
Physiology, Diagnosis and Treatment of Cardiac Light Chain Amyloidosis

 , , ,
, , ,  ,
,  and
and
Abstract
:1. Introduction
2. Characteristics of Amyloidosis
3. Light-Chain (AL) Amyloidosis
3.1. Molecular Mechanisms
3.2. Symptoms of AL Amyloidosis
3.3. Cardiac AL Amyloidosis
4. Diagnostics
- Presence of clinical syndromes related to the affected organs
- Positive result Congo red staining of tissue specimens
- Identification of light chains in amyloid deposits
- Presence of deviations in additional studies resulting from the proliferation of monoclonal plasma cells
4.1. Diagnosis of Cardiac Amyloidosis
4.2. Red Flags
5. Prognosis
6. Treatment
6.1. Supportive Therapy
6.2. Therapy to Inhibit the Production of the Corresponding Precursor Protein
6.3. Latest Treatment for AL Amyloidosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Martinez-Naharro, A.; Hawkins, P.N.; Fontana, M. Cardiac Amyloidosis. Clin. Med. 2018, 18, s30–s35. [Google Scholar] [CrossRef]
- Monsellier, E.; Ramazzotti, M.; Taddei, N.; Chiti, F. Aggregation Propensity of the Human Proteome. PLoS Comput. Biol. 2008, 4, e1000199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banypersad, S.M.; Moon, J.C.; Whelan, C.; Hawkins, P.N.; Wechalekar, A.D. Updates in Cardiac Amyloidosis: A Review. J. Am. Heart Assoc. 2012, 1, e000364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, J.P.; Hanna, M. Cardiac Amyloidosis: An Update on Diagnosis and Treatment. Clevel. Clin. J. Med. 2017, 84, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Quarta, C.C.; Kruger, J.L.; Falk, R.H. Cardiac Amyloidosis. Circulation 2012, 126, e178–e182. [Google Scholar] [CrossRef]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.R. Techniques to Study Amyloid Fibril Formation in Vitro. Methods 2004, 34, 151–160. [Google Scholar] [CrossRef]
- Muchtar, E.; Dispenzieri, A.; Magen, H.; Grogan, M.; Mauermann, M.; McPhail, E.D.; Kurtin, P.J.; Leung, N.; Buadi, F.K.; Dingli, D.; et al. Systemic Amyloidosis from A (AA) to T (ATTR): A Review. J. Intern. Med. 2021, 289, 268–292. [Google Scholar] [CrossRef]
- Riek, R.; Eisenberg, D.S. The Activities of Amyloids from a Structural Perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Merlini, G. AL Amyloidosis: From Molecular Mechanisms to Targeted Therapies. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Bellotti, V. Molecular Mechanisms of Amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaxman, I.; Gertz, M. Recent Advances in the Diagnosis, Risk Stratification, and Management of Systemic Light-Chain Amyloidosis. Acta Haematol. 2019, 141, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Quock, T.P.; Yan, T.; Chang, E.; Guthrie, S.; Broder, M.S. Epidemiology of AL Amyloidosis: A Real-World Study Using US Claims Data. Blood Adv. 2018, 2, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Sipe, J.D.; Cohen, A.S. Review: History of the Amyloid Fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Stevens, F.J.; Kisilevsky, R. Immunoglobulin Light Chains, Glycosaminoglycans, and Amyloid. Cell. Mol. Life Sci. CMLS 2000, 57, 441–449. [Google Scholar] [CrossRef]
