


Global Incidence and Prevalence of Gaucher Disease: A Targeted Literature Review

Abstract
:1. Introduction
2. Methods
2.1. Literature Searches
2.2. Study Selection
3. Results
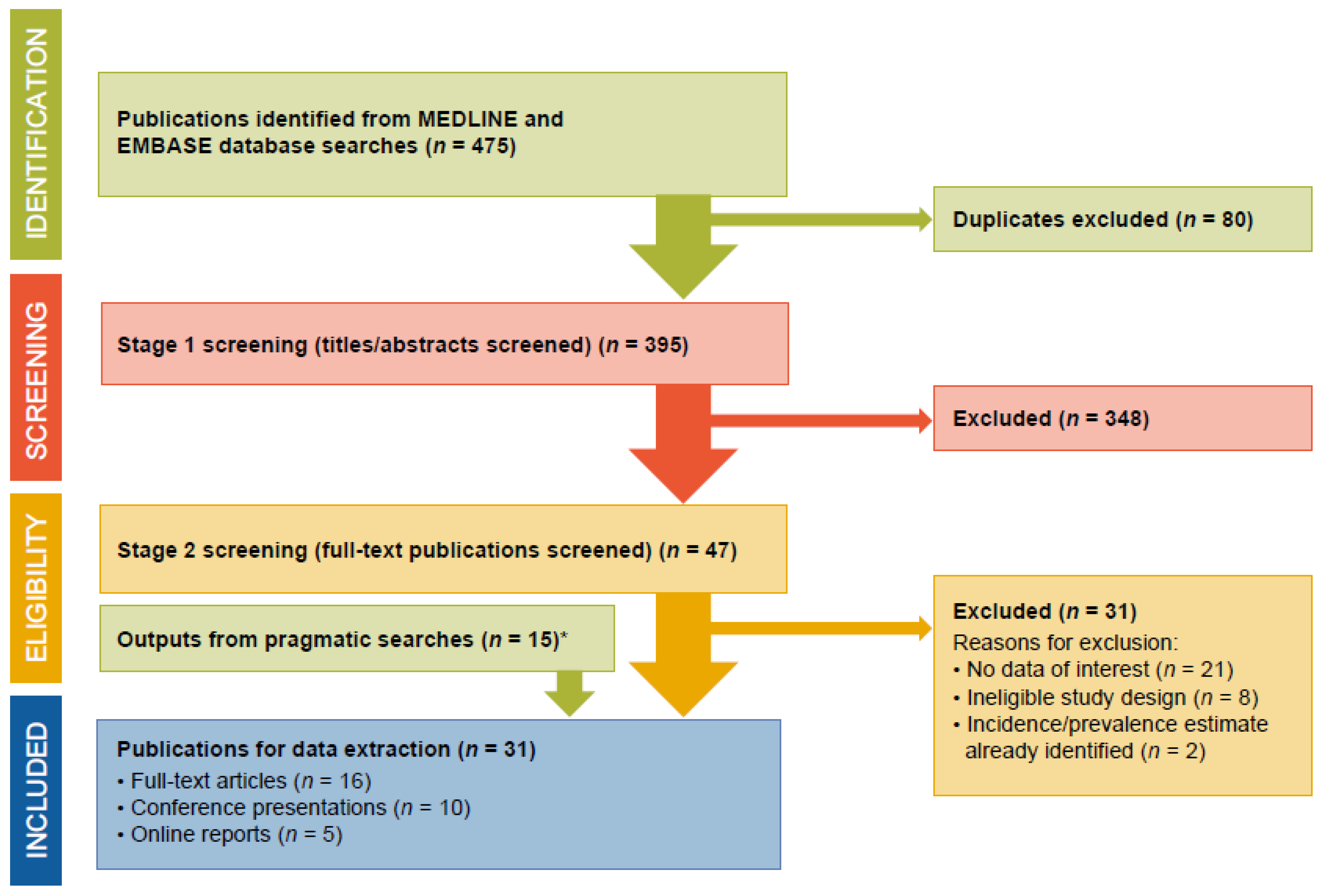
3.1. Search Outputs
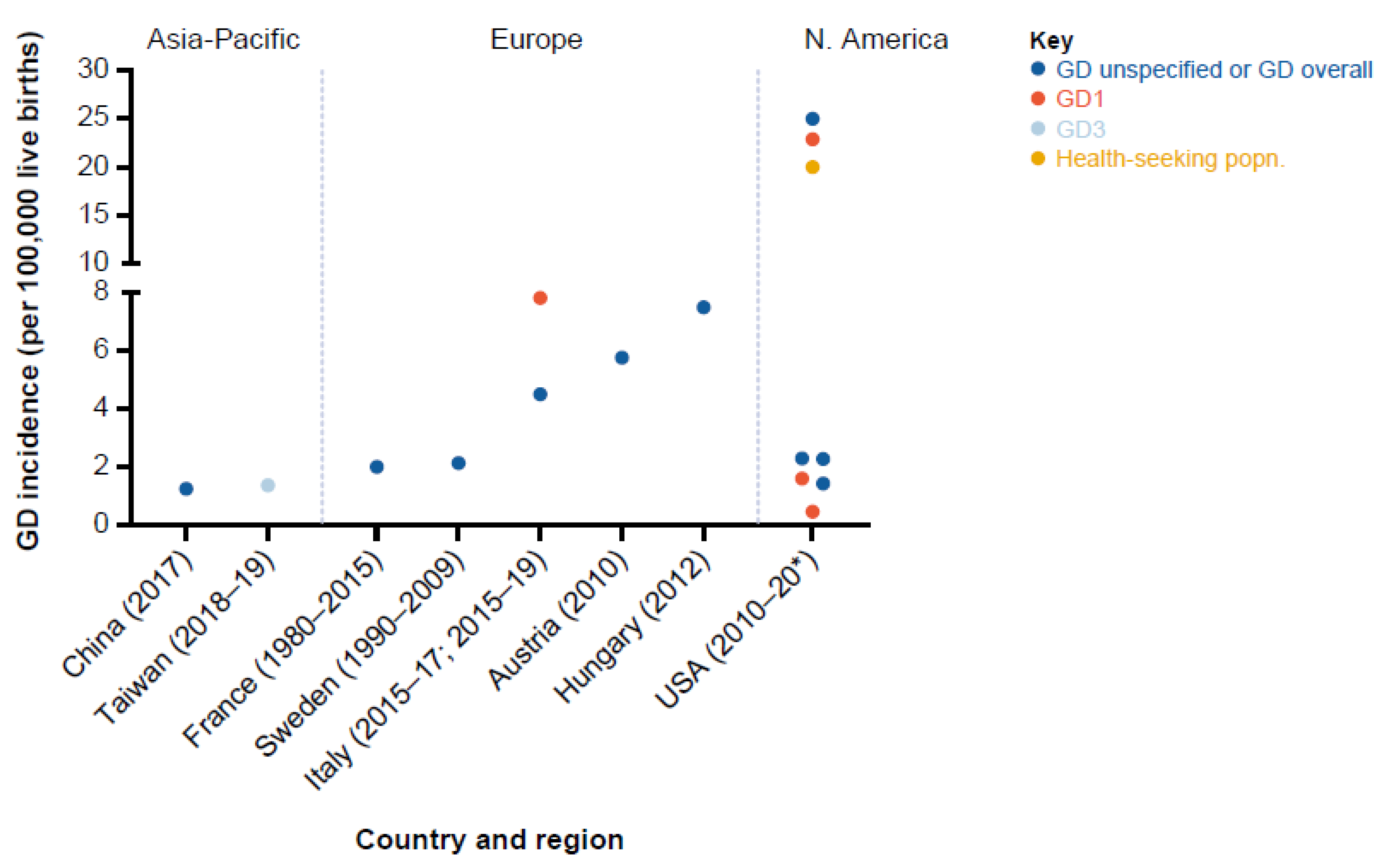
3.2. GD Incidence
3.2.1. Europe
3.2.2. North America
3.2.3. Asia-Pacific
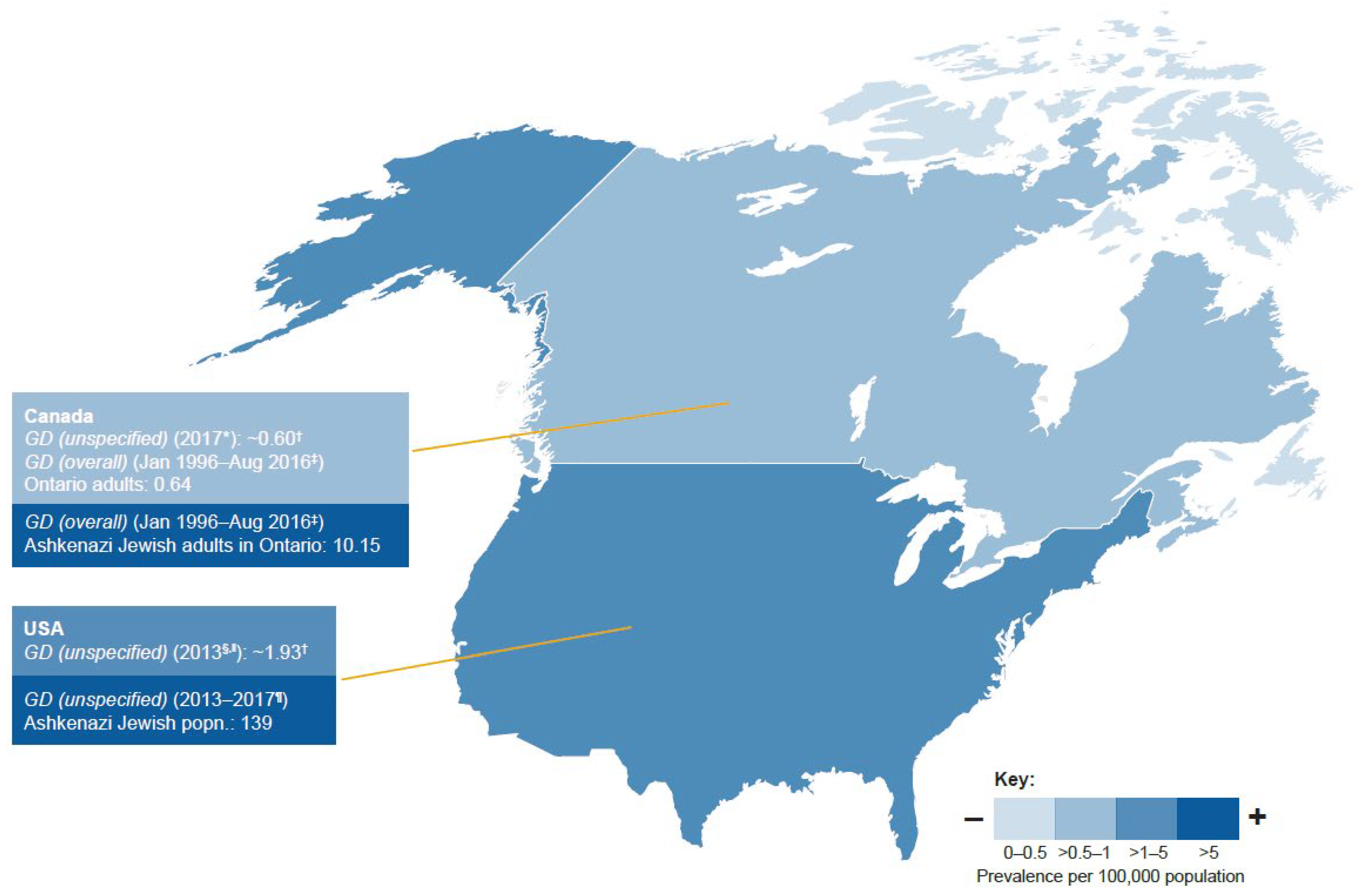
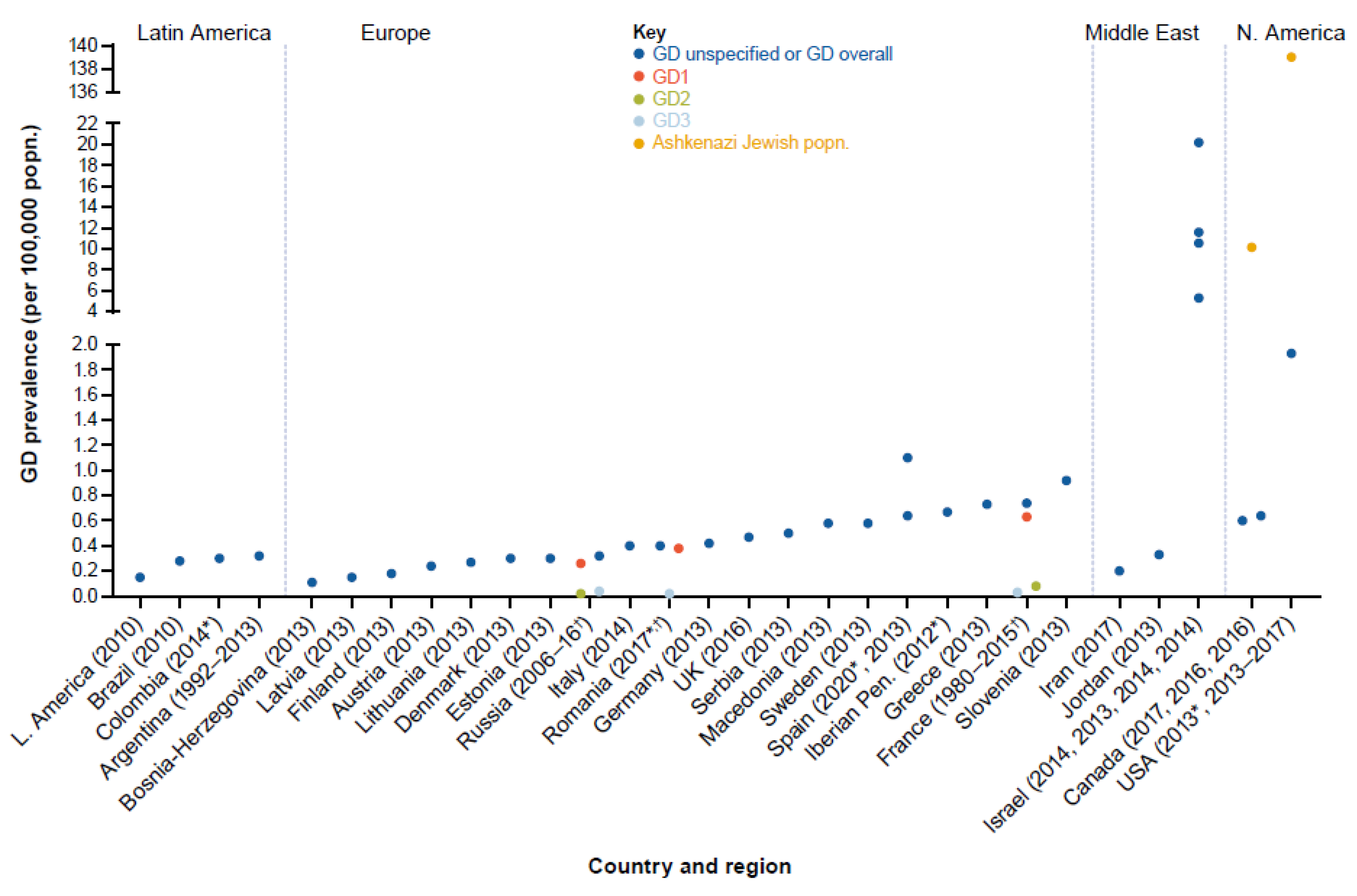
3.3. GD Prevalence
3.3.1. Europe
3.3.2. North America
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Li, F.; Zhang, J.; Lu, C.; Kong, W. Global Epidemiology of Gaucher Disease: An Updated Systematic Review and Meta-analysis. J. Pediatr. Hematol. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E. Gaucher disease: Insights from a rare Mendelian disorder. Discov. Med. 2012, 14, 273–281. [Google Scholar]
- Grabowski, G.A.; Zimran, A.; Ida, H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am. J. Hematol. 2015, 90 (Suppl. 1), S12–S18. [Google Scholar] [CrossRef]
- Rosenbloom, B.E.; Weinreb, N.J. Gaucher disease: A comprehensive review. Crit. Rev. Oncog. 2013, 18, 163–175. [Google Scholar] [CrossRef]
