
The Leading Role of the Immune Microenvironment in Multiple Myeloma: A New Target with a Great Prognostic and Clinical Value

 ,
,  ,
, 
 and
and 
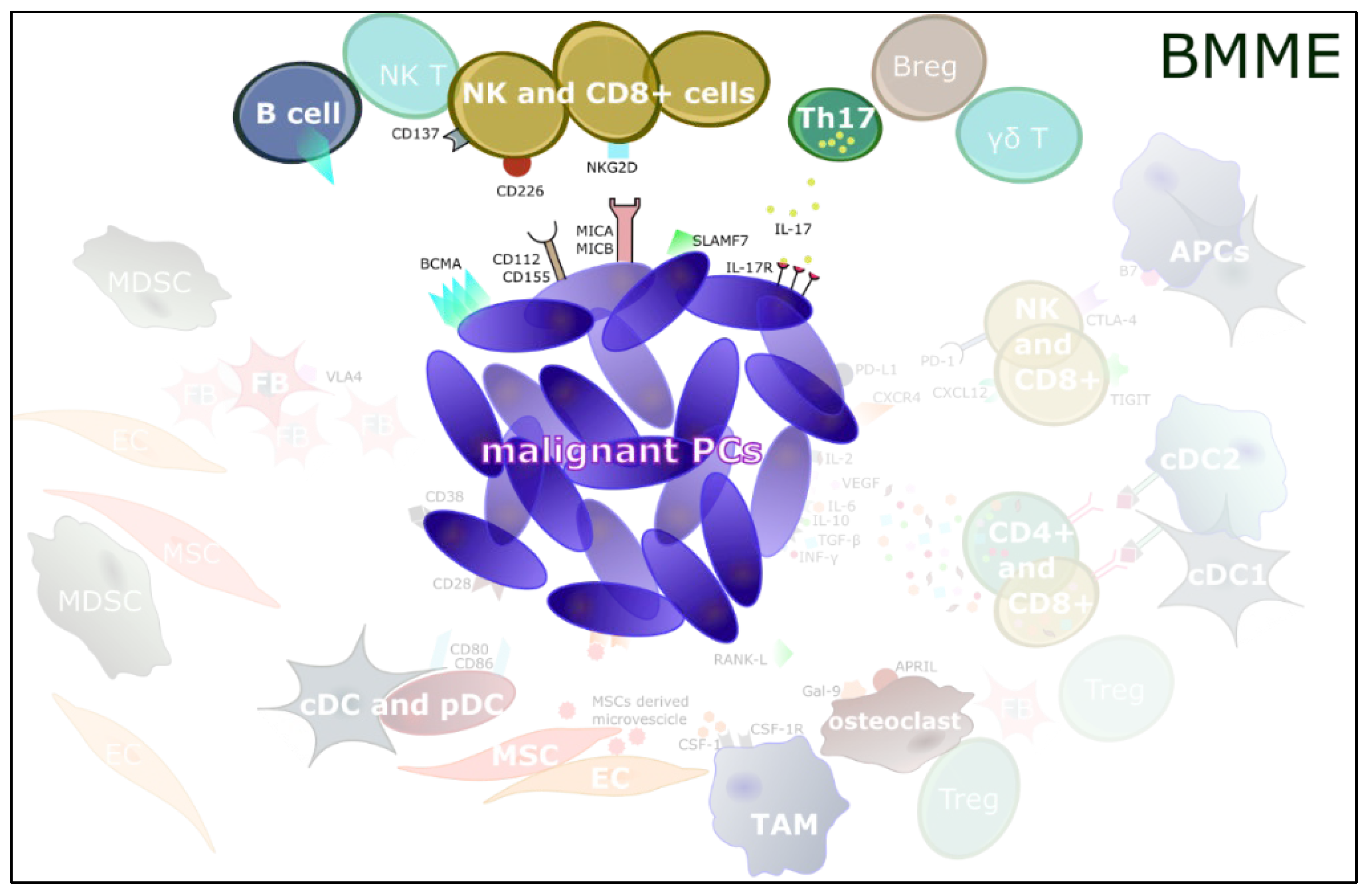{kind=link}
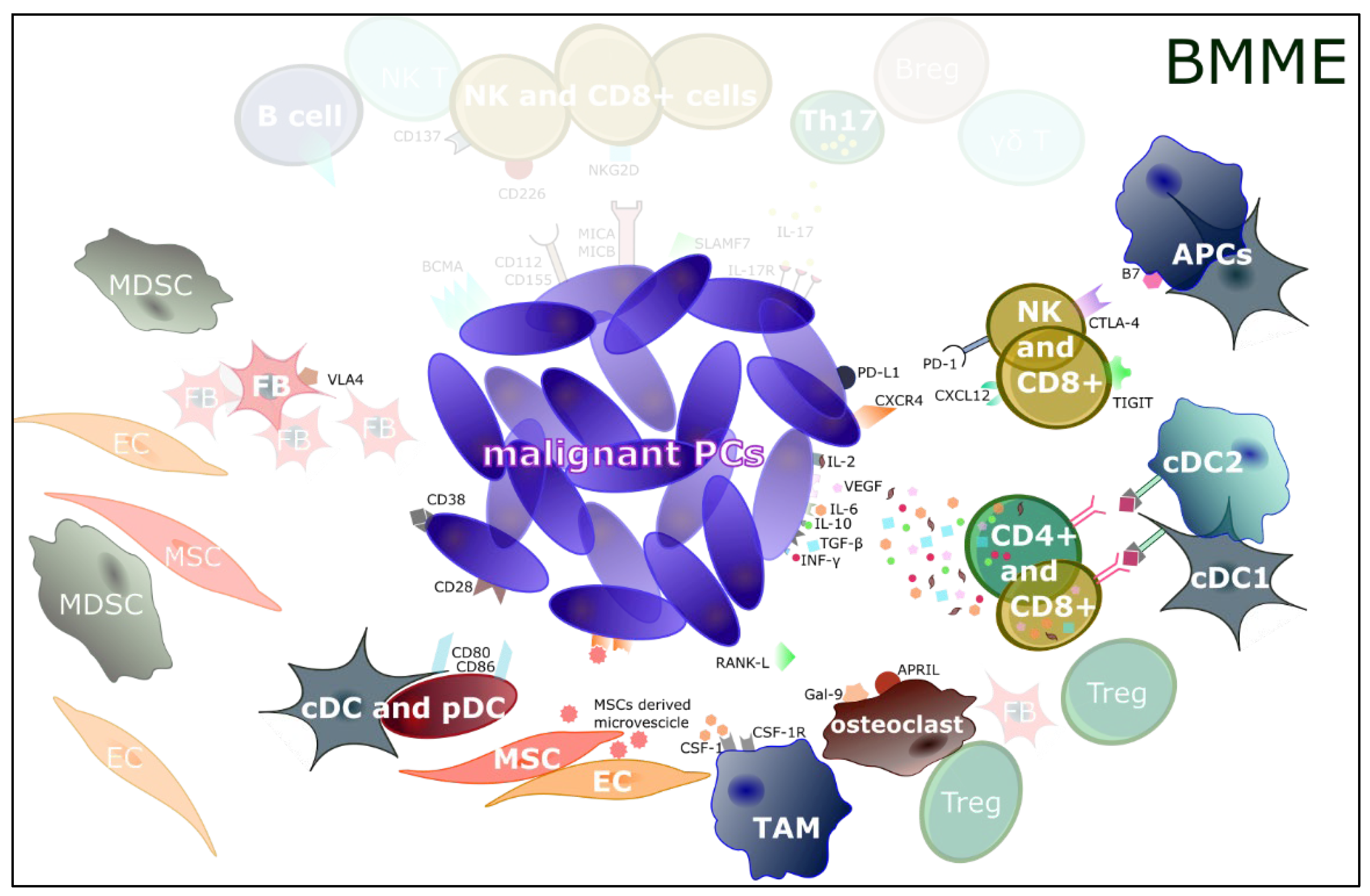{kind=link}
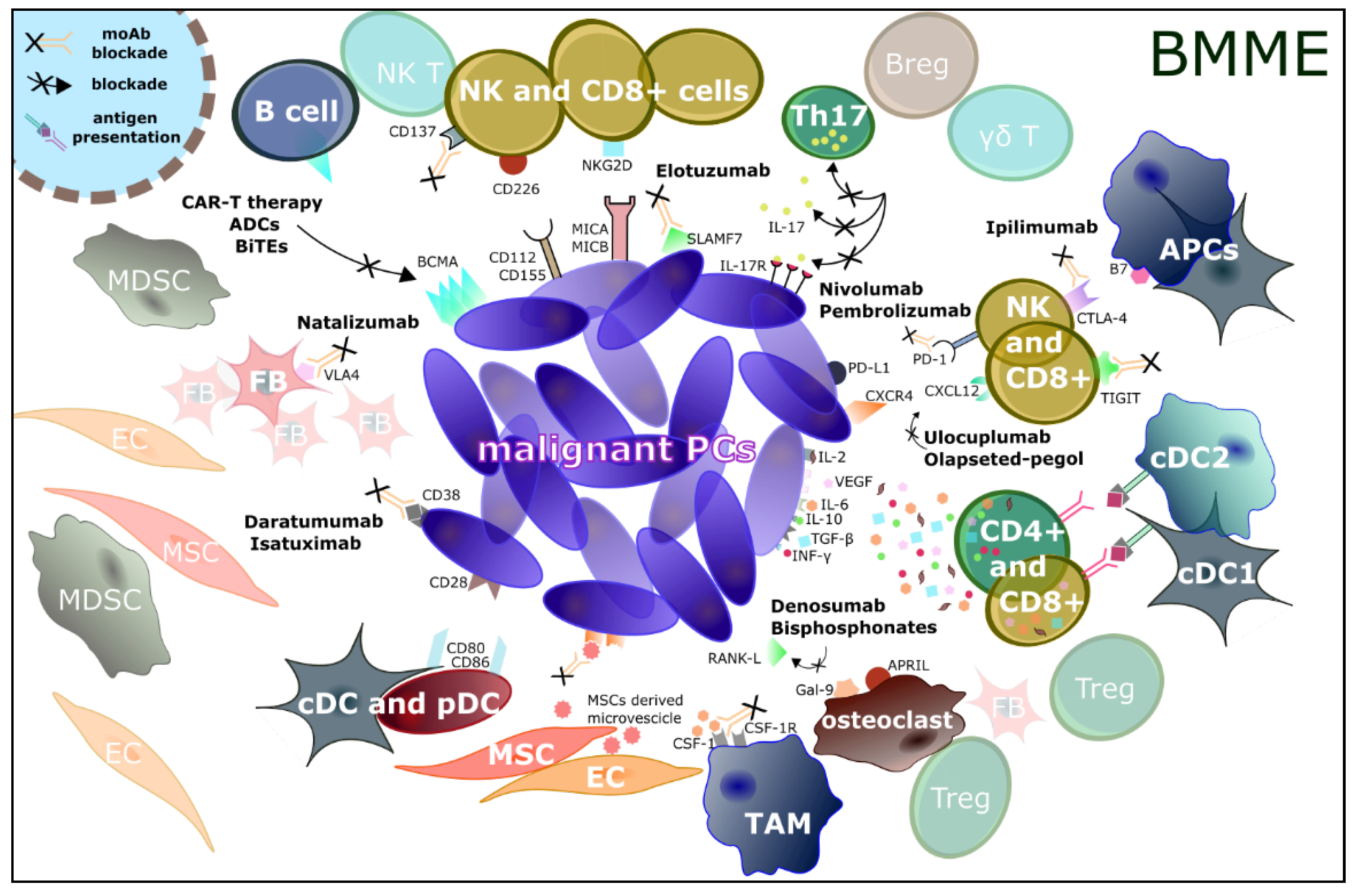{kind=link}
Abstract
:1. Introduction
2. Immunoediting in MM Progression
3. T Cell Dysfunction Occurs in MM Progression
4. Dendritic Cells as Important Players in MM Immune Response
5. Immune Checkpoints and MM Progression
5.1. CTLA-4
5.2. PD-1/PD-L1
6. Immunotherapy in MM
6.1. ImiDs and mAbs
6.2. Immune Checkpoints Inhibitors
6.3. CAR-T Cells
6.4. Ab-Drug Conjugates (ADCs)
6.5. Bispecific mAbs
7. Final Remarks and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| ADCC | Antibody-dependent cellular cytotoxicity |
| ADCP | Antibody-dependent cellular phagocytosis |
| ADCs | Antibody–drug conjugates |
| Ag | Antigen |
| APCs | Antigen-presenting cells |
| APRIL | A proliferation-inducing ligand |
| B7-H1 | B7-homologue 1 |
| BCL-2 | B-cell lymphoma-2 |
| BCMA | B-cell maturation antigen |
| BiTEs | Bispecific T cell engager mAbs |
| BM | Bone marrow |
| BMME | Bone marrow microenvironment |
| BMSCs | Bone marrow stromal cells |
| BMT | Bone marrow transplantation |
| Bregs | Regulatory B cells |
| CAR-T | Chimeric antigen receptor–T cell therapy |
| cDCs | Conventional dendritic cells |
| CCL | C-C motif ligand |
| CSF-1 | Colony-stimulating factor 1 |
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| CTLs | Cytotoxic T lymphocytes |
| CXCL | C-X-C motif Chemokine ligand |
| CXCR | C-X-C motif Chemokine receptor |
| DAMPs | Damage-associated molecular patterns |
| Dara-VTD | Daratumumab plus bortezomib, thalidomide, and dexamethasone |
| DCs | Dendritic cells |
| ECM | Extracellular matrix |
| ECs | Endothelial cells |
| EMA | European Medicines Agency |
| EPCs | Endothelial progenitor cells |
| ERK | Extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| FBs | Fibroblasts |
| Foxp3 | Forkhead box P3 |
| HLA | Human leukocyte antigen |
| IL | Interleukin |
| imDCs | Immature DCs |
| ImiDs | Immunomodulatory drugs |
| INF-γ | Interferon-γ |
| JAM-A | Junctional adhesion molecule A |
| mAb | Monoclonal antibodies |
| mDCs | Mature DCs |
| MDSCs | Myeloid-derived suppressor cells |
| MGUS | Monoclonal gammopathy of undetermined significance |
| MHC | Major histocompatibility complex |
| MM | Multiple myeloma |
| MSCs | Mesenchymal stromal cells |
| NK | Natural killer |
| OS | Overall survival |
| PCs | Plasma cells |
| PD-1 | Programmed death-1 |
| pDCs | Plasmacytoid dendritic cells |
| PD-L1 | PD ligand 1 |
| PFS | Progression-free survival |
| PGE2 | Prostaglandin E2 |
| PRRs | Pattern recognition receptors |
| RANKL | Receptor activator of nuclear factor kappa-B ligand |
| RR | Response rate |
| SMM | Smoldering multiple myeloma |
| TACI | Transmembrane activator CAML interactor |
| TAMs | Tumor-associated macrophages |
| TFH | T follicular helper |
| TGF-β | Transforming growth factor-β |
| Th | T helper |
| TIGIT | T cell immunoreceptor with immunoglobulin and ITIM domains |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| Tregs | Regulatory T cells |
| TTEs | Terminal effector CD8+ T cells |
| ULBPs | UL-binding proteins |
| VEGF | Vascular endothelial growth factor |
References