- Usnarska-Zubkiewicz, L.; Hołojda, J.; Kuliczkowski, K. Wolne łańcuchy lekkie w surowicy-znaczenie diagnostyczne i prognostyczne w dyskrazjach plazmocytowych. Acta Haematol. Pol. 2009, 40, 349–361. [Google Scholar]
- Xi, D.; Luo, T.; Xiong, H.; Liu, J.; Lu, H.; Li, M.; Hou, Y.; Guo, Z. SAP: Structure, Function, and Its Roles in Immune-Related Diseases. Int. J. Cardiol. 2015, 187, 20–26. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.-M.; Tycko, R. Molecular Structural Basis for Polymorphism in Alzheimer’s β-Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertz, M.A. Immunoglobulin Light Chain Amyloidosis: 2018 Update on Diagnosis, Prognosis, and Treatment. Am. J. Hematol. 2018, 93, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Rottenaicher, G.J.; Weber, B.; Rührnößl, F.; Kazman, P.; Absmeier, R.M.; Hitzenberger, M.; Zacharias, M.; Buchner, J. Molecular Mechanism of Amyloidogenic Mutations in Hypervariable Regions of Antibody Light Chains. J. Biol. Chem. 2021, 296, 100334. [Google Scholar] [CrossRef]
- Glenner, G.G.; Terry, W.D.; Isersky, C. Amyloidosis: Its Nature and Pathogenesis. Semin. Hematol. 1973, 10, 65–86. [Google Scholar]
- Solomon, A.; Frangione, B.; Franklin, E.C. Bence Jones Proteins and Light Chains of Immunoglobulins. Preferential Association of the V Lambda VI Subgroup of Human Light Chains with Amyloidosis AL (Lambda). J. Clin. Investig. 1982, 70, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Mazzullo, H.; Ross, F.M.; Cheung, K.L.; Gerrard, G.; Harewood, L.; Mehta, A.; Lachmann, H.J.; Hawkins, P.N.; Orchard, K.H. Translocations of 14q32 and Deletions of 13q14 Are Common Chromosomal Abnormalities in Systemic Amyloidosis. Br. J. Haematol. 2002, 117, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Bochtler, T.; Hegenbart, U.; Cremer, F.W.; Heiss, C.; Benner, A.; Hose, D.; Moos, M.; Bila, J.; Bartram, C.R.; Ho, A.D.; et al. Evaluation of the Cytogenetic Aberration Pattern in Amyloid Light Chain Amyloidosis as Compared with Monoclonal Gammopathy of Undetermined Significance Reveals Common Pathways of Karyotypic Instability. Blood 2008, 111, 4700–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayman, S.R.; Bailey, R.J.; Jalal, S.M.; Ahmann, G.J.; Dispenzieri, A.; Gertz, M.A.; Greipp, P.R.; Kyle, R.A.; Lacy, M.Q.; Rajkumar, S.V.; et al. Translocations Involving the Immunoglobulin Heavy-Chain Locus Are Possible Early Genetic Events in Patients with Primary Systemic Amyloidosis. Blood 2001, 98, 2266–2268. [Google Scholar] [CrossRef] [PubMed]
- Comenzo, R.L. How I Treat Amyloidosis. Blood 2009, 114, 3147–3157. [Google Scholar] [CrossRef] [Green Version]
- Kyle, R.A.; Linos, A.; Beard, C.M.; Linke, R.P.; Gertz, M.A.; O’Fallon, W.M.; Kurland, L.T. Incidence and Natural History of Primary Systemic Amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992, 79, 1817–1822. [Google Scholar] [CrossRef] [Green Version]
