
New Insights into Renal Failure in a Cohort of 317 Patients with Autosomal Dominant Forms of Alport Syndrome: Report of Two Novel Heterozygous Mutations in COL4A3

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Review of Reported Cases
2.2. Patients and Family Description
2.3. Informed Consent
2.4. Genetic Analysis
2.5. Statistical Analysis
3. Results
3.1. Review of Cases
3.2. Novel Missense Mutations in COL4A3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kashtan, C.E. Alport Syndrome: Achieving Early Diagnosis and Treatment. Am. J. Kidney Dis. 2021, 77, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Kamiyoshi, N.; Nozu, K.; Fu, X.J.; Morisada, N.; Nozu, Y.; Ye, M.J.; Imafuku, A.; Miura, K.; Yamamura, T.; Minamikawa, S.; et al. Genetic, Clinical, and Pathologic Backgrounds of Patients with Autosomal Dominant Alport Syndrome. Clin. J. Am. Soc. Nephrol. 2016, 11, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Nozu, K.; Nakanishi, K.; Abe, Y.; Udagawa, T.; Okada, S.; Okamoto, T.; Kaito, H.; Kanemoto, K.; Kobayashi, A.; Tanaka, E.; et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin. Exp. Nephrol. 2018, 23, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Papazachariou, L.; Demosthenous, P.; Pieri, M.; Papagregoriou, G.; Savva, I.; Stavrou, C.; Zavros, M.; Athanasiou, Y.; Ioannou, K.; Patsias, C.; et al. Frequency of COL4A3/COL4A4 Mutations amongst Families Segregating Glomerular Microscopic Hematuria and Evidence for Activation of the Unfolded Protein Response. Focal and Segmental Glomerulosclerosis Is a Frequent Development during Ageing. PLoS ONE 2014, 9, e115015. [Google Scholar] [CrossRef] [PubMed]
- Malone, A.F.; Phelan, P.J.; Hall, G.; Cetincelik, U.; Homstad, A.; Alonso, A.S.; Jiang, R.; Lindsey, T.B.; Wu, G.; Sparks, M.A.; et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014, 86, 1253–1259. [Google Scholar] [CrossRef]
- Stapleton, C.P.; Kennedy, C.; Fennelly, N.K.; Murray, S.L.; Connaughton, D.M.; Dorman, A.M.; Doyle, B.; Cavalleri, G.L.; Conlon, P.J. An Exome Sequencing Study of 10 Families with IgA Nephropathy. Nephron Exp. Nephrol. 2019, 144, 72–83. [Google Scholar] [CrossRef]