- Alrasheed, N.; Lee, L.; Ghorani, E.; Henry, J.Y.; Conde, L.; Chin, M.; Galas-Filipowicz, D.; Furness, A.J.S.; Chavda, S.J.; Richards, H.; et al. Marrow-Infiltrating Regulatory T Cells Correlate with the Presence of Dysfunctional CD4+ PD-1+ Cells and Inferior Survival in Patients with Newly Diagnosed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 3443–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgren, O.; Kyle, R.A.; Pfeiffer, R.M.; Katzmann, J.A.; Caporaso, N.E.; Hayes, R.B.; Dispenzieri, A.; Kumar, S.; Clark, R.J.; Baris, D.; et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: A prospective study. Blood 2009, 113, 5412–5417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oben, B.; Froyen, G.; Maclachlan, K.H.; Leongamornlert, D.; Abascal, F.; Zheng-Lin, B.; Yellapantula, V.; Derkach, A.; Geerdens, E.; Diamond, B.T.; et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat. Commun. 2021, 12, 1861. [Google Scholar] [CrossRef]
- Boyle, E.M.; Deshpande, S.; Tytarenko, R.; Ashby, C.; Wang, Y.; Bauer, M.A.; Johnson, S.K.; Wardell, C.P.; Thanendrarajan, S.; Zangari, M.; et al. The molecular make up of smoldering myeloma highlights the evolutionary pathways leading to multiple myeloma. Nat. Commun. 2021, 12, 293. [Google Scholar] [CrossRef] [PubMed]
- Laganà, A.; Perumal, D.; Melnekoff, D.; Readhead, B.; Kidd, B.A.; Leshchenko, V.; Kuo, P.-Y.; Keats, J.; DeRome, M.; Yesil, J.; et al. Integrative network analysis identifies novel drivers of pathogenesis and progression in newly diagnosed multiple myeloma. Leukemia 2018, 32, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef]
- Lopes, R.; Caetano, J.; Ferreira, B.; Barahona, F.; Carneiro, E.A.; João, C. The Immune Microenvironment in Multiple Myeloma: Friend or Foe? Cancers 2021, 13, 625. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Corre, J.; Mahtouk, K.; Attal, M.; Gadelorge, M.; Huynh, A.; Fleury-Cappellesso, S.; Danho, C.; Laharrague, P.; Klein, B.; Rème, T.; et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia 2007, 21, 1079–1088. [Google Scholar] [CrossRef]
- Botta, C.; Mendicino, F.; Martino, E.A.; Vigna, E.; Ronchetti, D.; Correale, P.; Morabito, F.; Neri, A.; Gentile, M. Mechanisms of Immune Evasion in Multiple Myeloma: Open Questions and Therapeutic Opportunities. Cancers 2021, 13, 3213. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J.; Martinet, L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood 2020, 136, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Bailur, J.K.; McCachren, S.S.; Doxie, D.B.; Shrestha, M.; Pendleton, K.; Nooka, A.K.; Neparidze, N.; Parker, T.L.; Bar, N.; Kaufman, J.L.; et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight 2019, 5, 127807. [Google Scholar] [CrossRef] [PubMed]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.-T.; Richardson, P.G.; et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef]
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, M.V.; Sexton, R.; Das, R.; Dhodapkar, K.M.; Zhang, L.; Sundaram, R.; Soni, S.; Crowley, J.J.; Orlowski, R.Z.; Barlogie, B. Prospective analysis of antigen-specific immunity, stem-cell antigens, and immune checkpoints in monoclonal gammopathy. Blood 2015, 126, 2475–2478. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br. J. Haematol. 2003, 121, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Guillerey, C.; Nakamura, K.; Vuckovic, S.; Hill, G.R.; Smyth, M.J. Immune responses in multiple myeloma: Role of the natural immune surveillance and potential of immunotherapies. Cell. Mol. Life Sci. 2016, 73, 1569–1589. [Google Scholar] [CrossRef]