- Muchtar, E.; Buadi, F.K.; Dispenzieri, A.; Gertz, M.A. Immunoglobulin Light-Chain Amyloidosis: From Basics to New Developments in Diagnosis, Prognosis and Therapy. Acta Haematol. 2016, 135, 172–190. [Google Scholar] [CrossRef]
- Pepys, M.B. Amyloidosis. Annu. Rev. Med. 2006, 57, 223–241. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Marin-Argany, M.; Lin, Y.; Misra, P.; Williams, A.; Wall, J.S.; Howell, K.G.; Elsbernd, L.R.; McClure, M.; Ramirez-Alvarado, M. Cell Damage in Light Chain Amyloidosis. J. Biol. Chem. 2016, 291, 19813–19825. [Google Scholar] [CrossRef] [Green Version]
- Perfetti, V.; Palladini, G.; Casarini, S.; Navazza, V.; Rognoni, P.; Obici, L.; Invernizzi, R.; Perlini, S.; Klersy, C.; Merlini, G. The Repertoire of λ Light Chains Causing Predominant Amyloid Heart Involvement and Identification of a Preferentially Involved Germline Gene, IGLV1-44. Blood 2012, 119, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Dasari, S.; Theis, J.D.; Ramirez-Alvarado, M.; Kurtin, P.J.; Gertz, M.A.; Zeldenrust, S.R.; Zenka, R.M.; Dogan, A.; Dispenzieri, A. Clarifying Immunoglobulin Gene Usage in Systemic and Localized Immunoglobulin Light-Chain Amyloidosis by Mass Spectrometry. Blood 2017, 129, 299–306. [Google Scholar] [CrossRef]
- Sher, T.; Hayman, S.R.; Gertz, M.A. Treatment of Primary Systemic Amyloidosis (AL): Role of Intensive and Standard Therapy. Clin. Adv. Hematol. Oncol. HO 2012, 10, 644–651. [Google Scholar]
- Shimazaki, C.; Hata, H.; Iida, S.; Ueda, M.; Katoh, N.; Sekijima, Y.; Ikeda, S.; Yazaki, M.; Fukushima, W.; Ando, Y. Nationwide Survey of 741 Patients with Systemic Amyloid Light-Chain Amyloidosis in Japan. Intern. Med. Tokyo Jpn. 2018, 57, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vora, M.; Kevil, C.G.; Herrera, G.A. Contribution of Human Smooth Muscle Cells to Amyloid Angiopathy in AL (Light-Chain) Amyloidosis. Ultrastruct. Pathol. 2017, 41, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.H.; Comenzo, R.L.; Skinner, M. The Systemic Amyloidoses. N. Engl. J. Med. 1997, 337, 898–909. [Google Scholar] [CrossRef]
- Keeling, J.; Teng, J.; Herrera, G.A. AL-Amyloidosis and Light-Chain Deposition Disease Light Chains Induce Divergent Phenotypic Transformations of Human Mesangial Cells. Lab. Investig. J. Tech. Methods Pathol. 2004, 84, 1322–1338. [Google Scholar] [CrossRef]
- Perkowska-Ptasinska, A.; Deborska-Materkowska, D.; Bartczak, A.; Stompor, T.; Liberek, T.; Bullo-Piontecka, B.; Wasinska, A.; Serwacka, A.; Klinger, M.; Chyl, J.; et al. Kidney Disease in the Elderly: Biopsy Based Data from 14 Renal Centers in Poland. BMC Nephrol. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Lan, P.; Feng, J.; Feng, X.; Xie, L. Renal Amyloidosis Complicated by Light Chain Deposition Nephropathy: A Case Report. Int. J. Clin. Exp. Pathol. 2019, 12, 2279–2283. [Google Scholar] [PubMed]
- Gertz, M.A.; Leung, N.; Lacy, M.Q.; Dispenzieri, A.; Zeldenrust, S.R.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Greipp, P.R.; Kumar, S.K.; et al. Clinical Outcome of Immunoglobulin Light Chain Amyloidosis Affecting the Kidney. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2009, 24, 3132–3137. [Google Scholar] [CrossRef]