- Marcocci, E.; Uliana, V.; Bruttini, M.; Artuso, R.; Silengo, M.C.; Zerial, M.; Bergesio, F.; Amoroso, A.; Savoldi, S.; Pennesi, M.; et al. Autosomal dominant Alport syndrome: Molecular analysis of the COL4A4 gene and clinical outcome. Nephrol. Dial. Transplant. 2009, 24, 1464–1471. [Google Scholar] [CrossRef]
- Matthaiou, A.; Poulli, T.; Deltas, C. Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: A systematic review. Clin. Kidney J. 2020, 13, 1025–1036. [Google Scholar] [CrossRef]
- Furlano, M.; Martínez, V.; Pybus, M.; Arce, Y.; Crespí, J.; Venegas, M.D.P.; Bullich, G.; Domingo, A.; Ayasreh, N.; Benito, S.; et al. Clinical and Genetic Features of Autosomal Dominant Alport Syndrome: A Cohort Study. Am. J. Kidney Dis. 2021, 78, 560–570. [Google Scholar] [CrossRef]
- Savige, J.; Lipska-Zietkiewicz, B.S.; Watson, E.; Hertz, J.M.; Deltas, C.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; Cerkauskaite, A.; et al. Guidelines for Genetic Testing and Management of Alport Syndrome. Clin. J. Am. Soc. Nephrol. 2021, 17, 143–154. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443, Erratum in Nature 2021, 590, E53; Erratum in Nature 2021, 597, E3–E4. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kleppel, M.; Fan, W.; Cheong, H.; Michael, A. Evidence for separate networks of classical and novel basement membrane collagen. Characterization of alpha 3(IV)-alport antigen heterodimer. J. Biol. Chem. 1992, 267, 4137–4142. [Google Scholar] [CrossRef]
- Gunwar, S.; Ballester, F.; Noelken, M.E.; Sado, Y.; Ninomiya, Y.; Hudson, B.G. Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of alpha3, alpha4, and alpha5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J. Biol. Chem. 1998, 273, 8767–8775. [Google Scholar] [CrossRef] [PubMed]
- Lemmink, H.H.; Nillesen, W.N.; Mochizuki, T.; Schröder, C.H.; Brunner, H.G.; Van Oost, B.A.; Monnens, L.A.; Smeets, H.J. Benign familial hematuria due to mutation of the type IV collagen alpha4 gene. J. Clin. Investig. 1996, 98, 1114–1118. [Google Scholar] [CrossRef]