- Minnie, S.A.; Hill, G.R. Immunotherapy of multiple myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Perumal, D.; Imai, N.; Laganà, A.; Finnigan, J.; Melnekoff, D.; Leshchenko, V.V.; Solovyov, A.; Madduri, D.; Chari, A.; Cho, H.J.; et al. Mutation-derived Neoantigen-specific T-cell Responses in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 450–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.; Asmann, Y.; Cattaneo, L.; Braggio, E.; Keats, J.; Auclair, D.; Lonial, S.; MMRF CoMMpass Network; Russell, S.J.; Stewart, A.K. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 2017, 7, e612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma-immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef]
- Guillerey, C.; Ferrari de Andrade, L.; Vuckovic, S.; Miles, K.; Ngiow, S.F.; Yong, M.C.R.; Teng, M.W.L.; Colonna, M.; Ritchie, D.S.; Chesi, M.; et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J. Clin. Investig. 2015, 125, 2077–2089. [Google Scholar] [CrossRef] [Green Version]
- Carbone, E.; Neri, P.; Mesuraca, M.; Fulciniti, M.T.; Otsuki, T.; Pende, D.; Groh, V.; Spies, T.; Pollio, G.; Cosman, D.; et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 2005, 105, 251–258. [Google Scholar] [CrossRef] [Green Version]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007, 67, 8444–8449. [Google Scholar] [CrossRef] [Green Version]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Jinushi, M.; Vanneman, M.; Munshi, N.C.; Tai, Y.-T.; Prabhala, R.H.; Ritz, J.; Neuberg, D.; Anderson, K.C.; Carrasco, D.R.; Dranoff, G. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc. Natl. Acad. Sci. USA 2008, 105, 1285–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, G.; Goodyear, O.; Moss, P. Immunodeficiency and immunotherapy in multiple myeloma. Br. J. Haematol. 2007, 138, 563–579. [Google Scholar] [CrossRef]
- Trotta, R.; Dal Col, J.; Yu, J.; Ciarlariello, D.; Thomas, B.; Zhang, X.; Allard, J.; Wei, M.; Mao, H.; Byrd, J.C.; et al. Immunodeficiency and immunotherapy in multiple myeloma. J. Immunol. 2008, 181, 3784–3792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.-T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousef, S.; Marvin, J.; Steinbach, M.; Langemo, A.; Kovacsovics, T.; Binder, M.; Kröger, N.; Luetkens, T.; Atanackovic, D. Immunomodulatory molecule PD-L1 is expressed on malignant plasma cells and myeloma-propagating pre-plasma cells in the bone marrow of multiple myeloma patients. Blood Cancer J. 2015, 5, e285. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, H.; Chen, H.; Wang, W.; Chen, X.-Q.; Geng, Q.-R.; Xia, Z.-J.; Lu, Y. Serum levels of soluble programmed death ligand 1 predict treatment response and progression free survival in multiple myeloma. Oncotarget 2015, 6, 41228–41236. [Google Scholar] [CrossRef] [Green Version]
- Tamura, H. Immunopathogenesis and immunotherapy of multiple myeloma. Int. J. Hematol. 2018, 107, 278–285. [Google Scholar] [CrossRef] [Green Version]
- Hope, C.; Foulcer, S.; Jagodinsky, J.; Chen, S.X.; Jensen, J.L.; Patel, S.; Leith, C.; Maroulakou, I.; Callander, N.; Miyamoto, S.; et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 2016, 128, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef]