- Ryšavá, R. AL Amyloidosis: Advances in Diagnostics and Treatment. Nephrol. Dial. Transplant. 2019, 34, 1460–1466. [Google Scholar] [CrossRef] [Green Version]
- Kastritis, E.; Gavriatopoulou, M.; Roussou, M.; Migkou, M.; Fotiou, D.; Ziogas, D.C.; Kanellias, N.; Eleutherakis-Papaiakovou, E.; Panagiotidis, I.; Giannouli, S.; et al. Renal Outcomes in Patients with AL Amyloidosis: Prognostic Factors, Renal Response and the Impact of Therapy. Am. J. Hematol. 2017, 92, 632–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumford, A.D.; O’Donnell, J.; Gillmore, J.D.; Manning, R.A.; Hawkins, P.N.; Laffan, M. Bleeding Symptoms and Coagulation Abnormalities in 337 Patients with AL-Amyloidosis. Br. J. Haematol. 2000, 110, 454–460. [Google Scholar] [CrossRef]
- Maleszewski, J.J. Cardiac Amyloidosis: Pathology, Nomenclature, and Typing. Cardiovasc. Pathol. 2015, 24, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Ardehali, H.; Qasim, A.; Cappola, T.; Howard, D.; Hruban, R.; Hare, J.M.; Baughman, K.L.; Kasper, E.K. Endomyocardial Biopsy Plays a Role in Diagnosing Patients with Unexplained Cardiomyopathy. Am. Heart J. 2004, 147, 919–923. [Google Scholar] [CrossRef]
- Miani, D.; Rocco, M.; Alberti, E.; Spedicato, L.; Fioretti, P.M. Amyloidosis of Epicardial and Intramural Coronary Arteries as an Unusual Cause of Myocardial Infarction and Refractory Angina Pectoris. Ital. Heart J. 2002, 3, 479–482. [Google Scholar]
- Guan, J.; Mishra, S.; Falk, R.H.; Liao, R. Current Perspectives on Cardiac Amyloidosis. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H544–H552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esplin, B.L.; Gertz, M.A. Current Trends in Diagnosis and Management of Cardiac Amyloidosis. Curr. Probl. Cardiol. 2013, 38, 53–96. [Google Scholar] [CrossRef]
- Dubrey, S.W.; Cha, K.; Anderson, J.; Chamarthi, B.; Reisinger, J.; Skinner, M.; Falk, R.H. The Clinical Features of Immunoglobulin Light-Chain (AL) Amyloidosis with Heart Involvement. QJM Mon. J. Assoc. Physicians 1998, 91, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, R.H. Diagnosis and Management of the Cardiac Amyloidoses. Circulation 2005, 112, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Berk, J.L.; Keane, J.; Seldin, D.C.; Sanchorawala, V.; Koyama, J.; Dember, L.M.; Falk, R.H. Persistent Pleural Effusions in Primary Systemic Amyloidosis: Etiology and Prognosis. Chest 2003, 124, 969–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, J.; Ray-Sequin, P.A.; Davidoff, R.; Falk, R.H. Usefulness of Pulsed Tissue Doppler Imaging for Evaluating Systolic and Diastolic Left Ventricular Function in Patients with AL (Primary) Amyloidosis. Am. J. Cardiol. 2002, 89, 1067–1071. [Google Scholar] [CrossRef]