- Artuso, R.; Fallerini, C.; Dosa, L.; Scionti, F.; Clementi, M.; Garosi, G.; Massella, L.; Epistolato, M.C.; Mancini, R.; Mari, F.; et al. Advances in Alport syndrome diagnosis using next-generation sequencing. Eur. J. Hum. Genet. 2011, 20, 50–57. [Google Scholar] [CrossRef]
- Domingo-Gallego, A.; Pybus, M.; Bullich, G.; Furlano, M.; Ejarque-Vila, L.; Lorente-Grandoso, L.; Ruiz, P.; Fraga, G.; González, M.L.; Piñero-Fernández, J.A.; et al. Clinical utility of genetic testing in early-onset kidney disease: Seven genes are the main players. Nephrol. Dial. Transplant. 2021, 37, 687–696. [Google Scholar] [CrossRef]
- Van Der Loop, F.T.; Heidet, L.; Timmer, E.D.; Bosch, B.J.V.D.; Leinonen, A.; Antignac, C.; Jefferson, J.A.; Maxwell, A.P.; Monnens, L.A.; Schröder, C.H.; et al. Autosomal dominant Alport syndrome caused by a COL4A3 splice site mutation. Kidney Int. 2000, 58, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, M.; Casu, D.; Wong, F.K.; Faedda, R.; Arvidsson, S.; Tonolo, G.; Luthman, H.; Satta, A. Identification of a new mutation in the α4(IV) collagen gene in a family with autosomal dominant Alport syndrome and hypercholesterolaemia. Nephrol. Dial. Transplant. 2001, 16, 2008–2012. [Google Scholar] [CrossRef]
- Rosado, C.; Bueno, E.; Felipe, C.; Valverde, S.; González-Sarmiento, R. Study of the True Clinical Progression of Autosomal Dominant Alport Syndrome in a European Population. Kidney Blood Press. Res. 2015, 40, 435–442. [Google Scholar] [CrossRef]
- Longo, I.; Porcedda, P.; Mari, F.; Giachino, D.F.; Meloni, I.; Deplano, C.; Brusco, A.; Bosio, M.; Massella, L.; Lavoratti, G.; et al. COL4A3/COL4A4 mutations: From familial hematuria to autosomal-dominant or recessive Alport syndrome. Kidney Int. 2002, 61, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- Pescucci, C.; Mari, F.; Longo, I.; Vogiatzi, P.; Caselli, R.; Scala, E.; Abaterusso, C.; Gusmano, R.; Seri, M.; Miglietti, N.; et al. Autosomal-dominant Alport