- Nakamura, K.; Kassem, S.; Cleynen, A.; Chrétien, M.-L.; Guillerey, C.; Putz, E.M.; Bald, T.; Förster, I.; Vuckovic, S.; Hill, G.R.; et al. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 2018, 33, 634–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, J.-Y.; Zhu, X.-D.; Chai, Z.-T.; Cai, H.; Zhang, Y.-Y.; Zhang, K.-Z.; Kong, L.-Q.; Zhang, N.; Ye, B.-G.; Ma, D.-N.; et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabbah, M.; Jarchowsky-Dolberg, O.; Attar-Schneider, O.; Tartakover Matalon, S.; Pasmanik-Chor, M.; Drucker, L.; Lishner, M. Multiple myeloma BM-MSCs increase the tumorigenicity of MM cells via transfer of VLA4-enriched microvesicles. Carcinogenesis 2020, 41, 100–110. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Najar, M.; Stamatopoulos, B.; Pieters, K.; Pradier, O.; Bron, D.; Meuleman, N.; Lagneaux, L. Immune impairments in multiple myeloma bone marrow mesenchymal stromal cells. Cancer Immunol. Immunother. 2015, 64, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.K.; Fink, J.L.; Grady, J.P.; Morgan, G.J.; Mullighan, C.G.; To, L.B.; Hewett, D.R.; Zannettino, A.C.W. Subclonal evolution in disease progression from MGUS/SMM to multiple myeloma is characterised by clonal stability. Leukemia 2019, 33, 457–468. [Google Scholar] [CrossRef]
- Xu, S.; De Veirman, K.; De Becker, A.; Vanderkerken, K.; Van Riet, I. Mesenchymal stem cells in multiple myeloma: A therapeutical tool or target? Leukemia 2018, 32, 1500–1514. [Google Scholar] [CrossRef]
- Menu, E.; Asosingh, K.; Indraccolo, S.; De Raeve, H.; Van Riet, I.; Van Valckenborgh, E.; Vande Broek, I.; Van de Broek, I.; Fujii, N.; Tamamura, H.; et al. The involvement of stromal derived factor 1alpha in homing and progression of multiple myeloma in the 5TMM model. Haematologica 2006, 91, 605–612. [Google Scholar]
- Alsayed, Y.; Ngo, H.; Runnels, J.; Leleu, X.; Singha, U.K.; Pitsillides, C.M.; Spencer, J.A.; Kimlinger, T.; Ghobrial, J.M.; Jia, X.; et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood 2007, 109, 2708–2717. [Google Scholar] [CrossRef]
- Murray, M.E.; Gavile, C.M.; Nair, J.R.; Koorella, C.; Carlson, L.M.; Buac, D.; Utley, A.; Chesi, M.; Bergsagel, P.L.; Boise, L.H.; et al. CD28-mediated pro-survival signaling induces chemotherapeutic resistance in multiple myeloma. Blood 2014, 123, 3770–3779. [Google Scholar] [CrossRef]
- Glatman Zaretsky, A.; Konradt, C.; Dépis, F.; Wing, J.B.; Goenka, R.; Atria, D.G.; Silver, J.S.; Cho, S.; Wolf, A.I.; Quinn, W.J.; et al. T Regulatory Cells Support Plasma Cell Populations in the Bone Marrow. Cell Rep. 2017, 18, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Palucka, A.K.; Blanck, J.-P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 2003, 19, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Song, Y.; Du, T.; Tai, Y.-T.; Chauhan, D.; Anderson, K.C. Targeting tryptophan catabolic kynurenine pathway enhances antitumor immunity and cytotoxicity in multiple myeloma. Leukemia 2020, 34, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.D.; Spencer, A.; Joy Ho, P.; Kennedy, N.; Kabani, K.; Yang, S.; Sze, D.M.; Aklilu, E.; Gibson, J.; Joshua, D.E. Prognostically significant cytotoxic T cell clones are stimulated after thalidomide therapy in patients with multiple myeloma. Leuk. Lymphoma 2009, 50, 1860–1864. [Google Scholar] [CrossRef] [PubMed]
