lett. 2020. [Google Scholar] [CrossRef]
- Labory, J.; Fierville, M.; Ait-El-Mkadem, S.; Bannwarth, S.; Paquis-Flucklinger, V.; Bottini, S. Multi-Omics Approaches to Improve Mitochondrial Disease Diagnosis: Challenges, Advances, and Perspectives. Front. Mol. Biosci. 2020, 7, 590842. [Google Scholar] [CrossRef]
- Kerr, M.; Hume, S.; Omar, F.; Koo, D.; Barnes, H.; Khan, M.; Aman, S.; Wei, X.C.; Alfuhaid, H.; McDonald, R.; et al. MITO-FIND: A study in 390 patients to determine a diagnostic strategy for mitochondrial disease. Mol. Genet. Metab. 2020, 131, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Zereg, E.; Chaussenot, A.; Morel, G.; Bannwarth, S.; Sacconi, S.; Soriani, M.H.; Attarian, S.; Cano, A.; Pouget, J.; Bellance, R.; et al. Single-fiber studies for assigning pathogenicity of eight mitochondrial DNA variants associated with mitochondrial diseases. Hum. Mutat. 2020, 41, 1394–1406. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| merrf[Title] OR melas[Title] OR narp[Title] OR leigh[Title] OR leber[Title] OR adoa[Title] OR mitochondrial disease * [Title] OR mitochondrial disorder * [Title] OR mitochondrial myopath * [Title] OR mitochondrial encephalomyopath * [Title] OR kearns-sayre[Title] OR progressive external ophthalmoplegia[Title] OR iosca[Title] OR miras[Title] OR alpers[Title] OR coenzyme Q10 deficienc * [Title] OR pearson[Title] OR mngie[Title] OR nshl[Title] OR mitochondrial encephalopath * [Title] OR mitochondrial cardiomyopath * [Title] |
| Filters applied: in the last 10 years, English |
| Syndrome | Relative Frequency | Typical Feature(s) | Associated Feature(s) | Inheritance | Most Frequent Genetic Findings | Treatment of Choice |
|---|---|---|---|---|---|---|
| Alpers syndrome | Very rare | Childhood myocerebrohepatopathy | Autosomal recessive | POLG mutations with secondary mtDNA depletion | Symptomatic (avoid valproate) | |
| Autosomal dominant optic atrophy (ADOA) | Rare | Optic neuropathy (blindness) | Autosomal dominant | OPA1 mutations | Symptomatic | |
| Coenzyme Q10 deficiency | Very rare | Ataxia or myopathy or multi-system disease | Autosomal recessive | Various nuclear genes | Coenzyme Q10 | |
| Kearns–Sayre Syndrome (KSS) | Frequent | Ocular myopathy (ptosis, ophthalmoparesis) | Ataxia, cardiac conduction defects | Sporadic | Single large-scale deletion of mtDNA | Symptomatic |
| Leber hereditary optic neuropathy (LHON) | Very frequent | Optic neuropathy (blindness) | Maternal (low penetrance, higher in male smokers) | Various mtDNA mutations | Idebenone | |
| Leigh syndrome | Frequent | Severe pediatric encephalopathy | Autosomal recessive, X-linked or maternal | Various nuclear or mtDNA mutations (e.g., m.8993T > G) | Symptomatic | |
| Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) | Frequent | Stroke-like episodes | Cardiac involvement, hearing loss, diabetes | Maternal | m.3243A > G | Symptomatic |
| Myoclonic encephalopathy with ragged-red fiber (MERRF) | Frequent | Myoclonus | Ataxia, myopathy | Maternal | m.8344A > G | Symptomatic (e.g., Levetiracetam) |
| Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE) | Very rare | Gastrointestinal dysmotility | Leukodystrophy, ocular myopathy, peripheral neuropathy | Autosomal recessive | TYMP mutations | Liver transplantation |
| Neuropathy, Ataxia, Retinitis Pigmentosa (NARP) | Rare | Ataxia | Neuropathy, retinitis pigmentosa | Maternal | m.8993T > G | Symptomatic |
| Non syndromic hearing loss (NSHL) | Frequent | Hearing loss | Maternal | m.1555A > G | Symptomatic (avoid aminoglycosides) | |
| Progressive external ophthalmoplegia (PEO) | Very frequent | Ocular myopathy | Myopathy | Autosomal dominant, recessive, maternal, or sporadic | Various nuclear genes with secondary mtDNA multiple deletions, various mtDNA point mutations, mtDNA single large-scale deletion | Symptomatic |
| Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) Patients with histological, biochemical, and/or molecular evidence of mitochondrial disease who experience stroke-like episodes. Specifically associated features (at least in MELAS due to the m.3243A>G mutation) include lactic acidosis, generalized seizures, cognitive involvement, and hearing loss. |
| Myoclonic encephalomyopathy with ragged red fibers (MERRF) A mitochondrial syndrome where myoclonus is the prominent clinical feature, and which does not meet the criteria of other well-defined mitochondrial encephalopathic syndromes, including MELAS, Leigh, and Alpers syndromes. Ataxia is a specifically associated feature, differently from epileptic seizures. |
| Kearns-Sayre Syndrome (KSS) spectrum Ophthalmoparesis and/or ptosis due to a mtDNA single large-scale deletion and at least one of the following features:
|
| Progressive external ophthalmoplegia (PEO) Ophthalmoparesis and/or ptosis, not fulfilling the criteria for Pearson syndrome nor “KSS spectrum” criteria or other encephalopathic syndromes. “Pure PEO”: isolated ocular myopathy. “PEO plus”: ocular myopathy with other features of neuromuscular involvement. |
| Primary mitochondrial myopathies (PMM) Genetically defined disorders leading to defects in oxidative phosphorylation affecting predominantly skeletal muscles (not fulfilling the criteria for other more complex syndromes). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orsucci, D.; Caldarazzo Ienco, E.; Rossi, A.; Siciliano, G.; Mancuso, M. Mitochondrial Syndromes Revisited. J. Clin. Med. 2021, 10, 1249. https://doi.org/10.3390/jcm10061249
Orsucci D, Caldarazzo Ienco E, Rossi A, Siciliano G, Mancuso M. Mitochondrial Syndromes Revisited. Journal of Clinical Medicine. 2021; 10(6):1249. https://doi.org/10.3390/jcm10061249
Chicago/Turabian StyleOrsucci, Daniele, Elena Caldarazzo Ienco, Andrea Rossi, Gabriele Siciliano, and Michelangelo Mancuso. 2021. "Mitochondrial Syndromes Revisited" Journal of Clinical Medicine 10, no. 6: 1249. https://doi.org/10.3390/jcm10061249
APA StyleOrsucci, D., Caldarazzo Ienco, E., Rossi, A., Siciliano, G., & Mancuso, M. (2021). Mitochondrial Syndromes Revisited. Journal of Clinical Medicine, 10(6), 1249. https://doi.org/10.3390/jcm10061249