syndrome: Natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int. 2004, 65, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Šlajpah, M.; Gorinšek, B.; Berginc, G.; Vizjak, A.; Ferluga, D.; Hvala, A.; Meglič, A.; Jakša, I.; Furlan, P.; Gregorič, A.; et al. Sixteen novel mutations identified in COL4A3, COL4A4, and COL4A5 genes in Slovenian families with Alport syndrome and benign familial hematuria. Kidney Int. 2007, 71, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Chen, Y.; Ding, J.; Li, G.; Zhang, H. A Novel Mutation of COL4A3 Presents a Different Contribution to Alport Syndrome and Thin Basement Membrane Nephropathy. Am. J. Nephrol. 2007, 27, 538–544. [Google Scholar] [CrossRef]
- Hoefele, J.; Lange-Sperandio, B.; Ruessmann, D.; Glöckner-Pagel, J.; Alberer, M.; Benz, M.R.; Nagel, M.; Weber, L.T. Novel heterozygous COL4A3 mutation in a family with late-onset ESRD. Pediatr. Nephrol. 2010, 25, 1539–1542. [Google Scholar] [CrossRef]
- Fallerini, C.; Dosa, L.; Tita, R.; Del Prete, D.; Feriozzi, S.; Gai, G.; Clementi, M.; La Manna, A.; Miglietti, N.; Mancini, R.; et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin. Genet. 2013, 86, 252–257. [Google Scholar] [CrossRef]
- Lin, F.; Bian, F.; Zou, J.; Wu, X.; Shan, J.; Lu, W.; Yao, Y.; Jiang, G.; Gale, D.P. Whole exome sequencing reveals novel COL4A3 and COL4A4mutations and resolves diagnosis in Chinese families with kidney disease. BMC Nephrol. 2014, 15, 175. [Google Scholar] [CrossRef]
- Xie, J.; Wu, X.; Ren, H.; Wang, W.; Wang, Z.; Pan, X.; Hao, X.; Tong, J.; Ma, J.; Ye, Z.; et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J. Mol. Cell Biol. 2014, 6, 498–505. [Google Scholar] [CrossRef]
- Weber, S.; Strasser, K.; Rath, S.; Kittke, A.; Beicht, S.; Alberer, M.; Lange-Sperandio, B.; Hoyer, P.F.; Benz, M.R.; Ponsel, S.; et al. Identification of 47 novel mutations in patients with Alport syndrome and thin basement membrane nephropathy. Pediatr. Nephrol. 2016, 31, 941–955. [Google Scholar] [CrossRef]