- Mehta, A.; Belmatoug, N.; Bembi, B.; Deegan, P.; Elstein, D.; Göker-Alpan, Ö.; Lukina, E.; Mengel, E.; Nakamura, K.; Pastores, G.M.; et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians. Mol. Genet. Metab. 2017, 122, 122–129. [Google Scholar] [CrossRef]
- Mistry, P.K.; Cappellini, M.D.; Lukina, E.; Ozsan, H.; Mach Pascual, S.; Rosenbaum, H.; Helena Solano, M.; Spigelman, Z.; Villarrubia, J.; Watman, N.P.; et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am. J. Hematol. 2011, 86, 110–115. [Google Scholar] [CrossRef]
- Mistry, P.K.; Sadan, S.; Yang, R.; Yee, J.; Yang, M. Consequences of diagnostic delays in type 1 Gaucher disease: The need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am. J. Hematol. 2007, 82, 697–701. [Google Scholar] [CrossRef]
- Gonzalez, D.E.; Turkia, H.B.; Lukina, E.A.; Kisinovsky, I.; Dridi, M.F.; Elstein, D.; Zahrieh, D.; Crombez, E.; Bhirangi, K.; Barton, N.W.; et al. Enzyme replacement therapy with velaglucerase alfa in Gaucher disease: Results from a randomized, double-blind, multinational, Phase 3 study. Am. J. Hematol. 2013, 88, 166–171. [Google Scholar] [CrossRef]
