
Dystonia Diagnosis: Clinical Neurophysiology and Genetics

 ,
,  ,
,
Abstract
:1. Introduction
- Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both.
- Dystonic movements are typically patterned, twisting, and may be tremulous.
- Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation.
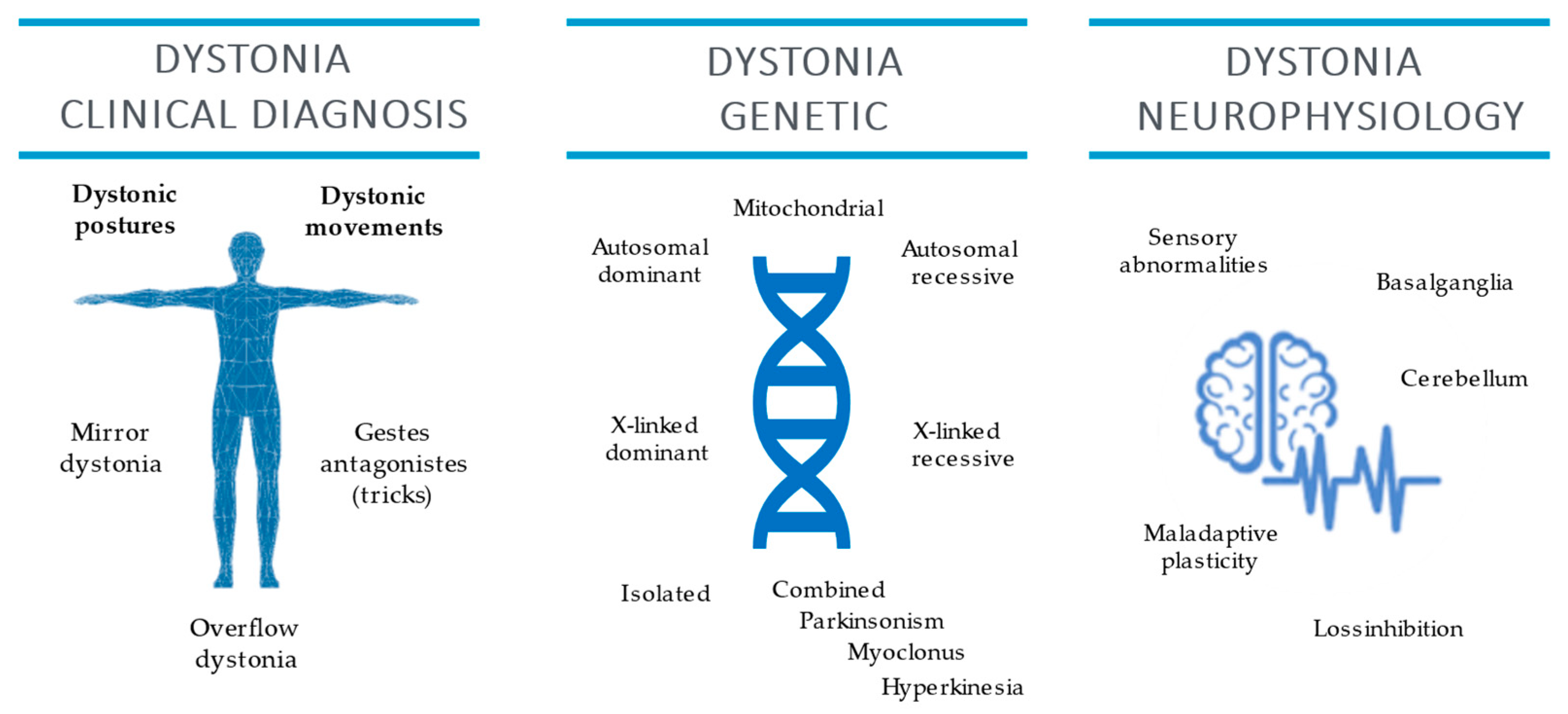
2. Clinical Neurophysiology
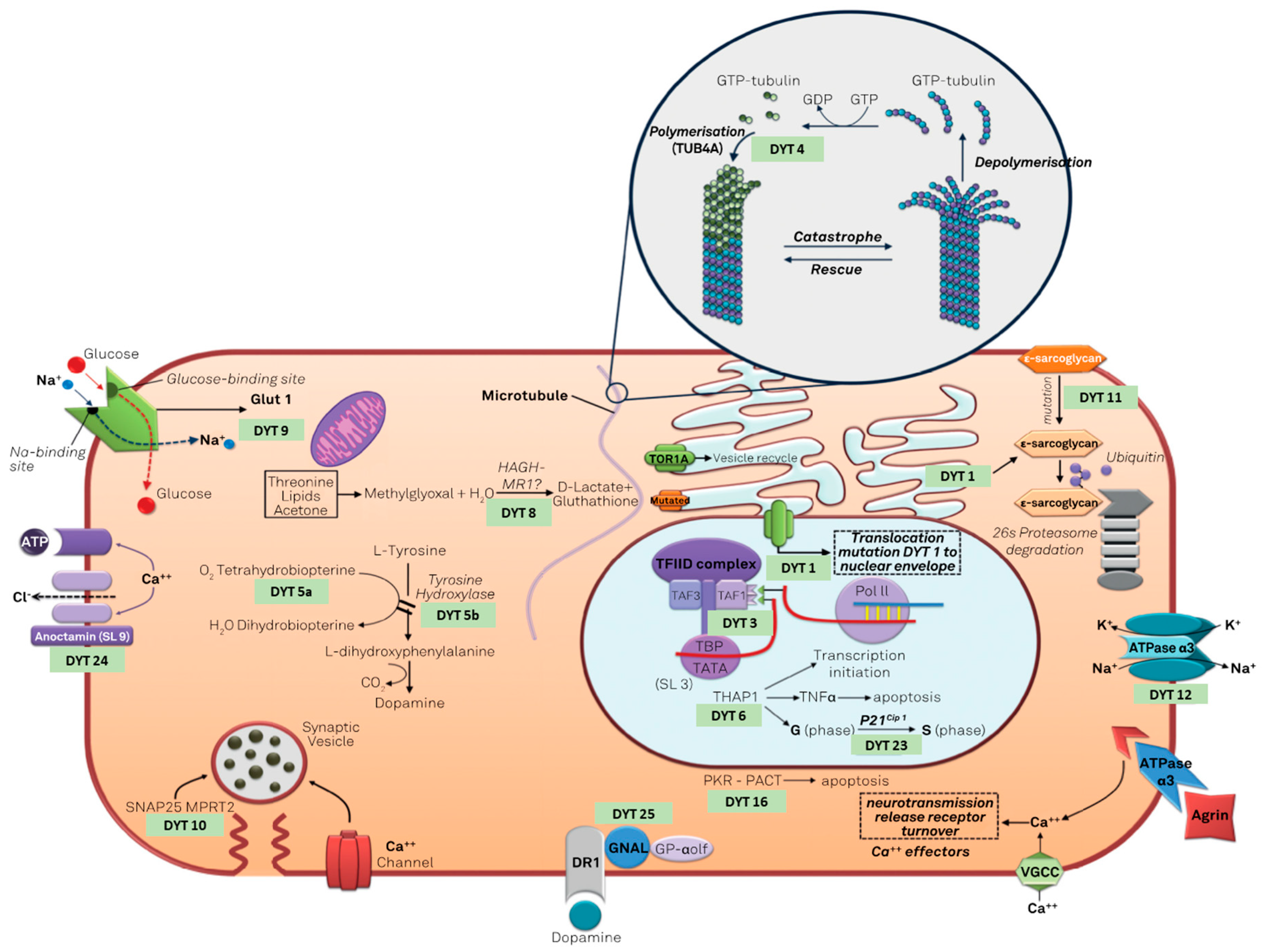
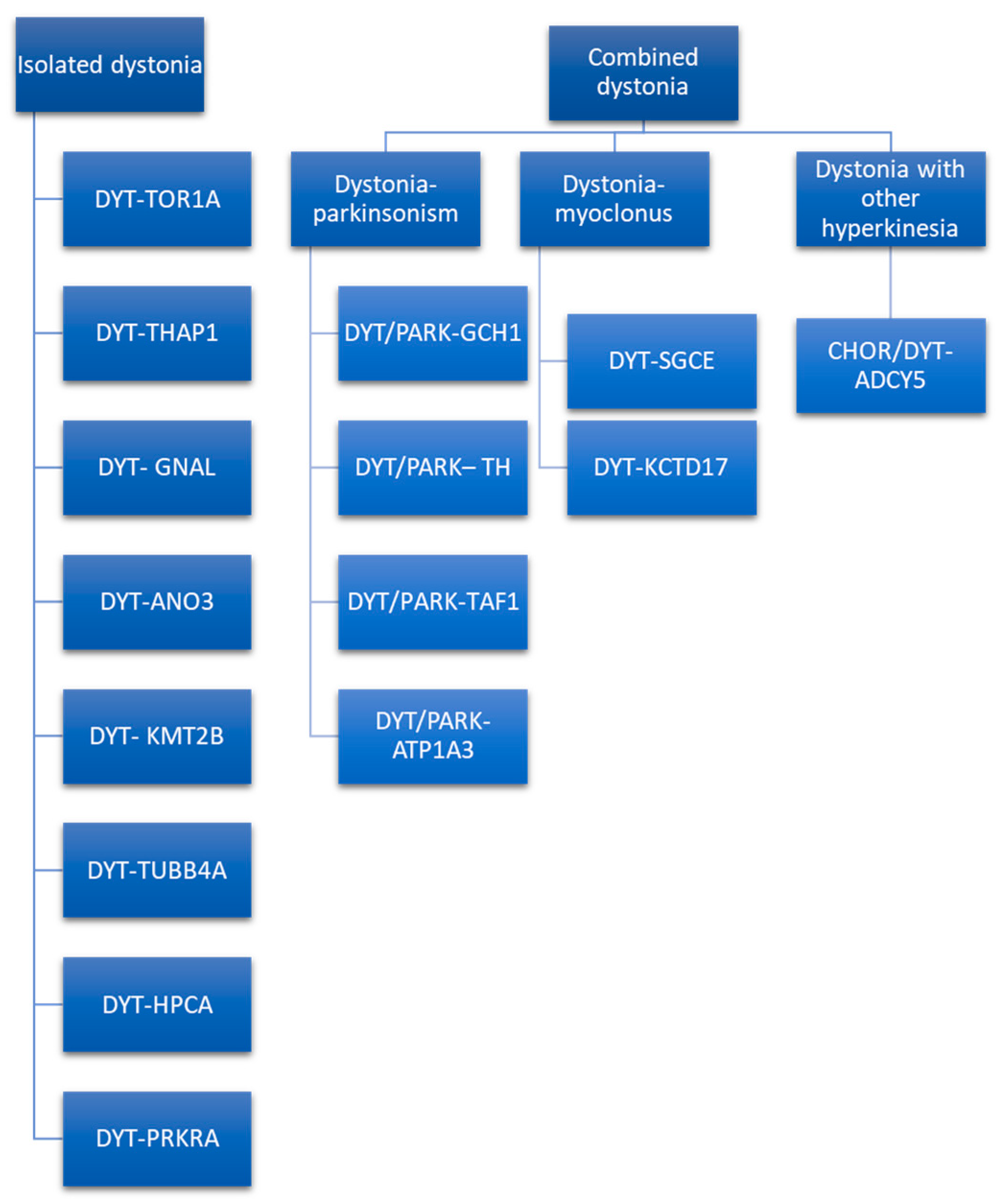
3. Dystonia Genetics
Genetic Testing and Genetic Counseling
- -
- Age at onset (0–20 years: score 2; >21 years: score 0),
- -
- Body distribution (generalized or segmental: score 1; focal: score 0),
- -
- Dystonia category (complex dystonia: score 2; combined dystonia: score 1; isolated dystonia: score 0).
- -
- DYT-SGCE dystonia has maternal imprinting of the gene, meaning that the dystonia-myoclonus only manifests when SGCE pathogenic variants are paternally inherited [103].
- -
- DYT-TOR1A has a reduced penetrance of the GAG deletion in TOR1A, from about 35% to 3% in individuals who also have a heterozygous NM_000113.2:646G>C (p.Asp216His) variant in TOR1A on the other allele [72].