- Xu, Y.; Guo, M.; Dong, H.; Jiang, W.; Ma, R.; Liu, S.; Li, S. A Novel COL4A4 Mutation Identified in a Chinese Family with Thin Basement Membrane Nephropathy. Sci. Rep. 2016, 6, 20244. [Google Scholar] [CrossRef]
- Kovács, G.; Kalmár, T.; Endreffy, E.; Ondrik, Z.; Iványi, B.; Rikker, C.; Haszon, I.; Túri, S.; Sinkó, M.; Bereczki, C.; et al. Efficient Targeted Next Generation Sequencing-Based Workflow for Differential Diagnosis of Alport-Related Disorders. PLoS ONE 2016, 11, e0149241. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Deng, S.; Xu, H.; Yuan, J.; Xiao, J.; Yuan, L.; Deng, X.; Guan, L.; Zhu, A.; Rong, P.; et al. Identification of a novel collagen type IV alpha-4 (COL4A4) mutation in a Chinese family with autosomal dominant Alport syndrome using exome sequencing. Indian J. Med Res. 2016, 144, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, D.; Dong, S.; Wang, D.; Yang, B.; Huang, Y. Mutation analysis of COL4A3 and COL4A4 genes in a Chinese autosomal-dominant Alport syndrome family. J. Genet. 2017, 96, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Cervera-Acedo, C.; Coloma, A.; Huarte-Loza, E.; Sierra-Carpio, M.; Domínguez-Garrido, E. Phenotype variability in a large Spanish family with Alport syndrome associated with novel mutations in COL4A3 gene. BMC Nephrol. 2017, 18, 325. [Google Scholar] [CrossRef]
- Imafuku, A.; Nozu, K.; Sawa, N.; Hasegawa, E.; Hiramatsu, R.; Kawada, M.; Hoshino, J.; Tanaka, K.; Ishii, Y.; Takaichi, K.; et al. Autosomal dominant form of type IV collagen nephropathy exists among patients with hereditary nephritis difficult to diagnose clinicopathologically. Nephrology 2017, 23, 940–947. [Google Scholar] [CrossRef]
- Xia, L.; Cao, Y.; Guo, Y.; Ba, G.; Luo, Q.; Shi, H.; Feng, Y.; Yin, S. A Novel Heterozygous Mutation of the COL4A3 Gene Causes a Peculiar Phenotype without Hematuria and Renal Function Impairment in a Chinese Family. Dis. Markers 2019, 2019, 8705989. [Google Scholar] [CrossRef]