- Hughes, D.A.; Gonzalez, D.E.; Lukina, E.A.; Mehta, A.; Kabra, M.; Elstein, D.; Kisinovsky, I.; Giraldo, P.; Bavdekar, A.; Hangartner, T.N.; et al. Velaglucerase alfa (VPRIV) enzyme replacement therapy in patients with Gaucher disease: Long-term data from phase III clinical trials. Am. J. Hematol. 2015, 90, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Mistry, P.K.; Deegan, P.; Vellodi, A.; Cole, J.A.; Yeh, M.; Weinreb, N.J. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: Effect on incidence of avascular necrosis. Br. J. Haematol. 2009, 147, 561–570. [Google Scholar] [CrossRef]
- Grabowski, G.A. Gaucher disease: Gene frequencies and genotype/phenotype correlations. Genet. Test 1997, 1, 5–12. [Google Scholar] [CrossRef]
- National Organization for Rare Disorders (NORD). Rare Disease Database. Available online: https://rarediseases.org/rare-diseases/gaucher-disease/ (accessed on 13 May 2022).
- Reynolds, T.M.; Wierzbicki, A.S.; Skrahina, V.; Beetz, C.; PATHFINDER Project Collaboration group. Screening for patients with Gaucher’s disease using routine pathology results: PATHFINDER (ferritin, alkaline phosphatase, platelets) study. Int. J. Clin. Pract. 2021, 75, e14422. [Google Scholar] [CrossRef]
- Silva García, R.; de Frutos, L.L.; Arreguin, E.; González, C.C.; Ortiz, J.E.G.; Ornelas, S.F.; Castellano, P.G.; Favela, F.B. Gaucher Disease: Identification and Novel Variants in Mexican and Spanish Patients. Arch. Med. Res. 2021, 52, 731–737. [Google Scholar] [CrossRef]
- Waggoner, D.J.; Tan, C.A. Expanding newborn screening for lysosomal disorders: Opportunities and challenges. Dev. Disabil. Res. Rev. 2011, 17, 9–14. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Orsini, J.J.; Goldenberg, A.; Caggana, M.; Levy, P.A.; Breilyn, M.; Gelb, M.H. The future of newborn screening for lysosomal disorders. Neurosci. Lett. 2021, 760, 136080. [Google Scholar] [CrossRef]
- Matern, D.; Oglesbee, D.; Tortorelli, S. Newborn screening for lysosomal storage disorders and other neuronopathic conditions. Dev. Disabil. Res. Rev. 2013, 17, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Institute of Medicine. Standards for Systematic Review. Available online: https://www.nationalacademies.org/publications (accessed on 25 September 2021).