- Oerlemans, M.I.F.J.; Rutten, K.H.G.; Minnema, M.C.; Raymakers, R.A.P.; Asselbergs, F.W.; de Jonge, N. Cardiac Amyloidosis: The Need for Early Diagnosis. Neth. Heart J. 2019, 27, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Falk, R.H.; Alexander, K.M.; Liao, R.; Dorbala, S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J. Am. Coll. Cardiol. 2016, 68, 1323–1341. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, F.; Zeppa, P.; Catalano, L.; Cozzolino, I.; Gargiulo, G.; Musto, P.; D’Auria, F.; Liso, V.; Rizzi, R.; Caruso, N.; et al. Amyloid in Bone Marrow Smears of Patients Affected by Multiple Myeloma. Ann. Hematol. 2010, 89, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Garcia, Y.; Collins, A.B.; Stone, J.R. Abdominal Fat Pad Excisional Biopsy for the Diagnosis and Typing of Systemic Amyloidosis. Hum. Pathol. 2018, 72, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Picken, M.M. Amyloidosis-Where Are We Now and Where Are We Heading? Arch. Pathol. Lab. Med. 2010, 134, 545–551. [Google Scholar] [CrossRef]
- Yadav, P.; Leung, N.; Sanders, P.W.; Cockwell, P. The Use of Immunoglobulin Light Chain Assays in the Diagnosis of Paraprotein-Related Kidney Disease. Kidney Int. 2015, 87, 692–697. [Google Scholar] [CrossRef] [Green Version]
- Bradwell, A.R.; Carr-Smith, H.D.; Mead, G.P.; Tang, L.X.; Showell, P.J.; Drayson, M.T.; Drew, R. Highly Sensitive, Automated Immunoassay for Immunoglobulin Free Light Chains in Serum and Urine. Clin. Chem. 2001, 47, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Kourelis, T.V.; Kumar, S.K.; Gertz, M.A.; Lacy, M.Q.; Buadi, F.K.; Hayman, S.R.; Zeldenrust, S.; Leung, N.; Kyle, R.A.; Russell, S.; et al. Coexistent Multiple Myeloma or Increased Bone Marrow Plasma Cells Define Equally High-Risk Populations in Patients with Immunoglobulin Light Chain Amyloidosis. J. Clin. Oncol. 2013, 31, 4319–4324. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Dispenzieri, A.; Katzmann, J.A.; Larson, D.R.; Colby, C.L.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Leung, N.; Zeldenrust, S.R.; et al. Serum Immunoglobulin Free Light-Chain Measurement in Primary Amyloidosis: Prognostic Value and Correlations with Clinical Features. Blood 2010, 116, 5126–5129. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Lacy, M.Q.; Katzmann, J.A.; Rajkumar, S.V.; Abraham, R.S.; Hayman, S.R.; Kumar, S.K.; Clark, R.; Kyle, R.A.; Litzow, M.R.; et al. Absolute Values of Immunoglobulin Free Light Chains Are Prognostic in Patients with Primary Systemic Amyloidosis Undergoing Peripheral Blood Stem Cell Transplantation. Blood 2006, 107, 3378–3383. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and Treatment of Cardiac Amyloidosis: A Position Statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2021, 42, 1554–1568. [Google Scholar] [CrossRef]
- Palladini, G.; Campana, C.; Klersy, C.; Balduini, A.; Vadacca, G.; Perfetti, V.; Perlini, S.; Obici, L.; Ascari, E.; d’Eril, G.M.; et al. Serum N-Terminal pro-Brain Natriuretic Peptide Is a Sensitive Marker of Myocardial Dysfunction in AL Amyloidosis. Circulation 2003, 107, 2440–2445. [Google Scholar] [CrossRef]