- Yang, C.; Song, Y.; Chen, Z.; Yuan, X.; Chen, X.; Ding, G.; Guan, Y.; McGrath, M.; Song, C.; Tong, Y.; et al. A Nonsense Mutation in COL4A4 Gene Causing Isolated Hematuria in Either Heterozygous or Homozygous State. Front. Genet. 2019, 10, 628. [Google Scholar] [CrossRef]
- Ersan, S.; Kirbiyik, O.; Sarikaya, T.; Guvenc, M.S.; Karadeniz, T. A novel COL4A4 gene variant (c.1856>a): From a focal segmental glomerulosclerosis case to a family with alport syndrome. Rev. Nefrol. Dislisis Transpl. 2019, 39, 120–125. [Google Scholar]
- Fan, L.; Liu, L.; Luo, F.; Du, R.; Wang, C.; Dong, Y.; Liu, J. A novel heterozygous variant of the COL4A4 gene in a Chinese family with hematuria and proteinuria leads to focal segmental glomerulosclerosis and chronic kidney disease. Mol. Genet. Genom. Med. 2020, 8, e1545. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Variables | Items | No. of Cases | Percentage (%) |
|---|---|---|---|
| Gene | COL4A3 | 136 | 46.70 |
| Mutation type | Nonsense | 0 | 0 |
| Frameshift | 0 | 0 | |
| Splicing | 1 | 1.09 | |
| Missense | 90 | 97.83 | |
| In-frame deletion | 1 | 1.09 | |
| Sex | Male | 52 | 53.26 |
| Female | 45 | 46.74 | |
| Presence of urine analytical alterations | Microhematuria | 18 | 19.57 |
| Hematuria | 36 | 39.13 | |
| Proteinuria | 55 | 59.78 | |
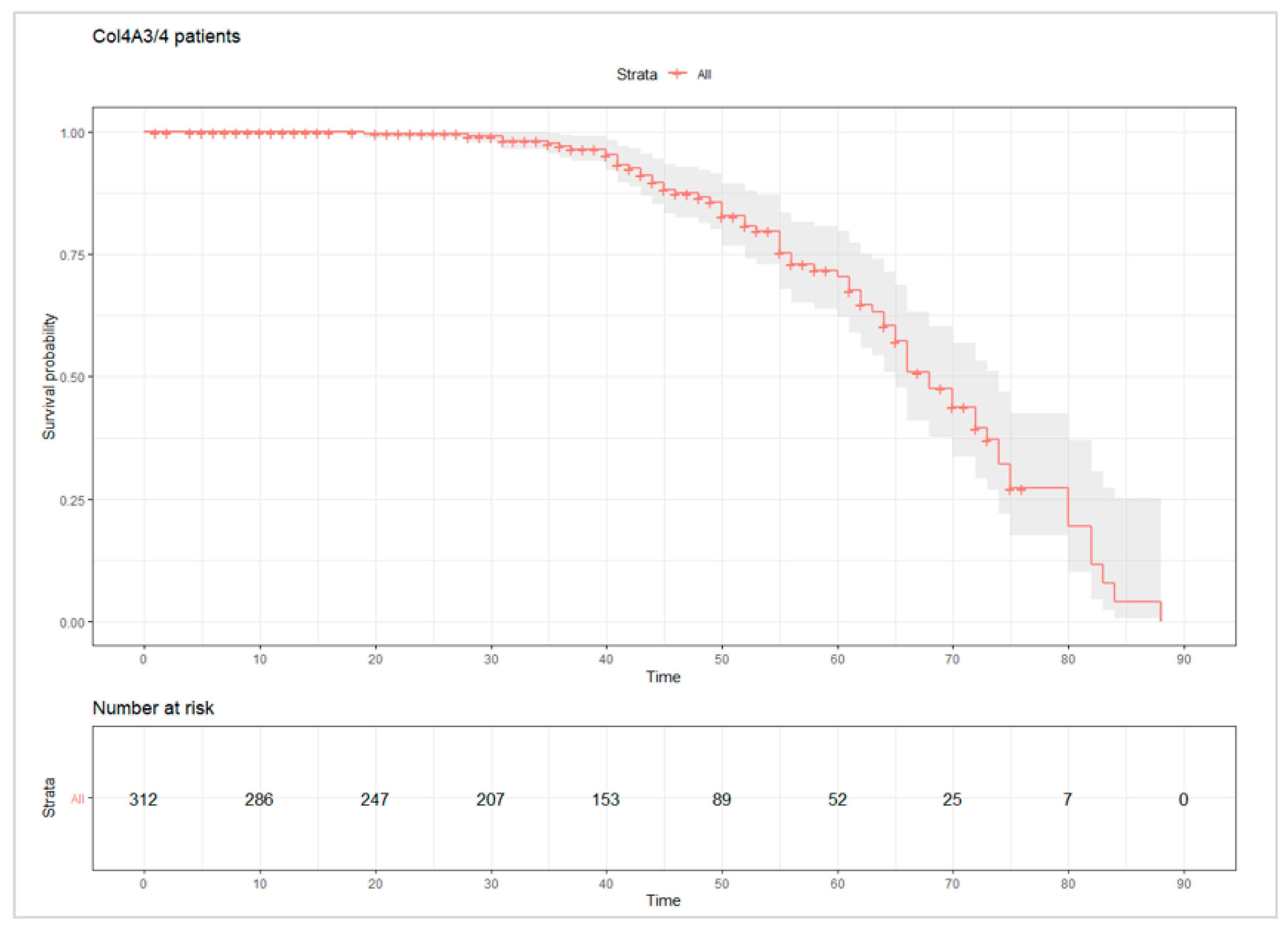
| Renal events | ESKD | 10 | 10.87 |
| CRD | 4 | 4.35 | |
| Dialysis | 6 | 6.52 | |
| Renal transplant | 4 | 4.35 | |
| Hearing impairment | Hypoacusia | 23 | 25 |
| Events | Ages (males) | Ages (females) | Total |
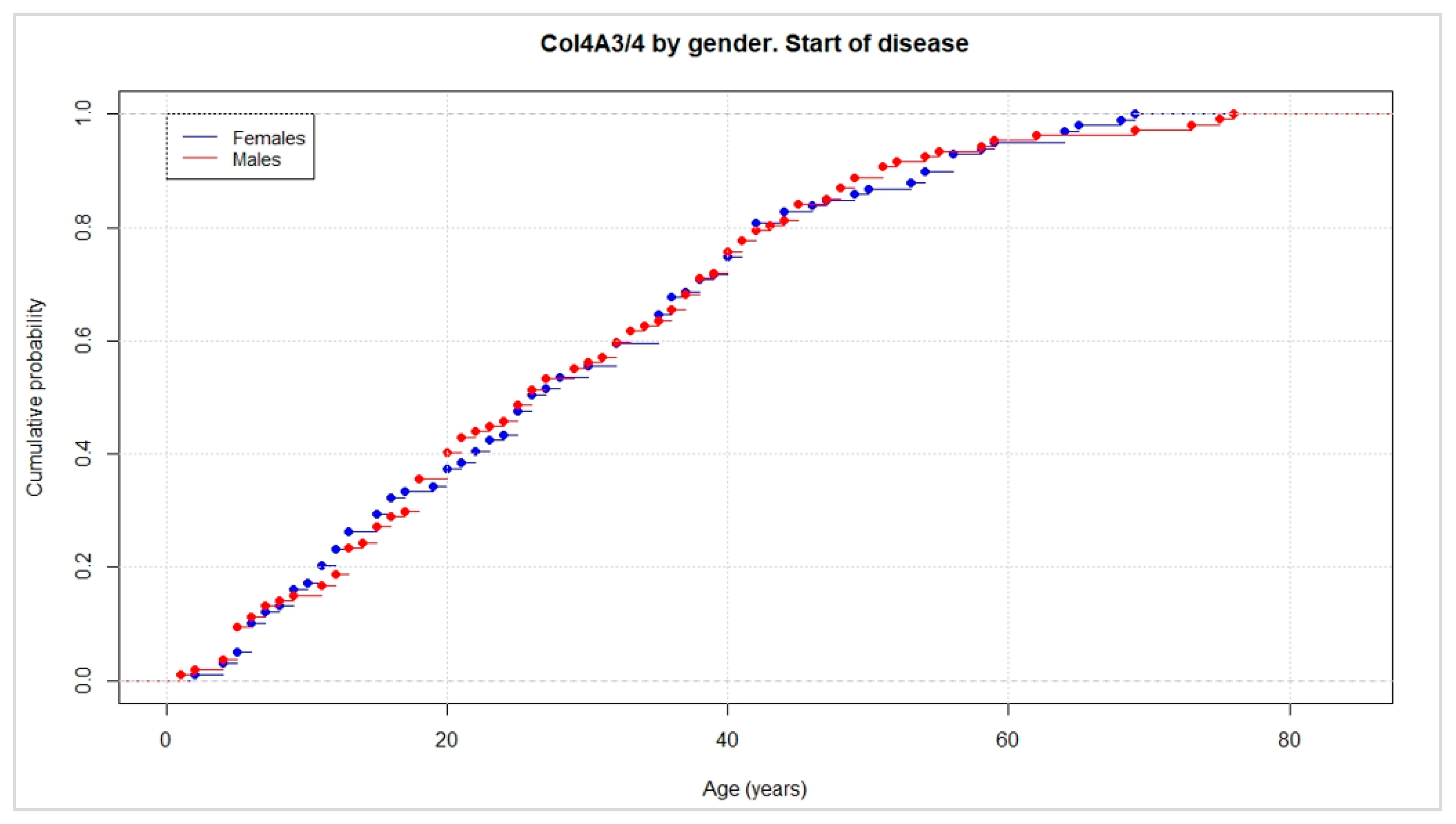
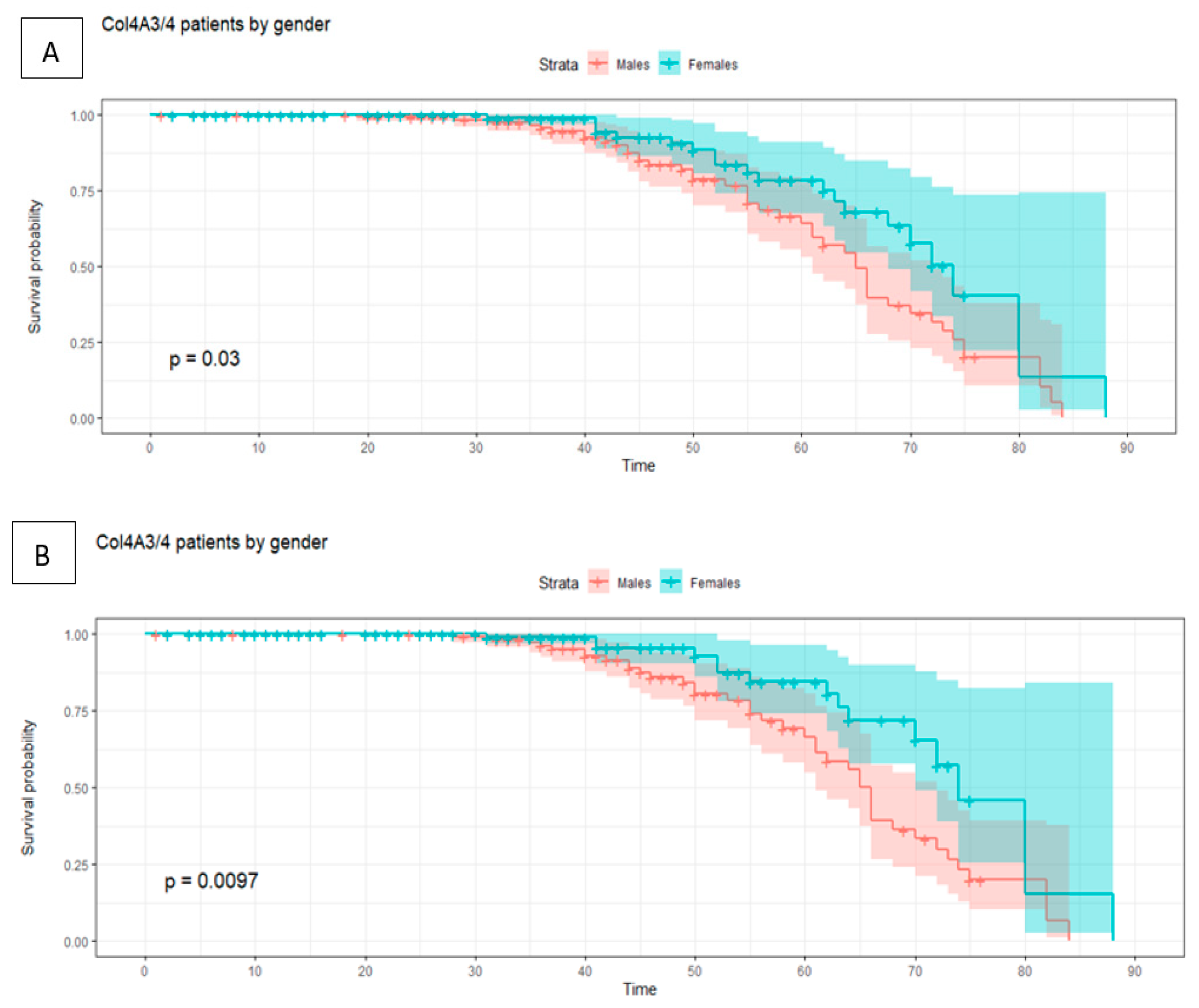
| Average of analytical alterations | 30 | 32 | 31 |
| Minimum age of onset of renal failure | 30 | 50 | 30 |
| Average of renal failure | 50 | 62 | 52 |
| Clinical Variables | Items | No. of Cases | Percentage (%) |
|---|---|---|---|
| Mutated gene | COL4A4 | 181 | 53.30 |
| Mutation type | Nonsense | 8 | 7.62 |
| Frameshift | 17 | 16.19 | |
| Splicing | 27 | 25.71 | |
| Missense | 51 | 48.57 | |
| In-frame deletion | 2 | 1.90 | |
| Sex | Male | 55 | 52.38 |
| Female | 50 | 47.62 | |
| Presence of urine analytical alterations | Microhematuria | 56 | 53.33 |