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Albanese, A.; Bhatia, K.; Bressman, S.B.; DeLong, M.R.; Fahn, S.; Fung, V.S.; Hallett, M.; Jankovic, J.; Jinnah, H.A.; Klein, C.; et al. Phenomenology and classification of dystonia: A consensus update. Mov. Disord. 2013, 28, 863–873. [Google Scholar] [CrossRef] [Green Version]
- Albanese, A.; Lalli, S. Is this dystonia? Mov. Disord. 2009, 24, 1725–1731. [Google Scholar] [CrossRef]
- Albanese, A.; Di Giovanni, M.; Lalli, S. Dystonia: Diagnosis and management. Eur. J. Neurol. 2018, 26, 5–17. [Google Scholar] [CrossRef]
- Di Biase, L.; Di Santo, A.; Caminiti, M.L.; Pecoraro, P.M.; Di Lazzaro, V. Classification of Dystonia. Life 2022, 12, 206. [Google Scholar] [CrossRef]
- Van Gerpen, J.A.; Matsumoto, J.Y.; Ahlskog, J.E.; Maraganore, D.M.; McManis, P.G. Utility of an EMG mapping study in treating cervical dystonia. Muscle Nerve 2000, 23, 1752–1756. [Google Scholar] [CrossRef]
- Edwards, M.J.; Talelli, P.; Rothwell, J.C. Clinical applications of transcranial magnetic stimulation in patients with movement disorders. Lancet Neurol. 2008, 7, 827–840. [Google Scholar] [CrossRef]
- Chen, R.; Cros, D.; Curra, A.; Di Lazzaro, V.; Lefaucheur, J.-P.; Magistris, M.R.; Mills, K.; Rösler, K.M.; Triggs, W.J.; Ugawa, Y.; et al. The clinical diagnostic utility of transcranial magnetic stimulation: Report of an IFCN committee. Clin. Neurophysiol. 2008, 119, 504–532. [Google Scholar] [CrossRef]
- Fregni, F.; Boggio, P.S.; Santos, M.C.; Lima, M.; Vieira, A.L.; Rigonatti, S.P.; Silva, M.T.A.; Barbosa, E.R.; Nitsche, M.A.; Pascual-Leone, A. Noninvasive cortical stimulation with transcranial direct current stimulation in Parkinson’s disease. Mov. Disord. 2006, 21, 1693–1702. [Google Scholar] [CrossRef]
- Ferrucci, R.; Mameli, F.; Ruggiero, F.; Priori, A. Transcranial direct current stimulation as treatment for Parkinson’s disease and other movement disorders. Basal Ganglia 2015, 6, 53–61. [Google Scholar] [CrossRef]
- Darrow, D.P. Focused Ultrasound for Neuromodulation. Neurotherapeutics 2019, 16, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Di Biase, L.; Falato, E.; Di Lazzaro, V. Transcranial Focused Ultrasound (tFUS) and Transcranial Unfocused Ultrasound (tUS) Neuromodulation: From Theoretical Principles to Stimulation Practices. Front. Neurol. 2019, 10, 549. [Google Scholar] [CrossRef] [Green Version]
- Di Biase, L.; Falato, E.; Caminiti, M.L.; Pecoraro, P.M.; Narducci, F.; Di Lazzaro, V. Focused Ultrasound (FUS) for Chronic Pain Management: Approved and Potential Applications. Neurol. Res. Int. 2021, 2021, 1–16. [Google Scholar] [CrossRef]
- Hallett, M. Chapter 1 Movement disorders: Overview. In Handbook of Clinical Neurophysiology; Elsevier: Amsterdam, The Netherlands, 2003; pp. 3–4. [Google Scholar]
- Bologna, M.; Suppa, A.; Di Stasio, F.; Conte, A.; Fabbrini, G.; Berardelli, A. Neurophysiological studies on atypical parkinsonian syndromes. Park. Relat. Disord. 2017, 42, 12–21. [Google Scholar] [CrossRef]
- Valls-Solé, J. Neurophysiological characterization of parkinsonian syndromes. Neurophysiol. Clin. Neurophysiol. 2000, 30, 352–367. [Google Scholar] [CrossRef]
- Valls-Solé, J.; Valldeoriola, F. Neurophysiological correlate of clinical signs in Parkinson’s disease. Clin. Neurophysiol. 2002, 113, 792–805. [Google Scholar] [CrossRef]
- Di Biase, L.; Brittain, J.-S.; Shah, S.A.; Pedrosa, D.; Cagnan, H.; Mathy, A.; Chen, C.C.; Martín-Rodríguez, J.F.; Mir, P.; Timmerman, L.; et al. Tremor stability index: A new tool for differential diagnosis in tremor syndromes. Brain 2017, 140, 1977–1986. [Google Scholar] [CrossRef]
- Di Pino, G.; Formica, D.; Melgari, J.M.; Taffoni, F.; Salomone, G.; di Biase, L.; Caimo, E.; Vernieri, F.; Guglielmelli, E. Neurophysiological bases of tremors and accelerometric parameters analysis. In Proceedings of the 2012 4th IEEE RAS & EMBS International Conference on Biomedical Robotics and Biomechatronics (BioRob), Rome, Italy, 24−27 June 2012; pp. 1820–1825. [Google Scholar]
- Deuschl, G.; Krack, P.; Lauk, M.; Timmer, J. Clinical Neurophysiology of Tremor. J. Clin. Neurophysiol. 1996, 13, 110–121. [Google Scholar] [CrossRef]
- Caviness, J.N. Chapter 32 The clinical neurophysiology of myoclonus. In Handbook of Clinical Neurophysiology; Hallett, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 521–548. [Google Scholar]
- Kaji, R. Chapter 28 Dystonia. In Handbook of Clinical Neurophysiology; Hallett, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 451–461. [Google Scholar]
- Hallett, M. Neurophysiology of dystonia: The role of inhibition. Neurobiol. Dis. 2011, 42, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, J.C.; Obeso, J.A.; Day, B.L.; Marsden, C.D. Pathophysiology of dystonias. Adv. Neurol. 1983, 39, 851–863. [Google Scholar]
- Nakashima, K.; Rothwell, J.C.; Day, B.L.; Thompson, P.D.; Shannon, K.; Marsden, C.D. Reciprocal inhibition between forearm muscles in patients with writer’s cramp and other occupational cramps, symptomatic hemidystonia and hemiparesis due to stroke. Brain 1989, 112, 681–697. [Google Scholar] [CrossRef]
- Berardelli, A.; Rothwell, J.; Day, B.L.; Marsden, C.D. Pathophysiology of blepharospasm and oromandibular dystonia. Brain 1985, 108, 593–608. [Google Scholar] [CrossRef]