- Palladini, G.; Dispenzieri, A.; Gertz, M.A.; Kumar, S.; Wechalekar, A.; Hawkins, P.N.; Schönland, S.; Hegenbart, U.; Comenzo, R.; Kastritis, E.; et al. New Criteria for Response to Treatment in Immunoglobulin Light Chain Amyloidosis Based on Free Light Chain Measurement and Cardiac Biomarkers: Impact on Survival Outcomes. J. Clin. Oncol. 2012, 30, 4541–4549. [Google Scholar] [CrossRef]
- Palladini, G.; Lavatelli, F.; Russo, P.; Perlini, S.; Perfetti, V.; Bosoni, T.; Obici, L.; Bradwell, A.R.; D’Eril, G.M.; Fogari, R.; et al. Circulating Amyloidogenic Free Light Chains and Serum N-Terminal Natriuretic Peptide Type B Decrease Simultaneously in Association with Improvement of Survival in AL. Blood 2006, 107, 3854–3858. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Gertz, M.A.; Kyle, R.A.; Lacy, M.Q.; Burritt, M.F.; Therneau, T.M.; Greipp, P.R.; Witzig, T.E.; Lust, J.A.; Rajkumar, S.V.; et al. Serum Cardiac Troponins and N-Terminal pro-Brain Natriuretic Peptide: A Staging System for Primary Systemic Amyloidosis. J. Clin. Oncol. 2004, 22, 3751–3757. [Google Scholar] [CrossRef]
- Suresh, R.; Grogan, M.; Maleszewski, J.J.; Pellikka, P.A.; Hanna, M.; Dispenzieri, A.; Pereira, N.L. Advanced Cardiac Amyloidosis Associated with Normal Interventricular Septal Thickness: An Uncommon Presentation of Infiltrative Cardiomyopathy. J. Am. Soc. Echocardiogr. 2014, 27, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.Y.; Kim, K.; Choi, J.-O.; Kim, S.J.; Kim, J.-S.; Choe, Y.H.; Grogan, M.A.; Jeon, E.-S. Cardiac Amyloidosis without Increased Left Ventricular Wall Thickness. Mayo Clin. Proc. 2014, 89, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Bellavia, D.; Pellikka, P.A.; Al-Zahrani, G.B.; Abraham, T.P.; Dispenzieri, A.; Miyazaki, C.; Lacy, M.; Scott, C.G.; Oh, J.K.; Miller, F.A. Independent Predictors of Survival in Primary Systemic (Al) Amyloidosis, Including Cardiac Biomarkers and Left Ventricular Strain Imaging: An Observational Cohort Study. J. Am. Soc. Echocardiogr. Off. Publ. Am. Soc. Echocardiogr. 2010, 23, 643–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic Cardiac Amyloidoses. Circulation 2009, 120, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Colby, C.; Laumann, K.; Zeldenrust, S.R.; Leung, N.; Dingli, D.; et al. Revised Prognostic Staging System for Light Chain Amyloidosis Incorporating Cardiac Biomarkers and Serum Free Light Chain Measurements. J. Clin. Oncol. 2012, 30, 989–995. [Google Scholar] [CrossRef] [Green Version]
- Kristen, A.V.; Dengler, T.J.; Hegenbart, U.; Schonland, S.O.; Goldschmidt, H.; Sack, F.-U.; Voss, F.; Becker, R.; Katus, H.A.; Bauer, A. Prophylactic Implantation of Cardioverter-Defibrillator in Patients with Severe Cardiac Amyloidosis and High Risk for Sudden Cardiac Death. Heart Rhythm 2008, 5, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Wechalekar, A.D.; Goodman, H.J.B.; Lachmann, H.J.; Offer, M.; Hawkins, P.N.; Gillmore, J.D. Safety and Efficacy of Risk-Adapted Cyclophosphamide, Thalidomide, and Dexamethasone in Systemic AL Amyloidosis. Blood 2007, 109, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhodapkar, M.V.; Hussein, M.A.; Rasmussen, E.; Solomon, A.; Larson, R.A.; Crowley, J.J.; Barlogie, B.; United States Intergroup Trial Southwest Oncology Group. Clinical Efficacy of High-Dose Dexamethasone with Maintenance Dexamethasone/Alpha Interferon in Patients with Primary Systemic Amyloidosis: Results of United States Intergroup Trial Southwest Oncology Group (SWOG) S9628. Blood 2004, 104, 3520–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, L.; Palladini, G.; Cerruti, F.; Pengo, N.; Cascio, P.; Merlini, G.; Sitia, R.; Cenci, S. Assessing Proteostasis and Proteasome Stress In Light Chain Amyloidosis. Blood 2010, 116, 3992. [Google Scholar] [CrossRef]
- Mikhael, J.R.; Schuster, S.R.; Jimenez-Zepeda, V.H.; Bello, N.; Spong, J.; Reeder, C.B.; Stewart, A.K.; Bergsagel, P.L.; Fonseca, R. The Combination of Cyclophosphamide-Bortezomib-Dexamethasone (CYBOR-D) Is a Highly Effective and Well Tolerated Regimen That Produces Rapid and Complete Hematological Response In Patients with AL Amyloidosis. Blood 2010, 116, 3063. [Google Scholar] [CrossRef]
- Zonder, J.A.; Sanchorawala, V.; Snyder, R.M.; Matous, J.; Terebelo, H.; Janakiraman, N.; Mapara, M.Y.; Lalo, S.; Tageja, N.; Webb, C.; et al. Melphalan and Dexamethasone Plus Bortezomib Induces Hematologic and Organ Responses in AL-Amyloidosis with Tolerable Neurotoxicity. Blood 2009, 114, 746. [Google Scholar] [CrossRef]
- D’Souza, A.; Dispenzieri, A.; Wirk, B.; Zhang, M.-J.; Huang, J.; Gertz, M.A.; Kyle, R.A.; Kumar, S.; Comenzo, R.L.; Peter Gale, R.; et al. Improved Outcomes After Autologous Hematopoietic Cell Transplantation for Light Chain Amyloidosis: A Center for International Blood and Marrow Transplant Research Study. J. Clin. Oncol. 2015, 33, 3741–3749. [Google Scholar] [CrossRef] [PubMed]
- Sidiqi, M.H.; Gertz, M. Daratumumab for the Treatment of AL Amyloidosis. Leuk. Lymphoma 2019, 60, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Jeryczynski, G.; Antlanger, M.; Duca, F.; Binder-Rodriguez, C.; Reiter, T.; Simonitsch-Klupp, I.; Bonderman, D.; Kain, R.; Krauth, M.-T.; Agis, H. First-Line Daratumumab Shows High Efficacy and Tolerability Even in Advanced AL Amyloidosis: The Real-World Experience. ESMO Open 2021, 6, 100065. [Google Scholar] [CrossRef] [PubMed]
- Sher, T.; Fenton, B.; Akhtar, A.; Gertz, M.A. First Report of Safety and Efficacy of Daratumumab in 2 Cases of Advanced Immunoglobulin Light Chain Amyloidosis. Blood 2016, 128, 1987–1989. [Google Scholar] [CrossRef] [Green Version]


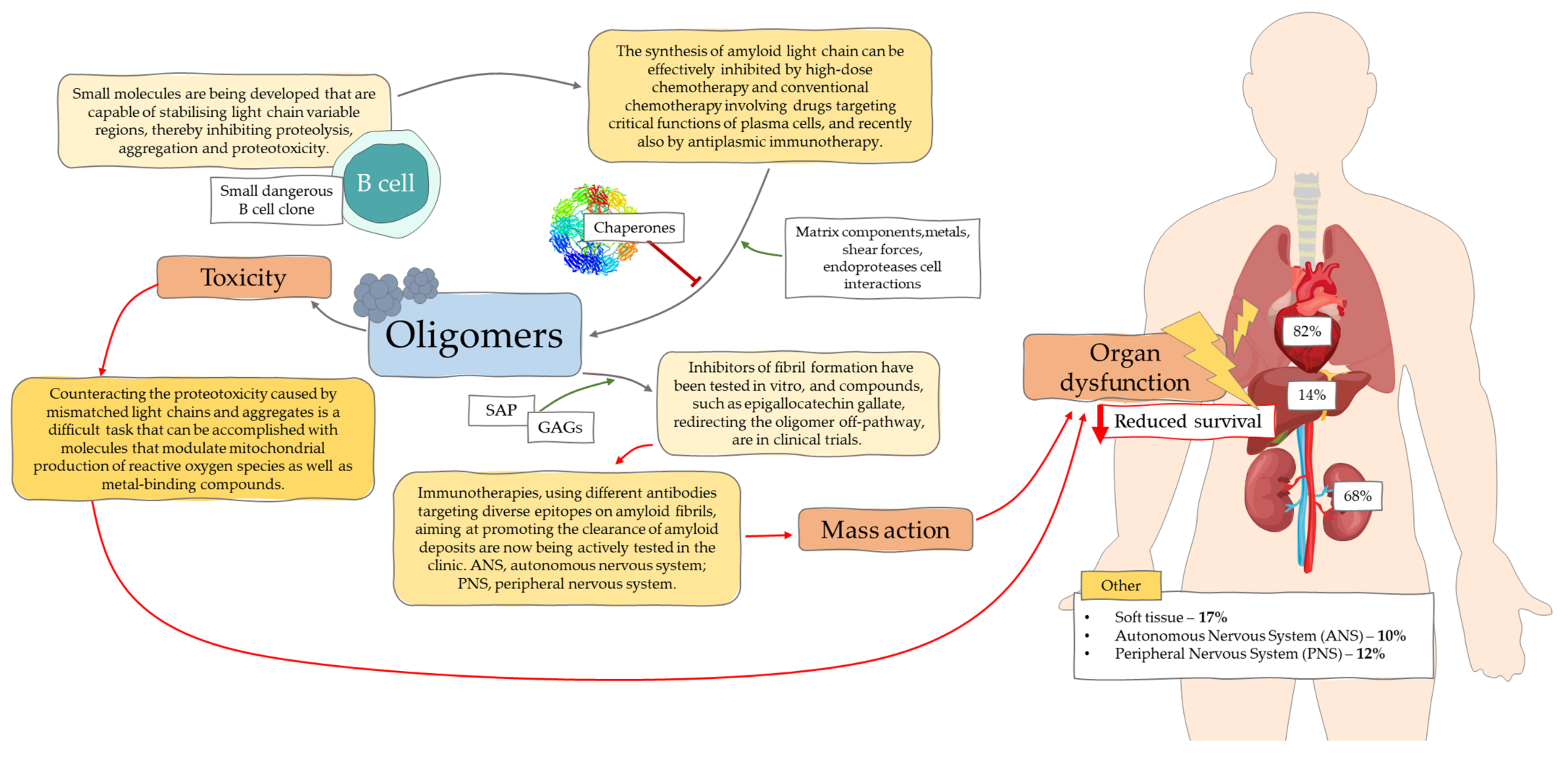{kind=link}
| Name of Protein | Type of Systemic Amyloidosis |
|---|---|
| Immunoglobulin light chain | Light chain |
| Transthyretin (wild-type) | TRwt |
| Transthyretin (mutant) | TTRv |
| Serum amyloid A (SAA) | AA |
| Leucocyte chemotactic factor 2 (LECT2) | ALECT2 |
| Gelsolin | AGel |
| Apolipoprotein AI (ApoAI) | AApoAI |
| Apolipoprotein AII (ApoAII) | AApoAII |
| Apolipoprotein AIV (ApoAIV) | AApoAIV |
| Apolipoprotein CII (ApoCII) | AApoCII |
