
Paracrine Interaction of Cholangiocellular Carcinoma with Cancer-Associated Fibroblasts and Schwann Cells Impact Cell Migration

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
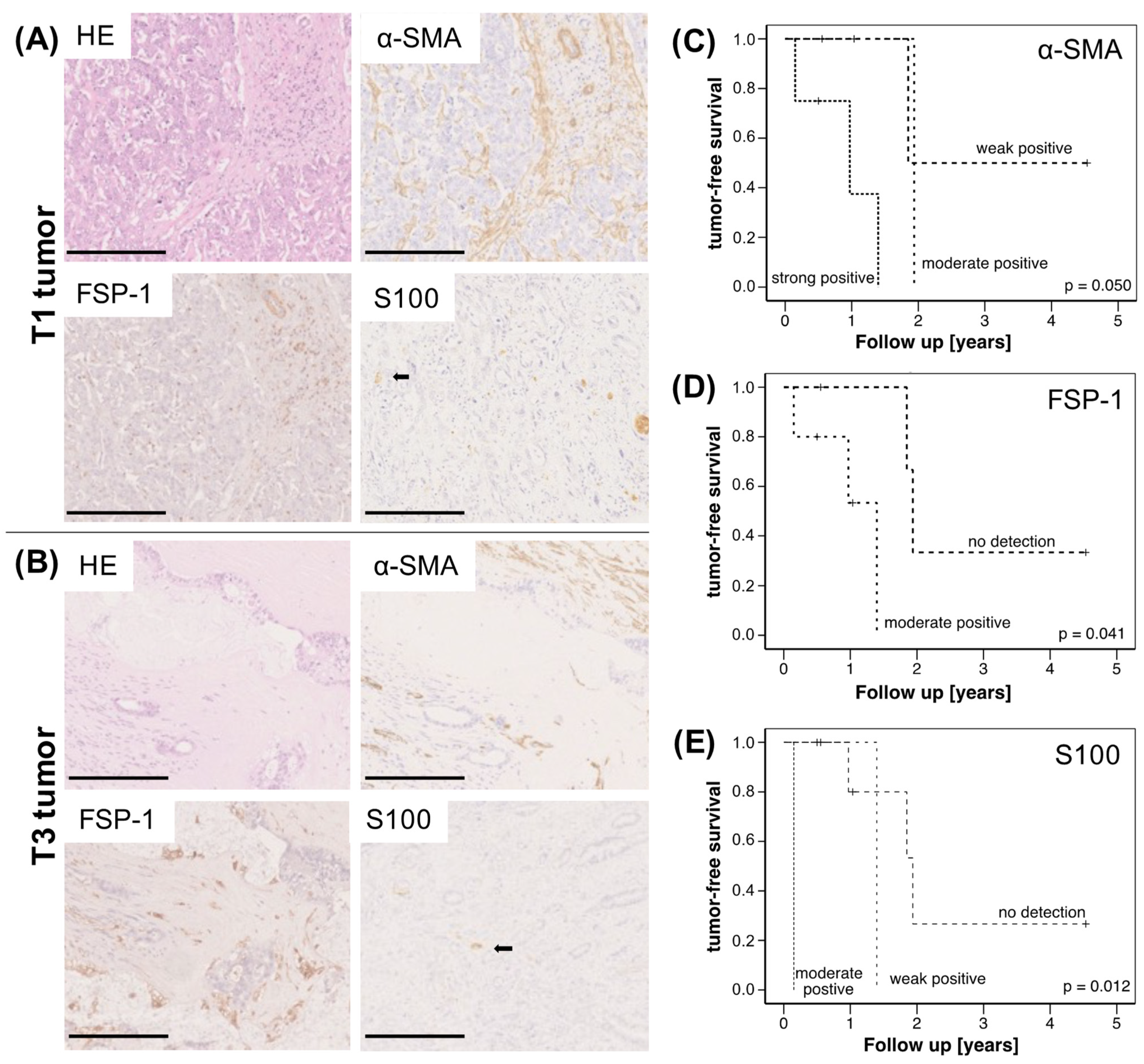
2.1. High Abundance of CAF and SC Is Associated with Reduced Survival of CCA Patients
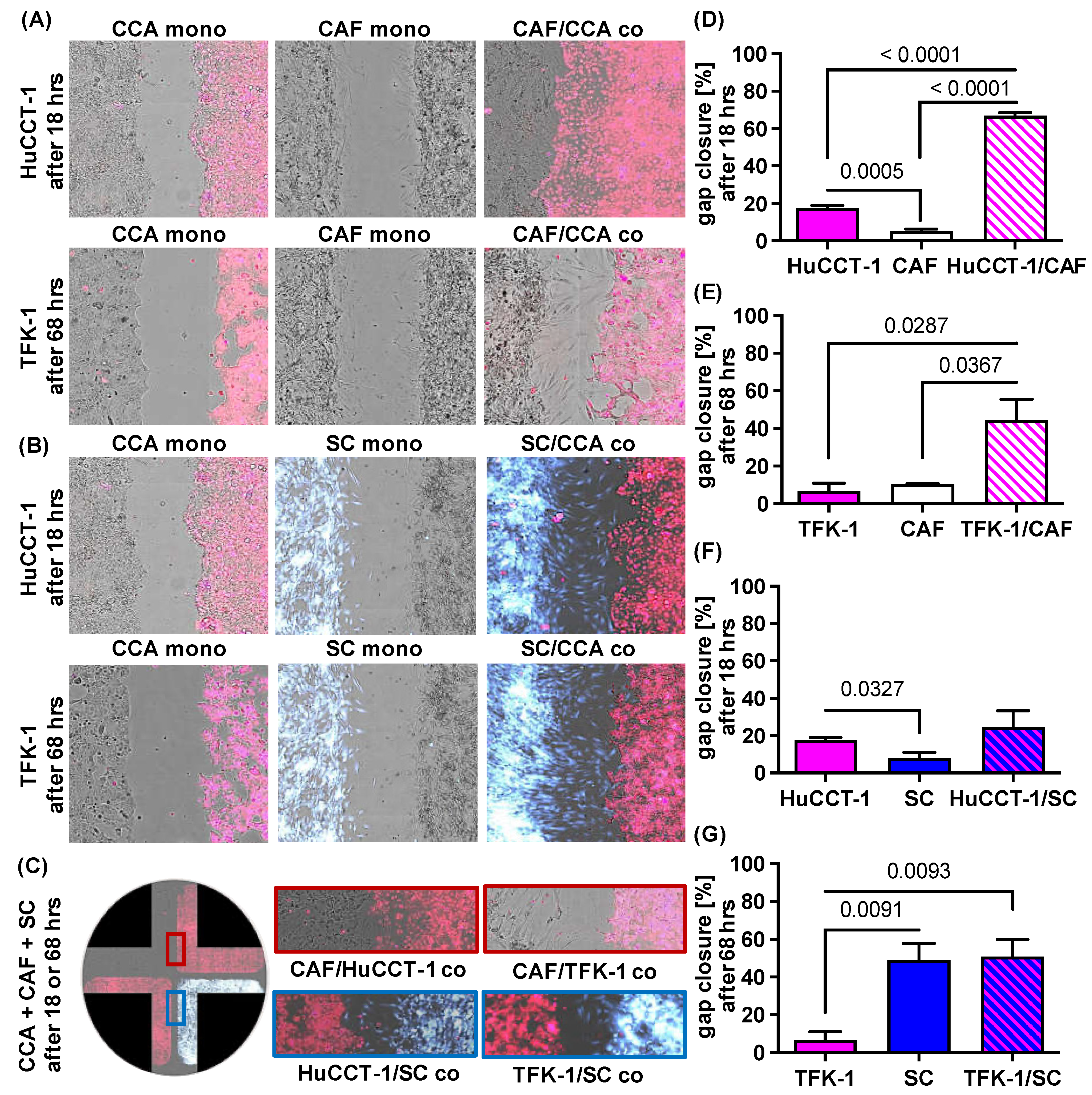
2.2. Impact of CAF and SC on Cell Migration of CCA Cells
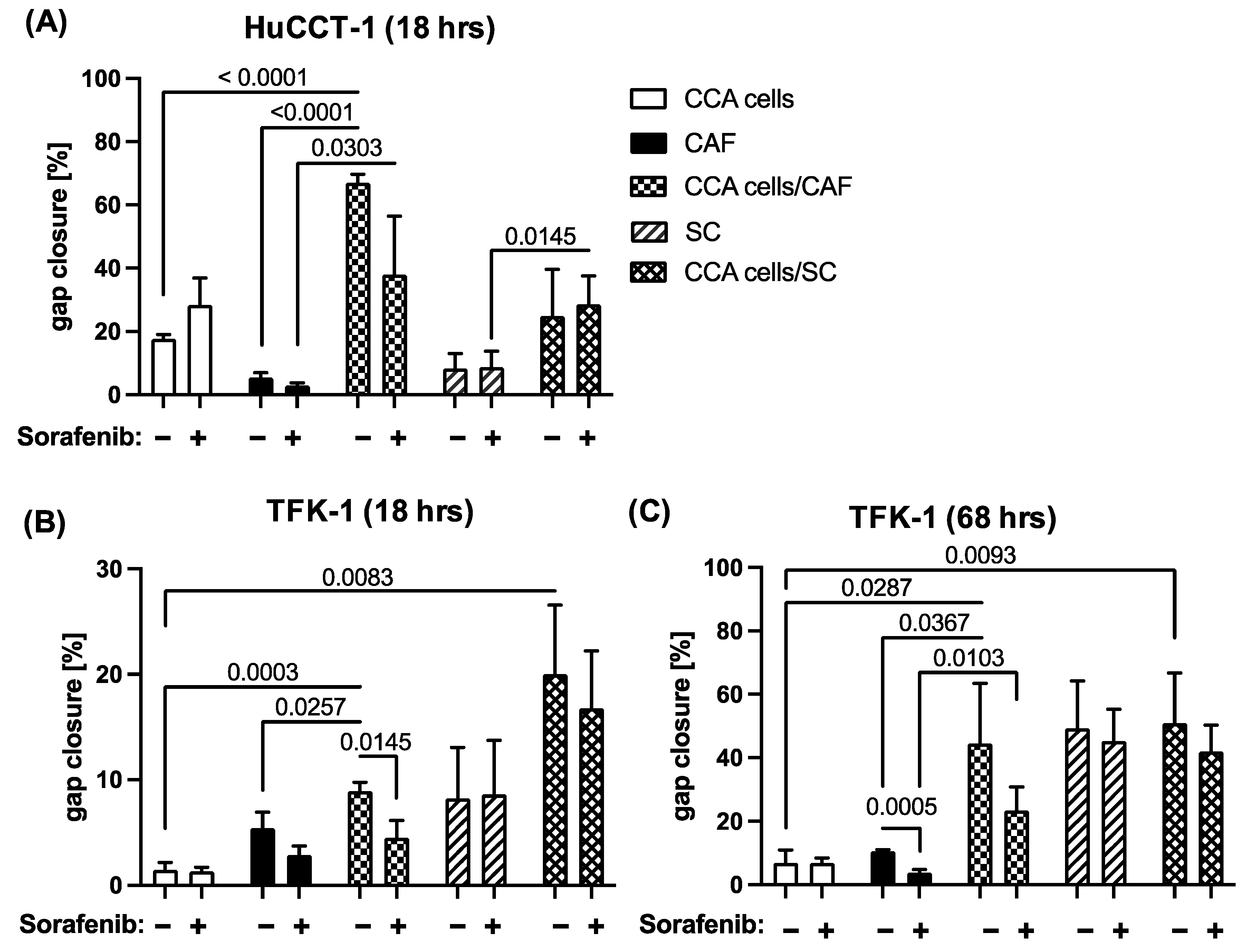
2.3. Sorafenib Inhibits Migration of CCA and Stromal Cells in a Context Dependent Manner
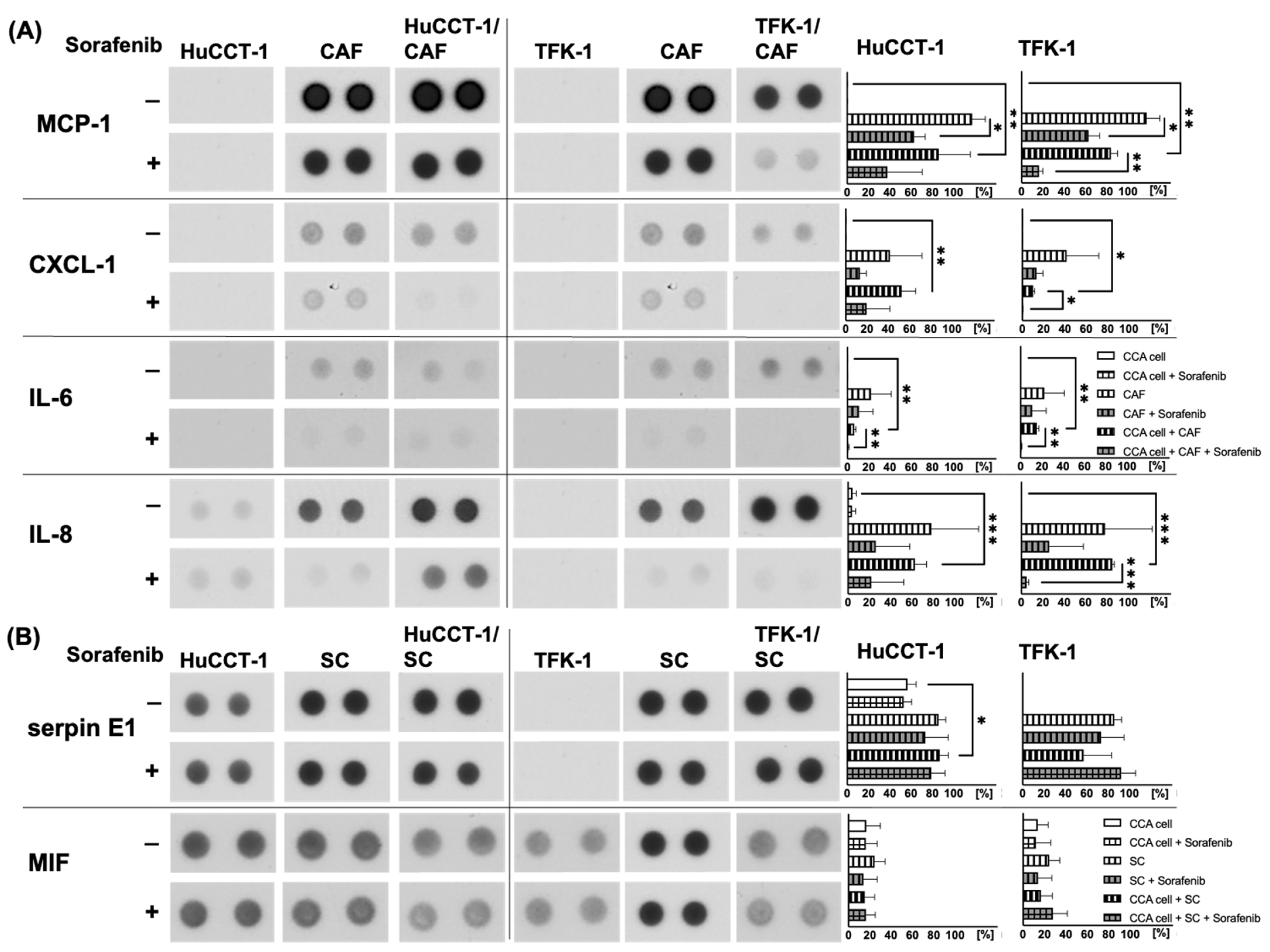
2.4. Analysis of Potential Migratory Inducing Factors in Co-Culture with CAF or SC
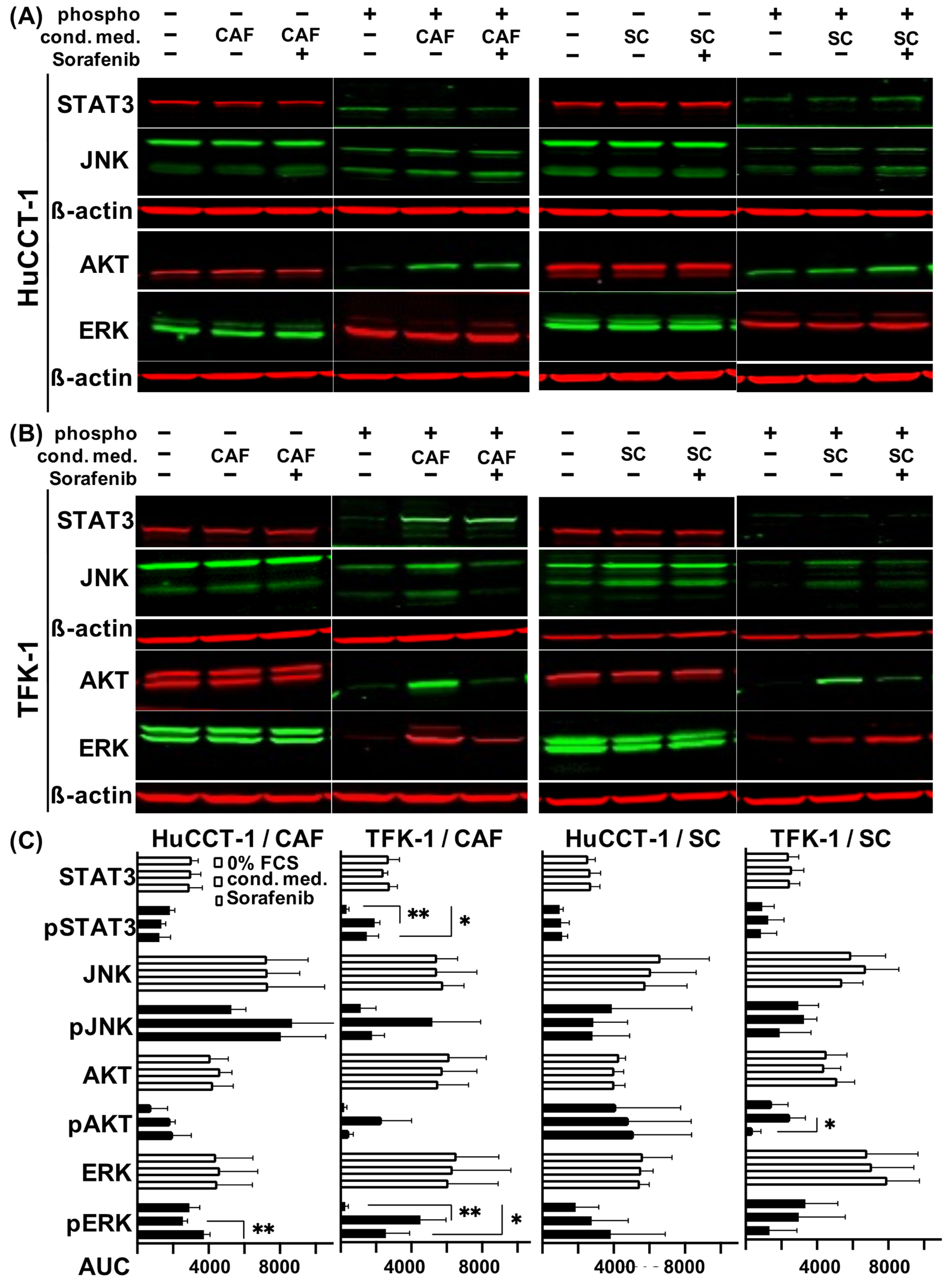
2.5. Altered Signal Transduction in CAF and SC Stimulated CCA Cells
3. Discussion
4. Materials and Methods
4.1. Immunohistochemical Stainings of CCA Tissues
4.2. Cell Lines and Generation of CAF
4.3. Paracrine Interaction Model to Analyze Cell Migration
4.4. Human Cytokine Array
4.5. Generation of Conditioned Medium of Stromal Cells
4.6. Western Blot to Analyze Activation of Signaling Pathways
4.7. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.S.; Dageforde, L.A. Cholangiocarcinoma. Surg. Clin. N. Am. 2019, 99, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Emadossadaty, S.; Ladep, N.G.; Thomas, H.C.; Elliott, P.; Taylor-Robinson, S.D.; Toledano, M.B. Rising trends in cholangiocarcinoma: Is the ICD classification system misleading us? J. Hepatol. 2012, 56, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [Green Version]
- Squadroni, M.; Tondulli, L.; Gatta, G.; Mosconi, S.; Beretta, G.; Labianca, R. Cholangiocarcinoma. Crit. Rev. Oncol. Hematol. 2017, 116, 11–31. [Google Scholar] [CrossRef]
- Labib, P.L.; Goodchild, G.; Pereira, S.P. Molecular Pathogenesis of Cholangiocarcinoma. BMC Cancer 2019, 19, 185. [Google Scholar] [CrossRef]