- Quartarone, A.; Girlanda, P.; Di Lazzaro, V.; Majorana, G.; Battaglia, F.; Messina, C. Short latency trigemi-no-sternocleidomastoid response in muscles in patients with spasmodic torticollis and blepharospasm. Clin. Neurophysiol. 2000, 111, 1672–1677. [Google Scholar] [CrossRef]
- Huang, Y.-Z.; Rothwell, J.; Lu, C.-S.; Wang, J.-J.; Chen, R.-S. Restoration of motor inhibition through an abnormal premotor-motor connection in dystonia. Mov. Disord. 2010, 25, 696–703. [Google Scholar] [CrossRef]
- Espay, A.; Morgante, F.; Purzner, J.; Gunraj, C.A.; Lang, A.; Chen, R. Cortical and spinal abnormalities in psychogenic dystonia. Ann. Neurol. 2006, 59, 825–834. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Oliviero, A.; Profice, P.; Dileone, M.; Pilato, F.; Insola, A.; Della Marca, G.; Tonali, P.; Mazzone, P. Reduced cerebral cortex inhibition in dystonia: Direct evidence in humans. Clin. Neurophysiol. 2009, 120, 834–839. [Google Scholar] [CrossRef]
- Beck, S.; Shamim, E.A.; Richardson, S.P.; Schubert, M.; Hallett, M. Inter-hemispheric inhibition is impaired in mirror dystonia. Eur. J. Neurosci. 2009, 29, 1634–1640. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Wassermann, E.M.; Caños, M.; Hallett, M. Impaired inhibition in writer’s cramp during voluntary muscle activation. Neurology 1997, 49, 1054–1059. [Google Scholar] [CrossRef]
- Tinazzi, M.; Farina, S.; Edwards, M.; Moretto, G.; Restivo, D.; Fiaschi, A.; Berardelli, A. Task−specific impairment of motor cortical excitation and inhibition in patients with writer’s cramp. Neurosci. Lett. 2005, 378, 55–58. [Google Scholar] [CrossRef]
- Kojovic, M.; Pareés, I.; Kassavetis, P.; Palomar, F.J.; Mir, P.; Teo, J.; Cordivari, C.; Rothwell, J.; Bhatia, K.; Edwards, M.J. Secondary and primary dystonia: Pathophysiological differences. Brain 2013, 136, 2038–2049. [Google Scholar] [CrossRef] [Green Version]
- Sadnicka, A.; Teo, J.; Kojovic, M.; Pareés, I.; Saifee, T.A.; Kassavetis, P.; Schwingenschuh, P.; Katschnig−Winter, P.; Stamelou, M.; Mencacci, N.E.; et al. All in the blink of an eye: New insight into cerebellar and brainstem function in DYT1 and DYT6 dystonia. Eur. J. Neurol. 2014, 22, 762–767. [Google Scholar] [CrossRef]
- Brighina, F.; Romano, M.; Giglia, G.; Saia, V.; Puma, A.; Giglia, F.; Fierro, B. Effects of cerebellar TMS on motor cortex of patients with focal dystonia: A preliminary report. Exp. Brain Res. 2008, 192, 651–656. [Google Scholar] [CrossRef]
- Molloy, F.M.; Zeuner, K.E.; Dambrosia, J.M.; Carr, T.D.; Hallett, M. Abnormalities of spatial discrimination in focal and generalized dystonia. Brain 2003, 126, 2175–2182. [Google Scholar] [CrossRef]
- Conte, A.; Ferrazzano, G.; Belvisi, D.; Manzo, N.; Battista, E.; Voti, P.L.; Nardella, A.; Fabbrini, G.; Berardelli, A. Somatosensory temporal discrimination in Parkinson’s disease, dystonia and essential tremor: Pathophysiological and clinical implications. Clin. Neurophysiol. 2018, 129, 1849–1853. [Google Scholar] [CrossRef]
- Grünewald, R.A.; Shipman, J.M.; Sagar, H.J.; Yoneda, Y. Idiopathic focal dystonia: A disorder of muscle spindle afferent processing? Brain 1997, 120, 2179–2185. [Google Scholar] [CrossRef]
- Quartarone, A.; Bagnato, S.; Rizzo, V.; Siebner, H.R.; Dattola, V.; Scalfari, A.; Morgante, F.; Battaglia, F.; Romano, M.; Girlanda, P. Abnormal associative plasticity of the human motor cortex in writer’s cramp. Brain 2003, 126, 2586–2596. [Google Scholar] [CrossRef]
- Erro, R.; Rocchi, L.; Antelmi, E.; Liguori, R.; Tinazzi, M.; Berardelli, A.; Rothwell, J.; Bhatia, K.P. High frequency somatosensory stimulation in dystonia: Evidence fordefective inhibitory plasticity. Mov. Disord. 2018, 33, 1902–1909. [Google Scholar] [CrossRef]
- Silberstein, P.; Kühn, A.A.; Kupsch, A.; Trottenberg, T.; Krauss, J.K.; Wöhrle, J.C.; Mazzone, P.; Insola, A.; Di Lazzaro, V.; Oliviero, A.; et al. Patterning of globus pallidus local field potentials differs between Parkinson’s disease and dystonia. Brain 2003, 126, 2597–2608. [Google Scholar] [CrossRef]
- Chen, C.C.; Kühn, A.A.; Trottenberg, T.; Kupsch, A.; Schneider, G.-H.; Brown, P. Neuronal activity in globus pallidus interna can be synchronized to local field potential activity over 3–12 Hz in patients with dystonia. Exp. Neurol. 2006, 202, 480–486. [Google Scholar] [CrossRef]
- Conte, A.; Rocchi, L.; Latorre, A.; Belvisi, D.; Rothwell, J.C.; Berardelli, A. Ten-Year Reflections on the Neurophysiological Abnormalities of Focal Dystonias in Humans. Mov. Disord. 2019, 34, 1616–1628. [Google Scholar] [CrossRef]
- Latorre, A.; Rocchi, L.; Bhatia, K.P. Delineating the electrophysiological signature of dystonia. Exp. Brain Res. 2020, 238, 1685–1692. [Google Scholar] [CrossRef]
- Bologna, M.; Berardelli, A. The cerebellum and dystonia. Handb. Clin. Neurol. 2018, 155, 259–272. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Molinari, M.; Restuccia, D.; Leggio, M.; Nardone, R.; Fogli, D.; Tonali, P. Cerebro-cerebellar interactions in man: Neurophysiological studies in patients with focal cerebellar lesions. Electroencephalogr. Clin. Neurophysiol. Potentials Sect. 1994, 93, 27–34. [Google Scholar] [CrossRef]
- Tinazzi, M.; Marotta, A.; Fasano, A.; Bove, F.; Bentivoglio, A.R.; Squintani, G.; Pozzer, L.; Fiorio, M. Aristotle’s illusion reveals interdigit functional somatosensory alterations in focal hand dystonia. Brain 2013, 136, 782–789. [Google Scholar] [CrossRef] [Green Version]