| Apolipoprotein CIII (ApoCIII) | AApoCIII |
| Fibrinogen | AFib |
| β2 microglobulin | Aβ2M |
| Lysozyme | ALys |
| Type | Red Flag |
|---|---|
| Extracardiac | Skin bruising |
| Macroglossia | |
| Renal insufficiency | |
| Proteinuria | |
| Cardiac | Hypotension or normotensive if previous hypertensive |
| Pseudoinfarct pattern in ECG | |
| Low/decreased QRS voltage to degree of LV thickness | |
| AV conduction disease | |
| Disproportionally elevated NT-proBNP to degree of HF | |
| Persisting elevated troponin levels | |
| Granular sparkling of myocardium in echocardiogram | |
| Increased right ventricular wall thickness | |
| Increased valve thickness | |
| Pericardial effusion | |
| Reduced longitudinal strain with apical sparing pattern | |
| Subendocardial late gadolinium enhancement | |
| Elevated native T1 values | |
| Increased extracellular volume | |
| Abnormal gadolinium kinetics |
| Stage | Amount of Parameters | 5-Year Survival |
|---|---|---|
| I | 0 parameters | 68% |
| II | 1 parameters | 60% |
| III | 2 parameters | 28% |
| IV | 3 parameters | 14% |
| Supportive Therapy | Therapy Specific to AL |
|---|---|
| patient education | dexamethasone with an alkylator |
| maintaining adequate blood pressure | cyclophosphamide with thalidomide |
| balancing peripheral edema and renal insufficiency | dexamethasone with thalidomide |
| balance between salt restriction and water supply | combinations with bortezomib |
| fludrocortisone enhances fluid retention | high-dose chemotherapy with autologous stem cell transplantation (ASCT) |
| midodrine and compression stockings for orthostatic hypotension | daratumumab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stelmach-Gołdyś, A.; Zaborek-Łyczba, M.; Łyczba, J.; Garus, B.; Pasiarski, M.; Mertowska, P.; Małkowska, P.; Hrynkiewicz, R.; Niedźwiedzka-Rystwej, P.; Grywalska, E. Physiology, Diagnosis and Treatment of Cardiac Light Chain Amyloidosis. J. Clin. Med. 2022, 11, 911. https://doi.org/10.3390/jcm11040911
Stelmach-Gołdyś A, Zaborek-Łyczba M, Łyczba J, Garus B, Pasiarski M, Mertowska P, Małkowska P, Hrynkiewicz R, Niedźwiedzka-Rystwej P, Grywalska E. Physiology, Diagnosis and Treatment of Cardiac Light Chain Amyloidosis. Journal of Clinical Medicine. 2022; 11(4):911. https://doi.org/10.3390/jcm11040911
Chicago/Turabian StyleStelmach-Gołdyś, Agnieszka, Monika Zaborek-Łyczba, Jakub Łyczba, Bartosz Garus, Marcin Pasiarski, Paulina Mertowska, Paulina Małkowska, Rafał Hrynkiewicz, Paulina Niedźwiedzka-Rystwej, and Ewelina Grywalska. 2022. "Physiology, Diagnosis and Treatment of Cardiac Light Chain Amyloidosis" Journal of Clinical Medicine 11, no. 4: 911. https://doi.org/10.3390/jcm11040911
APA StyleStelmach-Gołdyś, A., Zaborek-Łyczba, M., Łyczba, J., Garus, B., Pasiarski, M., Mertowska, P., Małkowska, P., Hrynkiewicz, R., Niedźwiedzka-Rystwej, P., & Grywalska, E. (2022). Physiology, Diagnosis and Treatment of Cardiac Light Chain Amyloidosis. Journal of Clinical Medicine, 11(4), 911. https://doi.org/10.3390/jcm11040911