
Role of the Renin Angiotensin Aldosterone System in the Pathogenesis of Sepsis-Induced Acute Kidney Injury: A Systematic Review


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Protocol Design and Registration
2.2. Eligibility Criteria
2.3. Study Selection
2.4. Quality Assessment
3. Results
3.1. Study Characteristics
3.2. Sepsis-Induced Acute Kidney Injury
3.2.1. Endothelial Cell Damage
3.2.2. Inflammatory and Oxidative Stress
3.2.3. Coagulative Dysfunction
3.3. Renin Angiotensin Aldosterone System
3.4. Role of the RAAS in S-AKI
3.4.1. Involvement of Angiotensin II
3.4.2. Use of RAAS Inhibitors
3.4.3. Impact of RAAS Inhibitors on Risk for S-AKI
3.4.4. Impact of RAAS Inhibitors on Outcomes of S-AKI
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- van der Poll, T.; van de Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Shibata, J.; Osawa, I.; Ito, H.; Soeno, S.; Hara, K.; Sonoo, T.; Nakamura, K.; Goto, T. Risk factors of sepsis among patients with qSOFA<2 in the emergency department. Am. J. Emerg. Med. 2021, 50, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Gerlach, H.; Vogelmann, T.; Preissing, F.; Stiefel, J.; Adam, D. Mortality in sepsis and septic shock in Europe, North America and Australia between 2009 and 2019- results from a systematic review and meta-analysis. Crit. Care 2020, 24, 239. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Suh, S.H.; Kim, C.S.; Choi, J.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. Acute kidney injury in patients with sepsis and septic shock: Risk factors and clinical outcomes. Yonsei Med. J. 2013, 54, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.C.; Hsu, C.W. Septic acute kidney injury patients in emergency department: The risk factors and its correlation to serum lactate. Am. J. Emerg. Med. 2019, 37, 204–208. [Google Scholar] [CrossRef]
- Thakar, C.V.; Christianson, A.; Freyberg, R.; Almenoff, P.; Render, M.L. Incidence and outcomes of acute kidney injury in intensive care units: A Veterans Administration study. Crit. Care Med. 2009, 37, 2552–2558. [Google Scholar] [CrossRef]
- Bagshaw, S.M.; Uchino, S.; Bellomo, R.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; Gibney, N.; et al. Septic acute kidney injury in critically ill patients: Clinical characteristics and outcomes. Clin. J. Am. Soc. Nephrol. 2007, 2, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Hoste, E.A.; Lameire, N.H.; Vanholder, R.C.; Benoit, D.D.; Decruyenaere, J.M.; Colardyn, F.A. Acute renal failure in patients with sepsis in a surgical ICU: Predictive factors, incidence, comorbidity, and outcome. J. Am. Soc. Nephrol. 2003, 14, 1022–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugan, R.; Karajala-Subramanyam, V.; Lee, M.; Yende, S.; Kong, L.; Carter, M.; Angus, D.C.; Kellum, J.A.; Genetic and Inflammatory Markers of Sepsis (GenIMS) Investigators. Acute kidney injury in non-severe pneumonia is associated with an increased immune response and lower survival. Kidney Int. 2010, 77, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Poukkanen, M.; Vaara, S.T.; Pettila, V.; Kaukonen, K.M.; Korhonen, A.M.; Hovilehto, S.; Inkinen, O.; Laru-Sompa, R.; Kaminski, T.; Reinikainen, M.; et al. Acute kidney injury in patients with severe sepsis in Finnish Intensive Care Units. Acta Anaesthesiol. Scand. 2013, 57, 863–872. [Google Scholar] [CrossRef]
- Bu, X.; Zhang, L.; Chen, P.; Wu, X. Relation of neutrophil-to-lymphocyte ratio to acute kidney injury in patients with sepsis and septic shock: A retrospective study. Int. Immunopharmacol. 2019, 70, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Munn, Z.; Barker, T.H.; Moola, S.; Tufanaru, C.; Stern, C.; McArthur, A.; Stephenson, M.; Aromataris, E. Methodological quality of case series studies: An introduction to the JBI critical appraisal tool. JBI Evid. Synth. 2020, 18, 2127–2133. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Rovers, M.M.; de Vries, R.B.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 43. [Google Scholar] [CrossRef] [Green Version]