- Wang, C.; Maass, T.; Krupp, M.; Thieringer, F.; Strand, S.; Worns, M.A.; Barreiros, A.P.; Galle, P.R.; Teufel, A. A systems biology perspective on cholangiocellular carcinoma development: Focus on MAPK-signaling and the extracellular environment. J. Hepatol. 2009, 50, 1122–1131. [Google Scholar] [CrossRef]
- Dokduang, H.; Juntana, S.; Techasen, A.; Namwat, N.; Yongvanit, P.; Khuntikeo, N.; Riggins, G.J.; Loilome, W. Survey of activated kinase proteins reveals potential targets for cholangiocarcinoma treatment. Tumour Biol. 2013, 34, 3519–3528. [Google Scholar] [CrossRef]
- Pang, R.W.; Poon, R.T. From molecular biology to targeted therapies for hepatocellular carcinoma: The future is now. Oncology 2007, 72 (Suppl. S1), 30–44. [Google Scholar] [CrossRef]
- Huether, A.; Hopfner, M.; Baradari, V.; Schuppan, D.; Scherubl, H. Sorafenib alone or as combination therapy for growth control of cholangiocarcinoma. Biochem. Pharmacol. 2007, 73, 1308–1317. [Google Scholar] [CrossRef]
- Yokoi, K.; Kobayashi, A.; Motoyama, H.; Kitazawa, M.; Shimizu, A.; Notake, T.; Yokoyama, T.; Matsumura, T.; Takeoka, M.; Miyagawa, S.I. Survival pathway of cholangiocarcinoma via AKT/mTOR signaling to escape RAF/MEK/ERK pathway inhibition by sorafenib. Oncol. Rep. 2018, 39, 843–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Gao, W.; Su, M.; Nice, E.C.; Zhang, W.; Lin, J.; Xie, N. Adaptive Mechanisms of Tumor Therapy Resistance Driven by Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 641469. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Aoudjehane, L.; Fouassier, L. Cancer-associated fibroblasts in cholangiocarcinoma. Curr. Opin. Gastroenterol. 2020, 36, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Mellody, K.T.; Orimo, A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin. Cell Dev. Biol. 2010, 21, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, A.M.; Liu, S.; Liu, W.; Li, T.J. Cancer-associated fibroblasts in digestive tumors. World J. Gastroenterol. 2014, 20, 17804–17818. [Google Scholar] [CrossRef] [PubMed]