| Hematuria | 36 | 34.29 | |
| Proteinuria | 45 | 42.86 | |
| Renal events | ESKD | 14 | 13.33 |
| CRD | 6 | 5.71 | |
| Dialysis * | 3 | 2.86 | |
| Renal transplant * | 1 | 0.95 | |
| Hearing impairment | Hypoacusia | 11 | 10.48 |
| Events | Age (males) | Age (females) | Total |
| Average of analytical alterations | 31 | 28 | 29 |
| Minimum age of onset of renal failure | 40 | 29 | 29 |
| Average of renal failure | 56 | 47 | 53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Aznar, J.M.; De la Higuera, L.; Besada Cerecedo, L.; Gandiaga, N.P.; Vega, A.I.; Fernández-Fresnedo, G.; González-Lamuño, D. New Insights into Renal Failure in a Cohort of 317 Patients with Autosomal Dominant Forms of Alport Syndrome: Report of Two Novel Heterozygous Mutations in COL4A3. J. Clin. Med. 2022, 11, 4883. https://doi.org/10.3390/jcm11164883
García-Aznar JM, De la Higuera L, Besada Cerecedo L, Gandiaga NP, Vega AI, Fernández-Fresnedo G, González-Lamuño D. New Insights into Renal Failure in a Cohort of 317 Patients with Autosomal Dominant Forms of Alport Syndrome: Report of Two Novel Heterozygous Mutations in COL4A3. Journal of Clinical Medicine. 2022; 11(16):4883. https://doi.org/10.3390/jcm11164883
Chicago/Turabian StyleGarcía-Aznar, José María, Luis De la Higuera, Lara Besada Cerecedo, Nerea Paz Gandiaga, Ana Isabel Vega, Gema Fernández-Fresnedo, and Domingo González-Lamuño. 2022. "New Insights into Renal Failure in a Cohort of 317 Patients with Autosomal Dominant Forms of Alport Syndrome: Report of Two Novel Heterozygous Mutations in COL4A3" Journal of Clinical Medicine 11, no. 16: 4883. https://doi.org/10.3390/jcm11164883
APA StyleGarcía-Aznar, J. M., De la Higuera, L., Besada Cerecedo, L., Gandiaga, N. P., Vega, A. I., Fernández-Fresnedo, G., & González-Lamuño, D. (2022). New Insights into Renal Failure in a Cohort of 317 Patients with Autosomal Dominant Forms of Alport Syndrome: Report of Two Novel Heterozygous Mutations in COL4A3. Journal of Clinical Medicine, 11(16), 4883. https://doi.org/10.3390/jcm11164883