- Tinazzi, M.; Priori, A.; Bertolasi, L.; Frasson, E.; Mauguière, F.; Fiaschi, A. Abnormal central integration of a dual somatosensory input in dystonia Evidence for sensory overflow. Brain 2000, 123, 42–50. [Google Scholar] [CrossRef]
- Edwards, M.J.; Huang, Y.-Z.; Mir, P.; Rothwell, J.; Bhatia, K.P. Abnormalities in motor cortical plasticity differentiate manifesting and nonmanifesting DYT1 carriers. Mov. Disord. 2006, 21, 2181–2186. [Google Scholar] [CrossRef]
- Quartarone, A.; Rizzo, V.; Terranova, C.; Morgante, F.; Schneider, S.; Ibrahim, N.; Girlanda, P.; Bhatia, K.P.; Rothwell, J.C. Abnormal sensorimotor plasticity in organic but not in psychogenic dystonia. Brain 2009, 132, 2871–2877. [Google Scholar] [CrossRef] [Green Version]
- Dileone, M.; Profice, P.; Pilato, F.; Alfieri, P.; Cesarini, L.; Mercuri, E.; Leoni, C.; Tartaglia, M.; Di Iorio, R.; Zampino, G.; et al. Enhanced human brain associative plasticity in Costello syndrome. J. Physiol. 2010, 588, 3445–3456. [Google Scholar] [CrossRef]
- Dileone, M.; Zampino, G.; Profice, P.; Pilato, F.; Leoni, C.; Ranieri, F.; Capone, F.; Tartaglia, M.; Brown, P.; Di Lazzaro, V. Dystonia in Costello syndrome. Park. Relat. Disord. 2012, 18, 798–800. [Google Scholar] [CrossRef]
- Piña-Fuentes, D.; Beudel, M.; Little, S.; van Zijl, J.; Elting, J.W.; Oterdoom, D.L.M.; van Egmond, M.E.; van Dijk, J.M.C.; Tijssen, M.A.J. Toward adaptive deep brain stimulation for dystonia. Neurosurg. Focus 2018, 45, E3. [Google Scholar] [CrossRef] [Green Version]
- Assenza, G.; Capone, F.; di Biase, L.; Ferreri, F.; Florio, L.; Guerra, A.; Marano, M.; Paolucci, M.; Ranieri, F.; Salomone, G. Oscillatory activities in neurological disorders of elderly: Biomarkers to target for neuromodulation. Front. Aging Neurosci. 2017, 9, 189. [Google Scholar] [CrossRef]
- Starr, P.A.; Rau, G.M.; Davis, V.; Marks, W.J.; Ostrem, J.L.; Simmons, D.; Lindsey, N.; Turner, R. Spontaneous Pallidal Neuronal Activity in Human Dystonia: Comparison With Parkinson’s Disease and Normal Macaque. J. Neurophysiol. 2005, 93, 3165–3176. [Google Scholar] [CrossRef] [Green Version]
- Vitek, J.L.; Delong, M.R.; Starr, P.A.; Hariz, M.I.; Metman, L.V. Intraoperative neurophysiology in DBS for dystonia. Mov. Disord. 2011, 26, S31–S36. [Google Scholar] [CrossRef]
- Little, S.; Pogosyan, A.; Neal, S.; Ba, B.Z.; Zrinzo, L.; Hariz, M.; Foltynie, T.; Limousin, P.; Ashkan, K.; FitzGerald, J.; et al. Adaptive deep brain stimulation in advanced Parkinson disease. Ann. Neurol. 2013, 74, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Little, S.; Pogosyan, A.; Neal, S.; Zrinzo, L.; Hariz, M.; Foltynie, T.; Limousin, P.; Brown, P. Controlling Parkinson’s Disease With Adaptive Deep Brain Stimulation. J. Vis. Exp. 2014, 89, e51403. [Google Scholar] [CrossRef] [Green Version]
- Tinkhauser, G.; Pogosyan, A.; Debove, I.; Nowacki, A.; Shah, S.A.; Seidel, K.; Tan, H.; Brittain, J.-S.; Petermann, K.; Di Biase, L.; et al. Directional local field potentials: A tool to optimize deep brain stimulation. Mov. Disord. 2017, 33, 159–164. [Google Scholar] [CrossRef]
- Di Biase, L.; Tinkhauser, G.; Martin Moraud, E.; Caminiti, M.L.; Pecoraro, P.M.; Di Lazzaro, V. Adaptive, personalized closed−loop therapy for Parkinson’s disease: Biochemical, neurophysiological, and wearable sensing systems. Expert Rev. Neurother. 2021, 21, 1371–1388. [Google Scholar] [CrossRef]
- Di Biase, L.; Di Santo, A.; Caminiti, M.L.; De Liso, A.; Shah, S.A.; Ricci, L.; Di Lazzaro, V. Gait Analysis in Parkinson’s Disease: An Overview of the Most Accurate Markers for Diagnosis and Symptoms Monitoring. Sensors 2020, 20, 3529. [Google Scholar] [CrossRef]
- Di Biase, L.; Summa, S.; Tosi, J.; Taffoni, F.; Marano, M.; Cascio Rizzo, A.; Vecchio, F.; Formica, D.; Di Lazzaro, V.; Di Pino, G.; et al. Quantitative Analysis of Bradykinesia and Rigidity in Parkinson’s Disease. Front. Neurol. 2018, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Raiano, L.; di Pino, G.; di Biase, L.; Tombini, M.; Tagliamonte, N.L.; Formica, D. PDMeter: A Wrist Wearable Device for an at−Home Assessment of the Parkinson’s Disease Rigidity. IEEE Trans. Neural Syst. Rehabil. Eng. 2020, 28, 1325–1333. [Google Scholar] [CrossRef]
- Di Biase, L.; Raiano, L.; Caminiti, M.L.; Pecoraro, P.M.; Di Lazzaro, V. Artificial intelligence in Parkinson’s disease-symptoms identification and monitoring. In Augmenting Neurological Disorder Prediction and Rehabilitation Using Artifi−cial Intelligence; Elsevier: Amsterdam, The Netherlands, 2022; pp. 35–52. [Google Scholar]
- Bressman, S.B. Dystonia genotypes, phenotypes, and classification. Adv. Neurol. 2004, 94, 101–107. [Google Scholar]
- Camargos, S.; Cardoso, F. Understanding dystonia: Diagnostic issues and how to overcome them. Arq. Neuropsiquiatr. 2016, 74, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.L.; De Leon, D.; Ozelius, L.; Risch, N.; Bressman, S.B.; Brin, M.F.; Schuback, D.E.; Burke, R.E.; Kwiatkowski, D.J.; Shale, H.; et al. Dystonia gene in Ashkenazi Jewish population is located on chromosome 9q32-34. Ann. Neurol. 1990, 27, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Lohmann, K.; Lang, A.; Klein, C. Fixing the broken system of genetic locus symbols: Parkinson disease and dystonia as examples. Neurology 2012, 78, 1016–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marras, C.; Lang, A.; Van De Warrenburg, B.P.; Sue, C.M.; Tabrizi, S.J.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 2016, 31, 436–457. [Google Scholar] [CrossRef]