- Chuaysri, C.; Thuwajit, P.; Paupairoj, A.; Chau-In, S.; Suthiphongchai, T.; Thuwajit, C. Alpha-smooth muscle actin-positive fibroblasts promote biliary cell proliferation and correlate with poor survival in cholangiocarcinoma. Oncol. Rep. 2009, 21, 957–969. [Google Scholar] [PubMed] [Green Version]
- Bunimovich, Y.L.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Schwann cells: A new player in the tumor microenvironment. Cancer Immunol. Immunother. 2017, 66, 959–968. [Google Scholar] [CrossRef]
- Liu, Z.; Jin, Y.Q.; Chen, L.; Wang, Y.; Yang, X.; Cheng, J.; Wu, W.; Qi, Z.; Shen, Z. Specific marker expression and cell state of Schwann cells during culture in vitro. PLoS ONE 2015, 10, e0123278. [Google Scholar] [CrossRef] [Green Version]
- Demir, I.E.; Boldis, A.; Pfitzinger, P.L.; Teller, S.; Brunner, E.; Klose, N.; Kehl, T.; Maak, M.; Lesina, M.; Laschinger, M.; et al. Investigation of Schwann cells at neoplastic cell sites before the onset of cancer invasion. J. Natl. Cancer Inst. 2014, 106, dju184. [Google Scholar] [CrossRef] [Green Version]
- Boilly, B.; Faulkner, S.; Jobling, P.; Hondermarck, H. Nerve Dependence: From Regeneration to Cancer. Cancer Cell 2017, 31, 342–354. [Google Scholar] [CrossRef] [Green Version]
- Deborde, S.; Omelchenko, T.; Lyubchik, A.; Zhou, Y.; He, S.; McNamara, W.F.; Chernichenko, N.; Lee, S.Y.; Barajas, F.; Chen, C.H.; et al. Schwann cells induce cancer cell dispersion and invasion. J. Clin. Investig. 2016, 126, 1538–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Chen, Z.; Guo, P.; Wang, Y.; Chen, G. Therapy for advanced cholangiocarcinoma: Current knowledge and future potential. J. Cell. Mol. Med. 2021, 25, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Guo, X.; Huang, L.; Ye, H.; Li, Z.; Lin, L.; Chen, R.; Zhou, Q. Tumor-neuroglia interaction promotes pancreatic cancer metastasis. Theranostics 2020, 10, 5029–5047. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Capanu, M.; O’Reilly, E.M.; Ma, J.; Chou, J.F.; Shia, J.; Katz, S.S.; Gansukh, B.; Reidy-Lagunes, D.; Segal, N.H.; et al. A phase II study of gemcitabine and cisplatin plus sorafenib in patients with advanced biliary adenocarcinomas. Br. J. Cancer 2013, 109, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Moehler, M.; Maderer, A.; Schimanski, C.; Kanzler, S.; Denzer, U.; Kolligs, F.T.; Ebert, M.P.; Distelrath, A.; Geissler, M.; Trojan, J.; et al. Gemcitabine plus sorafenib versus gemcitabine alone in advanced biliary tract cancer: A double-blind placebo-controlled multicentre phase II AIO study with biomarker and serum programme. Eur. J. Cancer 2014, 50, 3125–3135. [Google Scholar] [CrossRef] [Green Version]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Ha, T.Y.; Hwang, S.; Moon, K.M.; Won, Y.J.; Song, G.W.; Kim, N.; Tak, E.; Ryoo, B.Y.; Hong, H.N. Sorafenib inhibits migration and invasion of hepatocellular carcinoma cells through suppression of matrix metalloproteinase expression. Anticancer Res. 2015, 35, 1967–1976. [Google Scholar]
- Dattachoudhury, S.; Sharma, R.; Kumar, A.; Jaganathan, B.G. Sorafenib Inhibits Proliferation, Migration and Invasion of Breast Cancer Cells. Oncology 2020, 98, 478–486. [Google Scholar] [CrossRef]
- Harrisingh, M.C.; Perez-Nadales, E.; Parkinson, D.B.; Malcolm, D.S.; Mudge, A.W.; Lloyd, A.C. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 2004, 23, 3061–3071. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Zhu, L.; Li, C.; Sha, D.; Pan, H.; Wang, N.; Ma, S. ERK1/2 and AKT are vital factors in regulation of the migration of rat Schwann cells. J. Vet. Med. Sci. 2015, 77, 427–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammoun, S.; Flaiz, C.; Ristic, N.; Schuldt, J.; Hanemann, C.O. Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer Res. 2008, 68, 5236–5245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Lee, S.J.; Kim, S.; Cho, S.C.; Choi, Y.H.; Kim, W.J.; Moon, S.K. Interleukin-5 enhances the migration and invasion of bladder cancer cells via ERK1/2-mediated MMP-9/NF-kappaB/AP-1 pathway: Involvement of the p21WAF1 expression. Cell. Signal. 2013, 25, 2025–2038. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Du, X.; Xie, J.; Wang, J. Interleukin-6 regulates iron-related proteins through c-Jun N-terminal kinase activation in BV2 microglial cell lines. PLoS ONE 2017, 12, e0180464. [Google Scholar] [CrossRef]
- Fu, S.; Lin, J. Blocking Interleukin-6 and Interleukin-8 Signaling Inhibits Cell Viability, Colony-forming Activity, and Cell Migration in Human Triple-negative Breast Cancer and Pancreatic Cancer Cells. Anticancer Res. 2018, 38, 6271–6279. [Google Scholar] [CrossRef]
- Guo, N.; Shen, G.; Zhang, Y.; Moustafa, A.A.; Ge, D.; You, Z. Interleukin-17 Promotes Migration and Invasion of Human Cancer Cells through Upregulation of MTA1 Expression. Front. Oncol. 2019, 9, 546. [Google Scholar] [CrossRef]
- Xu, X.; Yang, C.; Chen, J.; Liu, J.; Li, P.; Shi, Y.; Yu, P. Interleukin-23 promotes the migration and invasion of gastric cancer cells by inducing epithelial-to-mesenchymal transition via the STAT3 pathway. Biochem. Biophys. Res. Commun. 2018, 499, 273–278. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, H.; Si, L.; Li, Q.; Zhu, X.; Yu, T.; Gang, X. MiR-206 inhibits proliferation and migration of prostate cancer cells by targeting CXCL11. Prostate 2018, 78, 479–490. [Google Scholar] [CrossRef]
- Arya, M.; Ahmed, H.; Silhi, N.; Williamson, M.; Patel, H.R. Clinical importance and therapeutic implications of the pivotal CXCL12-CXCR4 (chemokine ligand-receptor) interaction in cancer cell migration. Tumour Biol. 2007, 28, 123–131. [Google Scholar] [CrossRef]