- Joshua, D.E.; Vuckovic, S.; Favaloro, J.; Lau, K.H.A.; Yang, S.; Bryant, C.E.; Gibson, J.; Ho, P.J. Treg and Oligoclonal Expansion of Terminal Effector CD8+ T Cell as Key Players in Multiple Myeloma. Front. Immunol. 2021, 12, 620596. [Google Scholar] [CrossRef]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [Green Version]
- Favaloro, J.; Brown, R.; Aklilu, E.; Yang, S.; Suen, H.; Hart, D.; Fromm, P.; Gibson, J.; Khoo, L.; Ho, P.J.; et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk. Lymphoma 2014, 55, 1090–1098. [Google Scholar] [CrossRef]
- Brown, R.; Suen, H.; Favaloro, J.; Yang, S.; Ho, P.J.; Gibson, J.; Joshua, D. Trogocytosis generates acquired regulatory T cells adding further complexity to the dysfunctional immune response in multiple myeloma. Oncoimmunology 2012, 1, 1658–1660. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.-J.; Yuan, Z.-H.; Liu, Y.-X.; Hu, G.-Y. Increased numbers of T helper 17 cells and the correlation with clinicopathological characteristics in multiple myeloma. J. Int. Med. Res. 2012, 40, 556–564. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, Y.; Xin, V.W.; Wang, X.-W.; Peng, X.-C.; Liu, X.-Q.; Wang, D.; Li, N.; Cheng, J.-T.; Lyv, Y.-N.; et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 2020, 13, 107. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Nowis, D.; Golab, J.; Vandenabeele, P.; Krysko, D.V.; Agostinis, P. Immunogenic cell death, DAMPs and anticancer therapeutics: An emerging amalgamation. Biochim. Biophys. Acta 2010, 1805, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.M.; Martin, S.; Garg, A.D.; Agostinis, P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a DC-Cancer Cells Interface That Augments Anticancer Immunity. Front. Immunol. 2013, 4, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilligan, K.L.; Ronchese, F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell. Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Annu. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef]
- Tussiwand, R.; Everts, B.; Grajales-Reyes, G.E.; Kretzer, N.M.; Iwata, A.; Bagaitkar, J.; Wu, X.; Wong, R.; Anderson, D.A.; Murphy, T.L.; et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 2015, 42, 916–928. [Google Scholar] [CrossRef] [Green Version]
- Do, T.H.; Johnsen, H.E.; Kjaersgaard, E.; Taaning, E.; Svane, I.M. Impaired circulating myeloid DCs from myeloma patients. Cytotherapy 2004, 6, 196–203. [Google Scholar] [CrossRef]
- Leone, P.; Berardi, S.; Frassanito, M.A.; Ria, R.; De Re, V.; Cicco, S.; Battaglia, S.; Ditonno, P.; Dammacco, F.; Vacca, A.; et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood 2015, 126, 1443–1451. [Google Scholar] [CrossRef] [Green Version]
- Zelante, T.; Fric, J.; Wong, A.Y.W.; Ricciardi-Castagnoli, P. Interleukin-2 production by dendritic cells and its immuno-regulatory functions. Front. Immunol. 2012, 3, 161. [Google Scholar] [CrossRef] [Green Version]
- Rutella, S.; Locatelli, F. Targeting multiple-myeloma-induced immune dysfunction to improve immunotherapy outcomes. Clin. Dev. Immunol. 2012, 2012, 196063. [Google Scholar] [CrossRef]