- Lange, L.M.; Junker, J.; Loens, S.; Baumann, H.; Olschewski, L.; Schaake, S.; Madoev, H.; Petkovic, S.; Kuhnke, N.; Kasten, M.; et al. Genotype–Phenotype Relations for Isolated Dystonia Genes: MDSGene Systematic Review. Mov. Disord. 2021, 36, 1086–1103. [Google Scholar] [CrossRef]
- Weissbach, A.; Saranza, G.; Domingo, A. Combined dystonias: Clinical and genetic updates. J. Neural Transm. 2020, 128, 417–429. [Google Scholar] [CrossRef]
- Risch, N.J.; Bressman, S.B.; Senthil, G.; Ozelius, L.J. Intragenic Cis and Trans Modification of Genetic Susceptibility in DYT1 Torsion Dystonia. Am. J. Hum. Genet. 2007, 80, 1188–1193. [Google Scholar] [CrossRef] [Green Version]
- Djarmati, A.; A Schneider, S.; Lohmann, K.; Winkler, S.; Pawlack, H.; Hagenah, J.; Brüggemann, N.; Zittel, S.; Fuchs, T.; Raković, A.; et al. Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: A genetic screening study. Lancet Neurol. 2009, 8, 447–452. [Google Scholar] [CrossRef]
- Hersheson, J.; Mencacci, N.E.; Davis, M.; Macdonald, N.; Trabzuni, D.; Ryten, M.; Pittman, A.; Paudel, R.; Kara, E.; Fawcett, K.; et al. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann. Neurol. 2012, 73, 546–553. [Google Scholar] [CrossRef]
- Wilcox, R.A.; Winkler, S.; Lohmann, K.; Klein, C. Whispering dysphonia in an Australian family (DYT4): A clinical and genetic reappraisal. Mov. Disord. 2011, 26, 2404–2408. [Google Scholar] [CrossRef]
- Artusi, C.A.; Dwivedi, A.; Romagnolo, A.; Bortolani, S.; Marsili, L.; Imbalzano, G.; Sturchio, A.; Keeling, E.G.; Zibetti, M.; Contarino, M.F.; et al. Differential response to pallidal deep brain stimulation among monogenic dystonias: Systematic review and meta−analysis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Rajan, R.; Garg, K.; Saini, A.; Radhakrishnan, D.M.; Carecchio, M.; Bk, B.; Singh, M.; Srivastava, A.K. GPi-DBS for KMT2B-Associated Dystonia: Systematic Review and Meta-Analysis. Mov. Disord. Clin. Pract. 2021, 9, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.-T.; Li, L.-X.; Liu, Y.; Zhang, X.-L.; Pan, Y.-G.; Wang, L.; Wan, X.-H.; Jin, L.-J. The expanding clinical and genetic spectrum of ANO3 dystonia. Neurosci. Lett. 2020, 746, 135590. [Google Scholar] [CrossRef] [PubMed]
- Sarva, H.; Trosch, R.; Kiss, Z.H.; Furtado, S.; Luciano, M.S.; Glickman, A.; Ms, D.R.; Ozelius, L.J.; Bressman, S.B.; Saunders-Pullman, R. Deep Brain Stimulation in Isolated Dystonia With a GNAL Mutation. Mov. Disord. 2018, 34, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, H.; Ohye, T.; Takahashi, E.-I.; Seki, N.; Hori, T.-A.; Segawa, M.; Nomura, Y.; Endo, K.; Tanaka, H.; Tsuji, S.; et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat. Genet. 1994, 8, 236–242. [Google Scholar] [CrossRef]
- Salles, P.A.; Terán-Jimenez, M.; Vidal-Santoro, A.; Chaná-Cuevas, P.; Kauffman, M.; Espay, A.J. Recognizing Atypical Dopa−Responsive Dystonia and Its Mimics. Neurol. Clin. Pract. 2021, 11, e876–e884. [Google Scholar] [CrossRef]
- Dworniczak, B.; Lüdecke, B.; Bartholomé, K. A point mutation in the tyrosine hydroxylase gene associated with Segawa’s syndrome. Qual. Life Res. 1995, 95, 123–125. [Google Scholar] [CrossRef]
- Wevers, R.A.; Andel, J.F.D.R.-V.; Bräutigam, C.; Geurtz, B.; Heuvel, L.P.W.J.V.D.; Steenbergen-Spanjers, G.C.H.; Smeitink, J.A.M.; Hoffmann, G.F.; Gabreëls, F.J.M. A review of biochemical and molecular genetic aspects of tyrosine hydroxylase deficiency including a novel mutation (291delC). J. Inherit. Metab. Dis. 1999, 22, 364–373. [Google Scholar] [CrossRef]
- Stamelou, M.; Mencacci, N.E.; Cordivari, C.; Batla, A.; Wood, N.W.; Houlden, H.; Hardy, J.; Bhatia, K.P. Myoclonus−dystonia syndrome due to tyrosine hydroxylase deficiency. Neurology 2012, 79, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Weissbach, A.; Pauly, M.G.; Herzog, R.; Hahn, L.; Halmans, S.; Hamami, F.; Bolte, C.; Camargos, S.; Jeon, B.; Kurian, M.A.; et al. Relationship of Genotype, Phenotype, and Treatment in Dopa-Responsive Dystonia: MDSGene Review. Mov. Disord. 2021, 37, 237–252. [Google Scholar] [CrossRef]
- Makino, S.; Kaji, R.; Ando, S.; Tomizawa, M.; Yasuno, K.; Goto, S.; Matsumoto, S.; Tabuena, M.D.; Maranon, E.; Dantes, M.; et al. Reduced Neuron-Specific Expression of the TAF1 Gene Is Associated with X-Linked Dystonia-Parkinsonism. Am. J. Hum. Genet. 2007, 80, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.V.; Pascasio, F.M.; Fuentes, F.D.; Viterbo, G.H. Torsion dystonia in Panay, Philippines. Adv. Neurol. 1976, 14, 137–151. [Google Scholar]