- Huang, C.Y.; Fong, Y.C.; Lee, C.Y.; Chen, M.Y.; Tsai, H.C.; Hsu, H.C.; Tang, C.H. CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways. Biochem. Pharmacol. 2009, 77, 794–803. [Google Scholar] [CrossRef]
- Johrer, K.; Janke, K.; Krugmann, J.; Fiegl, M.; Greil, R. Transendothelial migration of myeloma cells is increased by tumor necrosis factor (TNF)-alpha via TNF receptor 2 and autocrine up-regulation of MCP-1. Clin. Cancer Res. 2004, 10, 1901–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, R.A.; Bucala, R. Tumor growth-promoting properties of macrophage migration inhibitory factor (MIF). Semin. Cancer Biol. 2000, 10, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Ma, L.; Zhu, Z. SERPINE1 as a cancer-promoting gene in gastric adenocarcinoma: Facilitates tumour cell proliferation, migration, and invasion by regulating EMT. J. Chemother. 2019, 31, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, Z. TNF-alpha promotes colon cancer cell migration and invasion by upregulating TROP-2. Oncol. Lett. 2018, 15, 3820–3827. [Google Scholar] [PubMed]
- Yoshimura, T. The production of monocyte chemoattractant protein-1 (MCP-1)/CCL2 in tumor microenvironments. Cytokine 2017, 98, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, S.; Wang, W.; Ning, B.F.; Chen, F.; Shen, W.; Ding, J.; Chen, W.; Xie, W.F.; Zhang, X. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-beta pathways. Cancer Lett. 2016, 379, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Zhu, C.; Wang, J.; Peng, S.; Shuai, C.; Gao, S.; Tang, Z.; Su, T. Focal adhesion kinase knockdown in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis via downregulating MCP-1/CCL2 expression. J. Biochem. Mol. Toxicol. 2015, 29, 70–76. [Google Scholar] [CrossRef]
- Haga, H.; Yan, I.K.; Takahashi, K.; Wood, J.; Zubair, A.; Patel, T. Tumour cell-derived extracellular vesicles interact with mesenchymal stem cells to modulate the microenvironment and enhance cholangiocarcinoma growth. J. Extracell. Vesicles 2015, 4, 24900. [Google Scholar] [CrossRef]