- Braga, W.M.T.; da Silva, B.R.; de Carvalho, A.C.; Maekawa, Y.H.; Bortoluzzo, A.B.; Rizzatti, E.G.; Atanackovic, D.; Colleoni, G.W.B. FOXP3 and CTLA4 overexpression in multiple myeloma bone marrow as a sign of accumulation of CD4(+) T regulatory cells. Cancer Immunol. Immunother. 2014, 63, 1189–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; Azpilikueta, A.; Puig, N.; Ocio, E.M.; Sharma, R.; Oyajobi, B.O.; Labiano, S.; San-Segundo, L.; Rodriguez, A.; Aires-Mejia, I.; et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia 2015, 29, 2110–2113. [Google Scholar] [CrossRef] [PubMed]
- Pianko, M.J.; Liu, Y.; Bagchi, S.; Lesokhin, A.M. Immune checkpoint blockade for hematologic malignancies: A review. Stem Cell Investig. 2017, 4, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Poma, S.M.; Salas-Benito, D.; Lozano, T.; Casares, N.; Riezu-Boj, J.-I.; Mancheño, U.; Elizalde, E.; Alignani, D.; Zubeldia, N.; Otano, I.; et al. Expansion of Tumor-Infiltrating CD8+ T cells Expressing PD-1 Improves the Efficacy of Adoptive T-cell Therapy. Cancer Res. 2017, 77, 3672–3684. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.J.; Pronschinske, K.B.; Shyer, J.A.; Sharma, S.; Leung, S.; Curran, S.A.; Lesokhin, A.M.; Devlin, S.M.; Giralt, S.A.; Young, J.W. T-cell Exhaustion in Multiple Myeloma Relapse after Autotransplant: Optimal Timing of Immunotherapy. Cancer Immunol. Res. 2016, 4, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Atanackovic, D.; Luetkens, T. Biomarkers for checkpoint inhibition in hematologic malignancies. Semin. Cancer Biol. 2018, 52, 198–206. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Itchaki, G.; Brown, J.R. Lenalidomide in the treatment of chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2017, 26, 633–650. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luptakova, K.; Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Stroopinsky, D.; Kufe, T.; Vasir, B.; Arnason, J.; Tzachanis, D.; Zwicker, J.I.; et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol. Immunother. 2013, 62, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Zhou, X.; Flüchter, P.; Nickel, K.; Meckel, K.; Messerschmidt, J.; Böckle, D.; Knorz, S.; Steinhardt, M.J.; Krummenast, F.; Danhof, S.; et al. Carfilzomib Based Treatment Strategies in the Management of Relapsed/Refractory Multiple Myeloma with Extramedullary Disease. Cancers 2020, 12, 1035. [Google Scholar] [CrossRef] [Green Version]
- Chari, A.; Martinez-Lopez, J.; Mateos, M.-V.; Bladé, J.; Benboubker, L.; Oriol, A.; Arnulf, B.; Rodriguez-Otero, P.; Pineiro, L.; Jakubowiak, A.; et al. Daratumumab plus carfilzomib and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood 2019, 134, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Rasche, L.; Hudecek, M.; Einsele, H. What is the future of immunotherapy in multiple myeloma? Blood 2020, 136, 2491–2497. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.-V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Gunaratne, J.; Cheong, L.L.; Liu, S.C.; Koh, T.L.; Huang, G.; Blackstock, W.P.; Chng, W.J. Plasma membrane proteomics identifies biomarkers associated with MMSET overexpression in T(4;14) multiple myeloma. Oncotarget 2013, 4, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Mishima, Y.; Sacco, A.; Moschetta, M.; Tai, Y.-T.; Shi, J.; Zhang, Y.; Reagan, M.R.; Huynh, D.; Kawano, Y.; et al. CXCR4 Regulates Extra-Medullary Myeloma through Epithelial-Mesenchymal-Transition-like Transcriptional Activation. Cell Rep. 2015, 12, 622–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roccaro, A.M.; Sacco, A.; Purschke, W.G.; Moschetta, M.; Buchner, K.; Maasch, C.; Zboralski, D.; Zöllner, S.; Vonhoff, S.; Mishima, Y.; et al. SDF-1 inhibition targets the bone marrow niche for cancer therapy. Cell Rep. 2014, 9, 118–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podar, K.; Zimmerhackl, A.; Fulciniti, M.; Tonon, G.; Hainz, U.; Tai, Y.