- Žnidar, I.; Collin-Histed, T.; Niemeyer, P.; Parkkinen, J.; Lauridsen, A.G.; Zariņa, S.; Cohen, Y.; Manuel, J. The European Gaucher Alliance: A survey of member patient organisations’ activities, healthcare environments and concerns. Orphanet. J. Rare Dis. 2014, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.; Charrow, J.; Angle, B.; Widera, S.; Waggoner, D. A Pilot Newborn Screening Program for Lysosomal Storage Disorders (LSD) in Illinois. Mol. Genet. Metab. 2012, 105, S23–S24. [Google Scholar] [CrossRef]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn Screening for Lysosomal Storage Disorders in Illinois: The Initial 15-Month Experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Chen, P.W.; Yeh, H.Y.; Gelb, M.H.; Chiu, P.C.; Chu, S.Y.; Lee, C.H.; Lee, A.R.; Hwu, W.L. Newborn screening for Morquio disease and other lysosomal storage diseases: Results from the 8-plex assay for 70,000 newborns. Orphanet. J. Rare Dis. 2020, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.V.; Klug, T.; Rogers, S.V.; Kiesling, J. State-wide newborn screening for four lysosomal diseases reveals high incidence rate for Pompe and Fabry diseases. Mol. Genet. Metab. 2017, 120, S66. [Google Scholar] [CrossRef]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef]
- Kang, L.; Zhan, X.; Gu, X.; Zhang, H. Successful newborn screening for Gaucher disease using fluorometric assay in China. J. Hum. Genet. 2017, 62, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Limgala, R.P.; Furtak, V.; Ivanova, M.M.; Fidelia-Lambert, M.N.; Gondré-Lewis, M.; Goker-Alpan, O. Selective screening for lysosomal disorders in a large cohort of minority groups shows higher incidence rates and novel variants. Mol. Genet. Metab. 2020, 129, S98. [Google Scholar] [CrossRef]
- Mechtler, T.P.; Stary, S.; Metz, T.F.; De Jesús, V.R.; Greber-Platzer, S.; Pollak, A.; Herkner, K.R.; Streubel, B.; Kasper, D.C. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet 2012, 379, 335–341. [Google Scholar] [CrossRef]
- Polo, G.; Gueraldi, D.; Rubert, L.; Cazzorla, C.; Giuliani, A.; Burlina, A. High incidence of Gaucher disease in northeast Italy: Results from lysosomal newborn screening. Mol. Genet. Metab. 2020, 129, S36. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet. Med. 2019, 21, 631–640. [Google Scholar] [CrossRef]
- Wittmann, J.; Karg, E.; Turi, S.; Legnini, E.; Wittmann, G.; Giese, A.K.; Lukas, J.; Gölnitz, U.; Klingenhäger, M.; Bodamer, O.; et al. Newborn screening for lysosomal storage disorders in Hungary. JIMD Rep. 2012, 6, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Burton, B.K.; Hoganson, G.E.; Charrow, J.; Tinkle, B.; Dimmock, D.; Waggoner, D.; Grange, D.; Nash, C.; Becker, J.; Shao, R.; et al. Newborn screening for lysosomal disorders in Illinois. Mol. Genet. Metab. 2016, 117, S31–S32. [Google Scholar] [CrossRef]
- Hult, M.; Darin, N.; von Döbeln, U.; Månsson, J.E. Epidemiology of lysosomal storage diseases in Sweden. Acta Paediatr. 2014, 103, 1258–1263. [Google Scholar] [CrossRef]
- Stirnemann, J.; Vigan, M.; Hamroun, D.; Heraoui, D.; Rossi-Semerano, L.; Berger, M.G.; Rose, C.; Camou, F.; de Roux-Serratrice, C.; Grosbois, B.; et al. The French Gaucher Disease Registry: Clinical characteristics, complications and treatment of 616 patients. Mol. Genet. Metab. 2016, 117, S25–S26. [Google Scholar] [CrossRef]
- Sociedad Española de Hematología y Hemoterapia (SEHH). El Retraso en el Diagnóstico Sigue Siendo un Reto Para el Abordaje de la Enfermedad de Gaucher. Available online: https://www.sehh.es/images/stories/recursos/2020/09/30/NdP_Di%CC%81a_Mundial_de_la_Enfermedad_de_Gaucher_2020.pdf. (accessed on 5 May 2021).