- Hwang, H.J.; Oh, M.S.; Lee, D.W.; Kuh, H.J. Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells. J. Exp. Clin. Cancer Res. 2019, 38, 258. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Garcia, E.; Diaz-Garcia, C.V.; Agudo-Lopez, A.; Pardo-Marques, V.; Garcia-Consuegra, I.; Asensio-Pena, S.; Alonso-Riano, M.; Perez, C.; Gomez, C.; Adeva, J.; et al. Tumor-Stromal Interactions in a Co-Culture Model of Human Pancreatic Adenocarcinoma Cells and Fibroblasts and Their Connection with Tumor Spread. Biomedicines 2021, 9, 364. [Google Scholar] [CrossRef]
- Nobre, C.C.; de Araujo, J.M.; Fernandes, T.A.; Cobucci, R.N.; Lanza, D.C.; Andrade, V.S.; Fernandes, J.V. Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathol. Oncol. Res. 2017, 23, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Conroy, H.; Mawhinney, L.; Donnelly, S.C. Inflammation and cancer: Macrophage migration inhibitory factor (MIF)—The potential missing link. QJM 2010, 103, 831–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Wei, Y. Antitumor activity of celastrol by inhibition of proliferation, invasion, and migration in cholangiocarcinoma via PTEN/PI3K/Akt pathway. Cancer Med. 2020, 9, 783–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabe, H.; Beppu, T.; Hayashi, H.; Horino, K.; Masuda, T.; Komori, H.; Ishikawa, S.; Watanabe, M.; Takamori, H.; Iyama, K.; et al. Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 2009, 16, 2555–2564. [Google Scholar] [CrossRef]
- Nakamura, T.; Matsumoto, K.; Kiritoshi, A.; Tano, Y.; Nakamura, T. Induction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: In vitro analysis of tumor-stromal interactions. Cancer Res. 1997, 57, 3305–3313. [Google Scholar]