-T.; Vallet, S.; Halama, N.; Jäger, D.; Olson, D.L.; et al. The selective adhesion molecule inhibitor Natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: Therapeutic implications. Br. J. Haematol. 2011, 155, 438–448. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients with Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.; Massard, C.; Soria, J.-C.; Deutsch, E. Concurrent irradiation with the anti-programmed cell death ligand-1 immune checkpoint blocker durvalumab: Single centre subset analysis from a phase 1/2 trial. Eur. J. Cancer 2016, 68, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Minnie, S.A.; Kuns, R.D.; Gartlan, K.H.; Zhang, P.; Wilkinson, A.N.; Samson, L.; Guillerey, C.; Engwerda, C.; MacDonald, K.P.; Smyth, M.J.; et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood 2018, 133, 1675–1688, Erratum in Blood 2019, 134, 1878. [Google Scholar] [CrossRef]
- Jing, W.; Gershan, J.A.; Weber, J.; Tlomak, D.; McOlash, L.; Sabatos-Peyton, C.; Johnson, B.D. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. J. Immunother. Cancer 2015, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Arce Vargas, F.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonezawa, A.; Dutt, S.; Chester, C.; Kim, J.; Kohrt, H.E. Boosting Cancer Immunotherapy with Anti-CD137 Antibody Therapy. Clin. Cancer Res. 2015, 21, 3113–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuckovic, S.; Minnie, S.A.; Smith, D.; Gartlan, K.H.; Watkins, T.S.; Markey, K.A.; Mukhopadhyay, P.; Guillerey, C.; Kuns, R.D.; Locke, K.R.; et al. Bone marrow transplantation generates T cell-dependent control of myeloma in mice. J. Clin. Investig. 2019, 129, 106–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKee, S.J.; Doff, B.L.; Soon, M.S.F.; Mattarollo, S.R. Therapeutic Efficacy of 4-1BB Costimulation Is Abrogated by PD-1 Blockade in a Model of Spontaneous B-cell Lymphoma. Cancer Immunol. Res. 2017, 5, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manieri, N.A.; Chiang, E.Y.; Grogan, J.L. TIGIT: A Key Inhibitor of the Cancer Immunity Cycle. Trends Immunol. 2017, 38, 20–28. [Google Scholar] [CrossRef]
- Garfall, A.L.; Maus, M.V.; Hwang, W.-T.; Lacey, S.F.; Mahnke, Y.D.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.D.; Weiss, B.M.; et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1040–1047. [Google Scholar] [CrossRef]
- Guo, B.; Chen, M.; Han, Q.; Hui, F.; Dai, H.; Zhang, W.; Zhang, Y.; Wang, Y.; Zhu, H.; Han, W. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. J. Cell. Immunother. 2016, 2, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Casucci, M.; Nicolis di Robilant, B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef]