- Drelichman, G.; Fernández Escobar, N.; Basack, N.; Kohan, R.; Watman, N.; Bolesina, M.; Elena, G.; Veber, S.E.; Dragosky, M.; Annetta, I.; et al. Enfermedad de Gaucher en Argentina: Un informe del Registro Internacional de Gaucher y del Grupo Argentino de Diagnóstico y Tratamiento de la Enfermedad de Gaucher. Hematología 2013, 17, 4–16. [Google Scholar]
- Grinzaid, K.A. Impact of education and the facilitation of carrier screening in a population at increased risk for lysosomal diseases. Mol. Genet. Metab. 2017, 120, S58–S59. [Google Scholar] [CrossRef]
- Drelichman, G.; Linares, A.; Vilalobos, J.; Cabello, J.F.; Kerstenetzky, M.; Kohan, R.M.; Martins, A.M. Enfermedad de Gaucher en Latinoamerica. Un informe del Registro Internacional y del Grupo Latinoamericano para la Enfermedad de Gaucher. Medicina 2012, 72, 273–282. [Google Scholar]
- Jaffe, D.H.; Flaks-Manov, N.; Benis, A.; Gabay, H.; DiBonaventura, M.; Rosenbaum, H.; Joseph, A.; Bachrach, A.; Leventer-Roberts, M. Population-based cohort of 500 patients with Gaucher disease in Israel. BMJ Open 2019, 9, e024251. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Wasim, S.; Amato, D. Gaucher Disease in Ontario, Canada: Clinical Manifestations, Natural Progression, and Treatment Response. J. Rare Dis. Res. Treat. 2018, 3, 7–16. [Google Scholar] [CrossRef]
- Bucerzan, S.; AlKhzouz, C.; Nascu, I.; Zimmerman, A.; Popp, R.; Lazea, C.; Grigorescu-Sido, P. OC-87 Gaucher disease in Romania–baseline characteristics, specific diagnosis, treatment and outcome. Arch. Dis. Child. 2017, 102, A34–A35. [Google Scholar] [CrossRef]
- Davari, M.; Nabizadeh, A.; Kadivar, M.; Asl, A.A.; Sarkheil, P. Healthcare resource utilization and cost of care for Gaucher patients in Iran. J. Diabetes Metab. Disord. 2019, 18, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, P.; Alfonso, P.; Irún, P.; Gort, L.; Chabás, A.; Vilageliu, L.; Grinberg, D.; CM, S.M.; Pocovi, M. Mapping the genetic and clinical characteristics of Gaucher disease in the Iberian Peninsula. Orphanet. J. Rare Dis. 2012, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Movsisyan, G.B.; Gundobina, O.S.; Namazova-Baranova, L.S.; Savostyanov, K.V.; Pushkov, A.A.; Chernikov, V.V.; Mazanova, N.N.; Romanyuk, A.M.; Smirnov, V.I. P234 Demographic, clinical and genetic characteristics of children with Gaucher disease: The data of paediatric registry in Russia. Arch. Dis. Child. 2017, 102, A125. [Google Scholar] [CrossRef]
- ACOPEL Asociación Colombiana de Pacientes con Enfermedad Lisosomal. Validez de la Prueba de Actividad Enzimática de la Glucocerebrosidasa Para el Diagnóstico de Enfermedad de Gaucher. 2014. Available online: https://docs.bvsalud.org/biblioref/2017/11/875828/validez-diagnostica-gaucher.pdf (accessed on 1 May 2021).
- Gauchers Association. GPnotebook. Gaucher’s Disease (GD). Available online: https://gpnotebook.com/simplepage.cfm?ID=-2147090425 (accessed on 4 May 2021).
- The National Organization for Rare Disorders. The Physician’s Guide to Gaucher Disease. Available online: http://www.filiere-g2m.fr/fileadmin/user_upload/webmasterfichiers/PNDS___RECO/NORD_Physician_Guides_Gaucher.pdf (accessed on 3 May 2021).
- L’Institut National D’excellence en Santé et en Services Sociaux (INESSS). Avis de Refus D’inscription Aux Listes des Médicaments–Valeur Thérapeutique. Available online: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Fevrier_2018/Cerdelga_2018_02.pdf (accessed on 3 May 2021).
- Jaffe, D.H.; Flaks-Manov, N.; Benis, A.; Gabay, H.; DiBonaventura, M.; Rosenbaum, H.; Joseph, A.; Leventer-Roberts, M. A Population-Based Cohort of Gaucher Disease Patients Identified Using EHR Data. Value Health 2016, 19, PA578. [Google Scholar] [CrossRef]
- Peake, R.W.A. Newborn Screening for Lysosomal Storage Diseases. In Lysosomal Storage Disorders: A Practical Guide, 2nd ed.; Mehta, A.B., Winchester, B., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2022; pp. 51–58. [Google Scholar]
- Moat, S.J.; George, R.S.; Carling, R.S. Use of Dried Blood Spot Specimens to Monitor Patients with Inherited Metabolic Disorders. Int. J. Neonatal. Screen 2020, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Sawada, T.; Kido, J.; Sugawara, K.; Yoshida, S.; Matsumoto, S.; Shimazu, T.; Matsushita, Y.; Inoue, T.; Hirose, S.; Endo, F.; et al. Newborn screening for Gaucher disease in Japan. Mol. Genet. Metab. Rep. 2022, 31, 100850. [Google Scholar] [CrossRef]
- Dinur, T.; Bauer, P.; Beetz, C.; Kramp, G.; Cozma, C.; Iurașcu, M.I.; Becker-Cohen, M.; Istaiti, M.; Rolfs, A.; Zimran, A.; et al. Gaucher Disease Diagnosis Using Lyso-Gb1 on Dry Blood Spot Samples: Time to Change the Paradigm? Int. J. Mol. Sci. 2022, 23, 1627. [Google Scholar] [CrossRef]
- Phetthong, T.; Tim-Aroon, T.; Khongkraparn, A.; Noojarern, S.; Kuptanon, C.; Wichajarn, K.; Sathienkijkanchai, A.; Suphapeetiporn, K.; Charoenkwan, P.; Tantiworawit, A.; et al. Gaucher disease: Clinical phenotypes and refining GBA mutational spectrum in Thai patients. Orphanet. J. Rare Dis. 2021, 16, 519. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [Green Version]
- Kadali, S.; Kolusu, A.; Gummadi, M.R.; Undamatla, J. The relative frequency of lysosomal storage disorders: A medical genetics referral laboratory’s experience from India. J. Child Neurol. 2014, 29, 1377–1382. [Google Scholar] [CrossRef]