- Heits, N.; Heinze, T.; Bernsmeier, A.; Kerber, J.; Hauser, C.; Becker, T.; Kalthoff, H.; Egberts, J.H.; Braun, F. Influence of mTOR-inhibitors and mycophenolic acid on human cholangiocellular carcinoma and cancer associated fibroblasts. BMC Cancer 2016, 16, 322. [Google Scholar] [CrossRef] [Green Version]
- Tawfik, D.; Groth, C.; Gundlach, J.P.; Peipp, M.; Kabelitz, D.; Becker, T.; Oberg, H.H.; Trauzold, A.; Wesch, D. TRAIL-Receptor 4 Modulates gammadelta T Cell-Cytotoxicity toward Cancer Cells. Front. Immunol. 2019, 10, 2044. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gundlach, J.-P.; Kerber, J.; Hendricks, A.; Bernsmeier, A.; Halske, C.; Röder, C.; Becker, T.; Röcken, C.; Braun, F.; Sebens, S.; et al. Paracrine Interaction of Cholangiocellular Carcinoma with Cancer-Associated Fibroblasts and Schwann Cells Impact Cell Migration. J. Clin. Med. 2022, 11, 2785. https://doi.org/10.3390/jcm11102785
Gundlach J-P, Kerber J, Hendricks A, Bernsmeier A, Halske C, Röder C, Becker T, Röcken C, Braun F, Sebens S, et al. Paracrine Interaction of Cholangiocellular Carcinoma with Cancer-Associated Fibroblasts and Schwann Cells Impact Cell Migration. Journal of Clinical Medicine. 2022; 11(10):2785. https://doi.org/10.3390/jcm11102785
Chicago/Turabian StyleGundlach, Jan-Paul, Jannik Kerber, Alexander Hendricks, Alexander Bernsmeier, Christine Halske, Christian Röder, Thomas Becker, Christoph Röcken, Felix Braun, Susanne Sebens, and et al. 2022. "Paracrine Interaction of Cholangiocellular Carcinoma with Cancer-Associated Fibroblasts and Schwann Cells Impact Cell Migration" Journal of Clinical Medicine 11, no. 10: 2785. https://doi.org/10.3390/jcm11102785
APA StyleGundlach, J. -P., Kerber, J., Hendricks, A., Bernsmeier, A., Halske, C., Röder, C., Becker, T., Röcken, C., Braun, F., Sebens, S., & Heits, N. (2022). Paracrine Interaction of Cholangiocellular Carcinoma with Cancer-Associated Fibroblasts and Schwann Cells Impact Cell Migration. Journal of Clinical Medicine, 11(10), 2785. https://doi.org/10.3390/jcm11102785