- Chu, J.; He, S.; Deng, Y.; Zhang, J.; Peng, Y.; Hughes, T.; Yi, L.; Kwon, C.-H.; Wang, Q.-E.; Devine, S.M.; et al. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clin. Cancer Res. 2014, 20, 3989–4000. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.A.; Savoldo, B.; Torrano, V.; Ballard, B.; Zhang, H.; Dakhova, O.; Liu, E.; Carrum, G.; Kamble, R.T.; Gee, A.P.; et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J. Clin. Investig. 2016, 126, 2588–2596. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, 133977. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Vaxman, I.; Abeykoon, J.; Dispenzieri, A.; Kumar, S.K.; Buadi, F.; Lacy, M.Q.; Dingli, D.; Hwa, Y.; Fonder, A.; Hobbs, M.; et al. “Real-life” data of the efficacy and safety of belantamab mafodotin in relapsed multiple myeloma-the Mayo Clinic experience. Blood Cancer J. 2021, 11, 196. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-H.; Liu, J.; Wang, B.-Y.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Moreau, P.; Sonneveld, P.; Boccadoro, M.; Cook, G.; Mateos, M.V.; Nahi, H.; Goldschmidt, H.; Dimopoulos, M.A.; Lucio, P.; Bladé, J.; et al. Chimeric antigen receptor T-cell therapy for multiple myeloma: A consensus statement from The European Myeloma Network. Haematologica 2019, 104, 2358–2360. [Google Scholar] [CrossRef]
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Tzogani, K.; Penttilä, K.; Lähteenvuo, J.; Lapveteläinen, T.; Lopez Anglada, L.; Prieto, C.; Garcia-Ochoa, B.; Enzmann, H.; Gisselbrecht, C.; Delgado, J.; et al. EMA Review of Belantamab Mafodotin (Blenrep) for the Treatment of Adult Patients with Relapsed/Refractory Multiple Myeloma. Oncologist 2021, 26, 70–76. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Demichelis-Gómez, R.; Pérez-Sámano, D.; Bourlon, C. Bispecific Antibodies in Hematologic Malignancies: When, to Whom, and How Should Be Best Used? Curr. Oncol. Rep. 2019, 21, 17. [Google Scholar] [CrossRef]
- Einsele, H.; Rasche, L.; Topp, M.S.; Martin Kortüm, K.; Duell, J. The use of bispecific antibodies to optimize the outcome of patients with acute leukemia, lymphoma and multiple myeloma after SCT. Bone Marrow Transplant. 2019, 54, 721–726. [Google Scholar] [CrossRef]
- Cho, S.-F.; Xing, L.; Anderson, K.C.; Tai, Y.-T. Promising Antigens for the New Frontier of Targeted Immunotherapy in Multiple Myeloma. Cancers 2021, 13, 6136. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desantis, V.; Savino, F.D.; Scaringella, A.; Potenza, M.A.; Nacci, C.; Frassanito, M.A.; Vacca, A.; Montagnani, M. The Leading Role of the Immune Microenvironment in Multiple Myeloma: A New Target with a Great Prognostic and Clinical Value. J. Clin. Med. 2022, 11, 2513. https://doi.org/10.3390/jcm11092513
Desantis V, Savino FD, Scaringella A, Potenza MA, Nacci C, Frassanito MA, Vacca A, Montagnani M. The Leading Role of the Immune Microenvironment in Multiple Myeloma: A New Target with a Great Prognostic and Clinical Value. Journal of Clinical Medicine. 2022; 11(9):2513. https://doi.org/10.3390/jcm11092513
Chicago/Turabian StyleDesantis, Vanessa, Francesco Domenico Savino, Antonietta Scaringella, Maria Assunta Potenza, Carmela Nacci, Maria Antonia Frassanito, Angelo Vacca, and Monica Montagnani. 2022. "The Leading Role of the Immune Microenvironment in Multiple Myeloma: A New Target with a Great Prognostic and Clinical Value" Journal of Clinical Medicine 11, no. 9: 2513. https://doi.org/10.3390/jcm11092513
APA StyleDesantis, V., Savino, F. D., Scaringella, A., Potenza, M. A., Nacci, C., Frassanito, M. A., Vacca, A., & Montagnani, M. (2022). The Leading Role of the Immune Microenvironment in Multiple Myeloma: A New Target with a Great Prognostic and Clinical Value. Journal of Clinical Medicine, 11(9), 2513. https://doi.org/10.3390/jcm11092513