- Ramdin, T.; Schapkaitz, E.; Varughese, S.; Sevitz, H. Gaucher disease: A cause of massive splenomegaly in a 15-year-old black African male. S. Afr. Med. J. 2022, 112, 13515. [Google Scholar] [CrossRef]
- Sevittz, H.; Laher, F.; Varughese, S.T.; Nel, M.; McMaster, A.; Jacobson, B.F. Baseline characteristics of 32 patients with Gaucher disease who were treated with imiglucerase: South African data from the International Collaborative Gaucher Group (ICGG) Gaucher Registry. S. Afr. Med. J. 2022, 112, 13518. [Google Scholar] [CrossRef]
- Kannauje, P.K.; Pandit, V.; Wasnik, P.N.; Gupta, A.K.; Venkatesan, N. Gaucher’s Disease in an Adult Female: A Rare Entity. Cureus 2021, 13, e17318. [Google Scholar] [CrossRef]
- Millington, D.; Norton, S.; Singh, R.; Sista, R.; Srinivasan, V.; Pamula, V. Digital microfluidics comes of age: High-throughput screening to bedside diagnostic testing for genetic disorders in newborns. Expert Rev. Mol. Diagn. 2018, 18, 701–712. [Google Scholar] [CrossRef]
- Verma, I.C.; El-Beshlawy, A.; Tylki-Szymańska, A.; Martins, A.; Duan, Y.L.; Collin-Histed, T.; van der Linde, M.S.; Mansour, R.; Dũng, V.C.; Mistry, P.K. Transformative effect of a Humanitarian Program for individuals affected by rare diseases: Building support systems and creating local expertise. Orphanet. J. Rare Dis. 2022, 17, 87. [Google Scholar] [CrossRef]
- Zimran, A.; Belmatoug, N.; Bembi, B.; Deegan, P.; Elstein, D.; Fernandez-Sasso, D.; Giraldo, P.; Goker-Alpan, O.; Lau, H.; Lukina, E.; et al. Demographics and patient characteristics of 1209 patients with Gaucher disease: Descriptive analysis from the Gaucher Outcome Survey (GOS). Am. J. Hematol. 2018, 93, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Savolainen, M.J.; Karlsson, A.; Rohkimainen, S.; Toppila, I.; Lassenius, M.I.; Falconi, C.V.; Uusi-Rauva, K.; Elomaa, K. The Gaucher earlier diagnosis consensus point-scoring system (GED-C PSS): Evaluation of a prototype in Finnish Gaucher disease patients and feasibility of screening retrospective electronic health record data for the recognition of potential undiagnosed patients in Finland. Mol. Genet. Metab. Rep. 2021, 27, 100725. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. Stage 1 Screening: Study Inclusion and Exclusion Criteria | |
|---|---|
| Inclusion Criteria | Exclusion Criteria |
| Studies conducted in humans | Case reports, letters to editors, editorials, opinions |
| Observational studies (e.g., cross-sectional, cohort, case–control, registries, case series *) | Literature reviews (systematic and non-systematic) and meta-analyses (used as reference source only) |
| Studies that included patients with GD (either as the study population or as a subgroup analysis) | Clinical trials, non-clinical or experimental (preclinical) studies |
| Studies that reported incidence and/or prevalence estimates | Studies reporting preliminary results (if later published as full text) |
| Studies published as full text, conference proceedings, or abstracts | |
| Studies published between 1 January 2011 and 6 May 2021 (date last searched) † | |
| Search was in English but outputs in French, Spanish, German, or Italian only were also considered when necessary | |
| B. Stage 2 Screening Criteria. | |
| Criteria (based on PICOTS) | Details |
| Population | Patients with GD of any type |
| Intervention | Receiving standard of care (including substrate replacement therapy and enzyme replacement therapy) |
| Outcomes | GD incidence and prevalence outcomes or data from which these could be derived |
| Time period | Published within the past 10 years: 1 January 2011 to 6 May 2021 |
| Setting | Real-world/observational studies |
| Assessment Criteria | Description |
|---|---|
| GD type | The availability of estimates for: GD overall (any GD or combining estimates where GD type was specified) GD unspecified (absence of any information on whether study targeted GD overall or a given GD type) GD type-specific (GD type 1–3 specific) |
| Number of regions collectively covered across studies | The availability of estimates for each region (North America, Europe, Asia-Pacific, Latin America, Middle East, and Africa) was determined |
| Countries covered | Listed countries were based on an assessment of the number and population size of countries per region: Asia-Pacific: If either China or India were covered, then the generalizability was considered as adequate Europe: If available studies collectively covered at least 4 countries among France, Germany, Italy, Spain, UK, then the findings were considered to have adequate generalizability. If only 2 or 3 of these countries were covered then generalizability was considered intermediate, and 0–1 was considered poor generalizability North America (includes the USA and Canada) *: If estimates were only available for either the USA or Canada, generalizability was deemed intermediate, otherwise, if both were covered both, then it was considered adequate Latin America: If available studies collectively covered at least 3 countries among Argentina, Brazil, Colombia, and Mexico, then the findings were considered to have adequate generalizability. If only 2 of the above-listed countries were covered, generalizability was considered intermediate, and 0–1 was considered poor Middle East: Generalizability was considered adequate if at least 3 countries among Egypt, Iran, Jordan, or Turkey were included, intermediate if 2 out of the 4 listed countries were included, and poor for 0 or 1 out of 4 countries Africa: Generalizability was considered adequate if at least 3 of the following countries were covered: Algeria or Morocco; South Africa; or any country from sub-Saharan Africa. If only some of those countries were covered then the generalizability was considered intermediate (2 out of 3 countries) or poor (0 or 1 out of 3) |