- Brüggemann, N.; Heldmann, M.; Klein, C.; Domingo, A.; Rasche, D.; Tronnier, V.; Rosales, R.L.; Jamora, R.D.G.; Lee, L.V.; Münte, T.F. Neuroanatomical changes extend beyond striatal atrophy in X-linked dystonia parkinsonism. Park. Relat. Disord. 2016, 31, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Song, P.C.; Le Bs, H.; Acuna, P.; De Guzman, J.K.P.; Sharma, N.; Ba, T.N.F.; Dy, M.E.; Go, C.L. Voice and swallowing dysfunction in X-linked dystonia parkinsonism. Laryngoscope 2019, 130, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Brashear, A.; Dobyns, W.; Aguiar, P.D.C.; Borg, M.; Frijns, C.J.M.; Gollamudi, S.; Green, A.; Guimaraes, J.; Haake, B.C.; Klein, C.; et al. The phenotypic spectrum of rapid−onset dystonia−parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 2007, 130, 828–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguiar, P.D.C.; Sweadner, K.J.; Penniston, J.T.; Zaremba, J.; Liu, L.; Caton, M.; Linazasoro, G.; Borg, M.; Tijssen, M.A.; Bressman, S.B.; et al. Mutations in the Na+/K+−ATPase α3 Gene ATP1A3 Are Associated with Rapid−Onset Dystonia Parkinsonism. Neuron 2004, 43, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Mencacci, N.E.; Rubio-Agusti, I.; Zdebik, A.; Asmus, F.; Ludtmann, M.H.; Ryten, M.; Plagnol, V.; Hauser, A.-K.; Bandres-Ciga, S.; Bettencourt, C.; et al. A Missense Mutation in KCTD17 Causes Autosomal Dominant Myoclonus−Dystonia. Am. J. Hum. Genet. 2015, 96, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Ferrini, A.; Steel, D.; Barwick, K.; Kurian, M.A. An Update on the Phenotype, Genotype and Neurobiology of ADCY5-Related Disease. Mov. Disord. 2021, 36, 1104–1114. [Google Scholar] [CrossRef]
- Méneret, A.; Gras, D.; McGovern, E.; Roze, E. Caffeine and the Dyskinesia Related to Mutations in the ADCY5 Gene. Ann. Intern. Med. 2019, 171, 439. [Google Scholar] [CrossRef]
- Albanese, A.; Asmus, F.; Bhatia, K.; Elia, A.E.; Elibol, B.; Filippini, G.; Gasser, T.; Krauss, J.K.; Nardocci, N.; Newton, A.; et al. EFNS guidelines on diagnosis and treatment of primary dystonias. Eur. J. Neurol. 2010, 18, 5–18. [Google Scholar] [CrossRef]
- Bressman, S.B.; Sabatti, C.; Raymond, D.; de Leon, D.; Klein, C.; Kramer, P.L.; Brin, M.F.; Fahn, S.; Breakefield, X.; Ozelius, L.J.; et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology 2000, 54, 1746–1753. [Google Scholar] [CrossRef] [PubMed]
- American Society of Human Genetics Board of Directors. Points to consider: Ethical, legal, and psychosocial implications of genetic testing in children and adolescents. Am. J. Hum. Genet. 1995, 57, 1233–1241. [Google Scholar]
- Klein, C.; Friedman, J.; Bressman, S.; Vieregge, P.; Brin, M.F.; Pramstaller, P.P.; de Leon, D.; Hagenah, J.; Sieberer, M.; Fleet, C.; et al. Genetic Testing for Early-Onset Torsion Dystonia (DYT1): Introduction of a Simple Screening Method, Experiences from Testing of a Large Patient Cohort, and Ethical Aspects. Genet. Test. 1999, 3, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.; McCarthy, G.T.; Bandmann, O.; Dobbie, M.; Surtees, R.; Wood, N. GTP cyclohydrolase deficiency; intrafamilial variation in clinical phenotype, including levodopa responsiveness. J. Neurol. Neurosurg. Psychiatry 1999, 66, 86–89. [Google Scholar] [CrossRef]
- Valente, E.M.; Edwards, M.J.; Mir, P.; DiGiorgio, A.; Salvi, S.; Davis, M.; Russo, N.; Bozi, M.; Kim, H.-T.; Pennisi, G.; et al. The epsilon−sarcoglycan gene in myoclonic syndromes. Neurology 2005, 64, 737–739. [Google Scholar] [CrossRef]
- Zech, M.; Jech, R.; Boesch, S.; Škorvánek, M.; Weber, S.; Wagner, M.; Zhao, C.; Jochim, A.; Necpál, J.; Dincer, Y.; et al. Monogenic variants in dystonia: An exome−wide sequencing study. Lancet Neurol. 2020, 19, 908–918. [Google Scholar] [CrossRef]
- Klein, C.; Lohmann, K.; Marras, C.; Münchau, A. Hereditary Dystonia Overview. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Müller, B.; Hedrich, K.; Kock, N.; Dragasevic, N.; Svetel, M.; Garrels, J.; Landt, O.; Nitschke, M.; Pramstaller, P.P.; Reik, W.; et al. Evidence That Paternal Expression of the ε-Sarcoglycan Gene Accounts for Reduced Penetrance in Myoclonus-Dystonia. Am. J. Hum. Genet. 2002, 71, 1303–1311. [Google Scholar] [CrossRef] [Green Version]
- Arlotti, M.; Colombo, M.; Bonfanti, A.; Mandat, T.; Lanotte, M.M.; Pirola, E.; Borellini, L.; Rampini, P.; Eleopra, R.; Rinaldo, S.; et al. A New Implantable Closed−Loop Clinical Neural Interface: First Application in Parkinson’s Disease. Front. Neurosci. 2021, 15, 763235. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Neurophysiological Test | Results | Accuracy | Ref. | |
|---|---|---|---|---|
| Loss inhibition | EMG | Prolonged bursts Co-contraction agonist and antagonist muscles Overflow to other muscles | NA | [23] |
| Spinal cord reciprocal inhibition | Reduced reciprocal inhibition | NA | [24] | |
| Blink reflex recovery cycle | Reduced inhibition of R2 component | NA | [25] | |
| Short latency trigemino-sternocleidomastoid response | Impairment of the trigemino-cervical reflex | NA | [26] | |
| SICI | Reduced in most studies | NA | [27,28,29] | |
| IHI | Loss of suppression | NA | [30] | |
| SP | Reduced | NA | [31,32] | |
| Cerebelum | EBCC | Impaired in primary focal dystonia Normal in DYT-TOR1A and DYT-THAP1 dystonia | NA | [33,34] |
| CBI | Absent | NA | [35] | |
| Sensory Abnormalities | GOT | Increased SD threshold in blepharospasm, CD, FHD Normal in DYT-TOR1A | NA | [36] |
| STDT | Abnormally increased STDT (higher in CD patients with tremor). No statistical differences between CD and PD | CD compared to ET:
100% NPV
| [37] | |
| TVR | Abnormally increased | NA | [38] | |
| Maladaptive Plasticity | PAS | Abnormally increased in dystonic patients Normal in functional dystonia and DYT-TOR1A carrier | NA | [39] |
| HF-RSS | Reduced inhibition | NA | [40] | |
| Basal Ganglia | LFP recordings (GPi) | Synchronized activities in 4–10 Hz band | NA | [41,42] |