- Prowle, J.R.; Molan, M.P.; Hornsey, E.; Bellomo, R. Measurement of renal blood flow by phase-contrast magnetic resonance imaging during septic acute kidney injury: A pilot investigation. Crit. Care Med. 2012, 40, 1768–1776. [Google Scholar] [CrossRef]
- Skytte Larsson, J.; Krumbholz, V.; Enskog, A.; Bragadottir, G.; Redfors, B.; Ricksten, S.E. Renal Blood Flow, Glomerular Filtration Rate, and Renal Oxygenation in Early Clinical Septic Shock. Crit. Care Med. 2018, 46, e560–e566. [Google Scholar] [CrossRef] [PubMed]
- Prowle, J.R.; Ishikawa, K.; May, C.N.; Bellomo, R. Renal blood flow during acute renal failure in man. Blood Purif. 2009, 28, 216–225. [Google Scholar] [CrossRef]
- Maiden, M.J.; Otto, S.; Brealey, J.K.; Finnis, M.E.; Chapman, M.J.; Kuchel, T.R.; Nash, C.H.; Edwards, J.; Bellomo, R. Structure and Function of the Kidney in Septic Shock. A Prospective Controlled Experimental Study. Am. J. Respir. Crit. Care Med. 2016, 194, 692–700. [Google Scholar] [CrossRef]
- Watchorn, J.; Huang, D.; Bramham, K.; Hutchings, S. Decreased renal cortical perfusion, independent of changes in renal blood flow and sublingual microcirculatory impairment, is associated with the severity of acute kidney injury in patients with septic shock. Crit. Care 2022, 26, 261. [Google Scholar] [CrossRef] [PubMed]
- Gomez, H.; Ince, C.; De Backer, D.; Pickkers, P.; Payen, D.; Hotchkiss, J.; Kellum, J.A. A unified theory of sepsis-induced acute kidney injury: Inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 2014, 41, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fels, B.; Kusche-Vihrog, K. It takes more than two to tango: Mechanosignaling of the endothelial surface. Pflugers Arch. 2020, 472, 419–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Yu, T.; Liu, D.; Shi, Y.; Yang, P.; Zhang, J.; Wang, J.; Liu, Y.; Zhang, X. Sepsis plasma-derived exosomal miR-1-3p induces endothelial cell dysfunction by targeting SERP1. Clin. Sci. 2021, 135, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Molema, G.; Zijlstra, J.G.; van Meurs, M.; Kamps, J. Renal microvascular endothelial cell responses in sepsis-induced acute kidney injury. Nat. Rev. Nephrol. 2022, 18, 95–112. [Google Scholar] [CrossRef]
- Smart, L.; Macdonald, S.P.J.; Burrows, S.; Bosio, E.; Arendts, G.; Fatovich, D.M. Endothelial glycocalyx biomarkers increase in patients with infection during Emergency Department treatment. J. Crit. Care 2017, 42, 304–309. [Google Scholar] [CrossRef]
- Piotti, A.; Novelli, D.; Meessen, J.; Ferlicca, D.; Coppolecchia, S.; Marino, A.; Salati, G.; Savioli, M.; Grasselli, G.; Bellani, G.; et al. Endothelial damage in septic shock patients as evidenced by circulating syndecan-1, sphingosine-1-phosphate and soluble VE-cadherin: A substudy of ALBIOS. Crit. Care 2021, 25, 113. [Google Scholar] [CrossRef]
- Yu, W.K.; McNeil, J.B.; Wickersham, N.E.; Shaver, C.M.; Bastarache, J.A.; Ware, L.B. Vascular endothelial cadherin shedding is more severe in sepsis patients with severe acute kidney injury. Crit. Care 2019, 23, 18. [Google Scholar] [CrossRef] [Green Version]
- Braun, L.J.; Stegmeyer, R.I.; Schafer, K.; Volkery, S.; Currie, S.M.; Kempe, B.; Nottebaum, A.F.; Vestweber, D. Platelets docking to VWF prevent leaks during leukocyte extravasation by stimulating Tie-2. Blood 2020, 136, 627–639. [Google Scholar] [CrossRef]
- Karakike, E.; Giamarellos-Bourboulis, E.J. Macrophage Activation-Like Syndrome: A Distinct Entity Leading to Early Death in Sepsis. Front. Immunol. 2019, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wang, W.; Xie, S.S.; Ma, W.X.; Fan, Q.W.; Chen, Y.; He, Y.; Wang, J.N.; Yang, Q.; Li, H.D.; et al. The Programmed Cell Death of Macrophages, Endothelial Cells, and Tubular Epithelial Cells in Sepsis-AKI. Front. Med. 2021, 8, 796724. [Google Scholar] [CrossRef]