| Size of study population | Within a country or region, the size of the studies (collective or individual) was also considered. For guidance purposes, studies with a sample size >200 patients with GD were considered arbitrarily to be large |
| Study Design | Study Period | Study Duration, Months | Study Population Size | Reference Population | Incidence Rate (Confirmed Cases/Screened Pts) | |
|---|---|---|---|---|---|---|
| France | ||||||
| Stirnemann et al. 2016 [37] | Retrospective cohort | 1980–2015 | - | 616 | Live births in corresponding years | GD (overall) 2.0/100,000 live births |
| Sweden | ||||||
| Hult et al. 2014 [36] | Retrospective cohort | 1990–2009 | 360 | 44/2,080,791 | Live births in corresponding years | GD (unspecified) 2.13/100,000 live births |
| Italy | ||||||
| Burlina et al. 2018 [23] | Prospective cohort | Sep 2015–Jan 2017 | 17 | 2/44,411 | Population-based newborn screening program in North-East Italy | GD1: 4.50/100,000 live births |
| Polo et al. 2020 [32] | Prospective cohort | Sep 2015–Aug 2019 | - | 2/127,869 | Population-based newborn screening program | GD (unspecified): 7.82/100,000 live births |
| Austria | ||||||
| Mechtler et al. 2012 [31] | Prospective cohort | Jan 2010–Jul 2010 | 7 | 2/34,736 | Population-based newborn screening program | GD (unspecified): 5.76/100,000 live births |
| Hungary | ||||||
| Wittmann et al. 2012 [34] | Prospective cohort | 2012 * | - | 3/40,024 | Population-based newborn screening program | GD (unspecified): 7.5/100,000 live births |
| Spain | ||||||
| SEHH 2020 [38] | Newsletter from the Spanish registry of GD | 2020 * | - | NA | Population of Spain in 2019 | GD (unspecified): 8–10 new cases/year |
| USA | Study Design | Study Period | Study Duration, Months | Study Population Size | Reference Population | Incidence Rate (Confirmed Cases/Screened Pts) |
|---|---|---|---|---|---|---|
| Hopkins et al. 2017 [27] | Prospective cohort | 2017 * | - | 4/282,500 | Missouri pilot newborn screening program | GD (unspecified) 1.42/100,000 live births |
| Burton et al. 2016 [35] | Prospective cohort | 2016 * | - | 1/63,007 | Illinois newborn screening program | GD1: 1.59/100,000 live births |
| Hopkins et al. 2015 [28] | Prospective cohort | Jan 2013–Jun 2013 | 6 | 1/43,701 | Missouri newborn screening program | GD (unspecified) 2.29/100,000 live births |
| Burton et al. 2017 [25] | Prospective cohort | Nov 2014–Aug 2016 | - | 5/219,793 | Illinois Department of Public Health in Chicago newborn screening program | GD (unspecified): 2.27/100,000 live births GD1: 0.45/100,000 live births (1/219,793) |
| Wasserstein et al. 2019 [33] | Prospective cohort | May 2013–Apr 2017 | 48 | 15/65,605 | New York pilot newborn screening program | GD1: 22.9/100,000 live births † |
| Burton et al. 2012 [24] | Prospective cohort | Nov 2010–Apr 2011 | 6 | 2/8012 | Illinois pilot newborn screening program | GD (unspecified): 25.0/100,000 live births |
| Limgala et al. 2020 [30] | Prospective cohort | 2020 * | - | 1/5000 | Patients (all ages) seeking healthcare for various health concerns: 85% African American 10% Hispanic 5% Caucasian/other | GD (unspecified): 20.0/100,000 healthcare-seeking patients |
| Study Design | Study Period | Study Duration, Months | Study Population Size | Reference Population | Incidence Rate (Confirmed Cases/Screened Pts) | |
|---|---|---|---|---|---|---|
| China | ||||||
| Kang et al. 2017 [29] | Prospective cohort | 2017 * | 12 | 1/80,855 | Newborns participating in the Neonatal Screening Center of Shanghai | GD (unspecified) 1.24/100,000 live births |
| Taiwan | ||||||
| Chien et al. 2020 [26] | Prospective cohort | Mar 2018–Apr 2019 | 12 | 1/73,743 | 35% of newborns in Taiwan | GD3: 1.36/100,000 live births |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillon, G.; Chang, S.-C.; Moride, Y. Global Incidence and Prevalence of Gaucher Disease: A Targeted Literature Review. J. Clin. Med. 2023, 12, 85. https://doi.org/10.3390/jcm12010085
Castillon G, Chang S-C, Moride Y. Global Incidence and Prevalence of Gaucher Disease: A Targeted Literature Review. Journal of Clinical Medicine. 2023; 12(1):85. https://doi.org/10.3390/jcm12010085
Chicago/Turabian StyleCastillon, Genaro, Shun-Chiao Chang, and Yola Moride. 2023. "Global Incidence and Prevalence of Gaucher Disease: A Targeted Literature Review" Journal of Clinical Medicine 12, no. 1: 85. https://doi.org/10.3390/jcm12010085
APA StyleCastillon, G., Chang, S. -C., & Moride, Y. (2023). Global Incidence and Prevalence of Gaucher Disease: A Targeted Literature Review. Journal of Clinical Medicine, 12(1), 85. https://doi.org/10.3390/jcm12010085