| Phenotype | Gene/ Locus | Inheritance/Penetrance | OMIM | Age of Onset | Body Distribution | Temporal Pattern | Associated Features | Drugs Response | DBS Response | Brain Imaging Findings | References | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dopa | Other Drugs | Alcohol | ||||||||||||
| Isolated | TOR1A/ DYT 1 | AD/ Reduced | 128100 | Childhood-Adolescence-Early adulthood | Generalized | Persistent | none | No | Anticholinergics | No | Good | None | [70] | |
| THAP1/ DYT 6 | AD/48% | 602629 | Childhood-Adolescence | Segmental-generalized | Persistent | Laryngeal dystonia/dysarthria/dysphonia | No | Anricholinergics | No | Variable | None | [70,73] | ||
| ANO3/ DYT 24 | AD/NA | 615034 | Infancy/childhood, early and late adulthood | Focal-Segmental | Persistent | Tremor | Yes | Anticholinergics/Antiepileptics | No | Good | None | [70,78] | ||
| GNAL/ DYT 25 | AD/High | 615073 | Early adulthood-Late adulthood | Focal-segmental-occasionaly generalized | Persistent | none | No | No | Yes | Good | None | [70] | ||
| KMT2B/ DYT 28 | AD/ Incomplete | 617284 | Infancy-Childhood-Adolescence | Generalized | Persistent | Nonmotor signs, neurodevelopemental disorders, Dysmorphisms, Psychiatric symptoms, | No | Anticholinergics | No | Good | Pallidal hypointensity | [70] | ||
| HPCA/ DYT2 | AR | 224500 | Infancy/childhood | Generalized | Persistent | Psychiatric features, cognitive impairment, dystonic tremor | No | Anticholinergics | No | Not know | None | [70] | ||
| TUBB4A/ DYT 4 | AD/High | 128101 | Childhood-Adolescence | Focal-generalized | Spasmodic dysphonia | Thin face-body habitus-hobby horse gait | No | No | Yes | Not known | None | [74,75] | ||
| PRKRA/ DYT 16 | AR/NA | 612067 | Infancy-Childhood-Adolescence | Generalized | Persistent | Parkinsonism, Hyperreflexia | No | No | No | Not known | None | [70] | ||
| CombinedKC | Parkinsonism | GCH1/ DYT 5a | AD/50% | 128230 | Infancy-Childhood | Mostly generalized | Diurnal fluctuations | Parkinsonism-spasticity-non motor features | Yes | None | No | Not known | None | [80,81] |
| TH/ DYT 5b | AR/NA | 605407 | Infancy | Mostly generalized | Diurnal fluctuations | Parkinsonism-ptosis- hypotonia-autonomic disturbances, oculogyric crises, developmental delay | Yes | None | No | Not known | None | [82,83,84,85] | ||
| TAF1/ DYT 3 | XL/Full | 314250 | Early adulthood-Late adulthood | Generalized | Persistent | Parkinsonism, jaw opening dystonia, bulbar involvement, striatal toe | No | None | No | Variable | Stiatal atrophy and pallidum volume loss in pallidum | [86,87,88,89] | ||
| ATP1A3/ DYT 12 | AD/ Incomplete | 128235 | Adolescence-Early adulthood | Generalized-Segmental | Persistent | Abrupt onset, Fluctuating course, Parkinsonism, Postural instability, Psychiatric features | No | None | No | Not known | None | [85,90,91] | ||
| Myoclonus | SGCE DYT 11 | AD/ Reduced (maternal imprinting) | 159900 | Childhood-Adolescence | Focal-segmental | Persistent | Myoclonic jerks mainly of the neck, prominent psychiatric features | No | None | Yes | Variable | None | [71] | |
| KCTD17 DYT26 | AD/NA | 616398 | Childhood-Adolescence | Focal-segmental | Persistent | Myoclonus of upper limbs, psychiatric features, | No | None | No | Good | None | [92] | ||
| Hyperkinesia | ADCY5 | AD/NA | 600293 | Childhood | Focal-segmental-generalized | Paroxysmal worsening | Generalized choreoathetosis, Facial dyskinesia, myoclonus, learning difficulties, behavioral abnormalities | No | Caffeine | No | Variable | None | [76,93,94] | |
| Autosomal Dominant | |
| Disease | OMIM Code |
| #128100 |
| #602629 |
| #128230 |
| #128235 |
| #159900 |
| #606159 |
| #125370 |
| #143100 |
| #109150 |
| #123400 |
| #213600 |
| #616398 |
| #617284 |
| #619291 |
| #619687 |
| #615073 |
| #615034 |
| #129101 |
| #616398 |
| #606703 |
| Autosomal recessive: | |
| #277900 |
| #234200 |
| #610217 |
| #604290 |
| #612319 |
| #608309 |
| #608643 |
| #612067 |
| #257220 |
| #204200 |
| #230500 |
| #272750 |
| #250100 |
| #277400 |
| #231670 |
| #251000 |
| #234500 |
| #208900 |
| #229300 |
| #200150 |
| #605407 |
| #603472 |
| #607259 |
| #270200 |
| #607483 |
| #224500 |
| #619565 |
| X-linked recessive: | |
| #314250 |
| #300322 |
| #304700 |
| X-linked dominant | |
| #312750 |
| Mitochondrial | |
| #256000 |
| #500001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
di Biase, L.; Di Santo, A.; Caminiti, M.L.; Pecoraro, P.M.; Carbone, S.P.; Di Lazzaro, V. Dystonia Diagnosis: Clinical Neurophysiology and Genetics. J. Clin. Med. 2022, 11, 4184. https://doi.org/10.3390/jcm11144184
di Biase L, Di Santo A, Caminiti ML, Pecoraro PM, Carbone SP, Di Lazzaro V. Dystonia Diagnosis: Clinical Neurophysiology and Genetics. Journal of Clinical Medicine. 2022; 11(14):4184. https://doi.org/10.3390/jcm11144184
Chicago/Turabian Styledi Biase, Lazzaro, Alessandro Di Santo, Maria Letizia Caminiti, Pasquale Maria Pecoraro, Simona Paola Carbone, and Vincenzo Di Lazzaro. 2022. "Dystonia Diagnosis: Clinical Neurophysiology and Genetics" Journal of Clinical Medicine 11, no. 14: 4184. https://doi.org/10.3390/jcm11144184
APA Styledi Biase, L., Di Santo, A., Caminiti, M. L., Pecoraro, P. M., Carbone, S. P., & Di Lazzaro, V. (2022). Dystonia Diagnosis: Clinical Neurophysiology and Genetics. Journal of Clinical Medicine, 11(14), 4184. https://doi.org/10.3390/jcm11144184