- Lerolle, N.; Nochy, D.; Guerot, E.; Bruneval, P.; Fagon, J.Y.; Diehl, J.L.; Hill, G. Histopathology of septic shock induced acute kidney injury: Apoptosis and leukocytic infiltration. Intensive Care Med. 2010, 36, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, L.; Meng, L.; Cao, G.; Wu, Y. Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell Cycle 2019, 18, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Li, J.; Mao, L.; Wu, J.; Deng, Z.; He, M.; An, S.; Zeng, Z.; Huang, Q.; Chen, Z. p53 Deacetylation Alleviates Sepsis-Induced Acute Kidney Injury by Promoting Autophagy. Front. Immunol. 2021, 12, 685523. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.; Dong, L.; Liu, J.; Ji, X.; Zhuang, H.; Duan, M. The study on role of endothelial cell autophagy in rats with sepsis-induced acute kidney injury. Heliyon 2023, 9, e13796. [Google Scholar] [CrossRef]
- van der Slikke, E.C.; Star, B.S.; van Meurs, M.; Henning, R.H.; Moser, J.; Bouma, H.R. Sepsis is associated with mitochondrial DNA damage and a reduced mitochondrial mass in the kidney of patients with sepsis-AKI. Crit. Care 2021, 25, 36. [Google Scholar] [CrossRef] [PubMed]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011, 107, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, M.M.; Brock, R.W.; Megyesi, J.K.; Kaushal, G.P.; Mayeux, P.R. Disruption of renal peritubular blood flow in lipopolysaccharide-induced renal failure: Role of nitric oxide and caspases. Am. J. Physiol. Renal Physiol. 2005, 289, F1324–F1332. [Google Scholar] [CrossRef] [Green Version]
- El-Awady, M.S.; Nader, M.A.; Sharawy, M.H. The inhibition of inducible nitric oxide synthase and oxidative stress by agmatine attenuates vascular dysfunction in rat acute endotoxemic model. Environ. Toxicol. Pharmacol. 2017, 55, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Van der Poll, T.; Parker, R.I. Platelet Activation and Endothelial Cell Dysfunction. Crit. Care Clin. 2020, 36, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Jesmin, S.; Gando, S.; Zaedi, S.; Prodhan, S.H.; Sawamura, A.; Miyauchi, T.; Hiroe, M.; Yamaguchi, N. Protease-activated receptor 2 blocking peptide counteracts endotoxin-induced inflammation and coagulation and ameliorates renal fibrin deposition in a rat model of acute renal failure. Shock 2009, 32, 626–632. [Google Scholar] [CrossRef]
- Fani, F.; Regolisti, G.; Delsante, M.; Cantaluppi, V.; Castellano, G.; Gesualdo, L.; Villa, G.; Fiaccadori, E. Recent advances in the pathogenetic mechanisms of sepsis-associated acute kidney injury. J. Nephrol. 2018, 31, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Mo, M.; Huang, A.; Li, S.; Luo, Y.; Li, X.; Wu, Q.; Yang, Z.; Liao, Y. Coagulation parameters may predict clinical outcomes in patients with septic acute kidney injury. Clin. Nephrol. 2021, 96, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Kudo, D.; Goto, T.; Uchimido, R.; Hayakawa, M.; Yamakawa, K.; Abe, T.; Shiraishi, A.; Kushimoto, S. Coagulation phenotypes in sepsis and effects of recombinant human thrombomodulin: An analysis of three multicentre observational studies. Crit. Care 2021, 25, 114. [Google Scholar] [CrossRef]
- Vincent, J.L.; Francois, B.; Zabolotskikh, I.; Daga, M.K.; Lascarrou, J.B.; Kirov, M.Y.; Pettila, V.; Wittebole, X.; Meziani, F.; Mercier, E.; et al. Effect of a Recombinant Human Soluble Thrombomodulin on Mortality in Patients With Sepsis-Associated Coagulopathy: The SCARLET Randomized Clinical Trial. JAMA 2019, 321, 1993–2002. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Ye, X.; Xu, H.; Liu, S.F. Activation of endothelial intrinsic NF-kappaB pathway impairs protein C anticoagulation mechanism and promotes coagulation in endotoxemic mice. Blood 2009, 114, 2521–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.; Nan, C.; Fei, D.; Meng, X.; Liu, W.; Zhang, W.; Jiang, L.; Zhao, M.; Pan, S.; Zhao, M. Heme oxygenase 1 modulates thrombomodulin and endothelial protein C receptor levels to attenuate septic kidney injury. Shock 2013, 40, 136–143. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, A.; Carrell, R.W.; Read, R.J. Structural basis for the specificity of renin-mediated angiotensinogen cleavage. J. Biol. Chem. 2019, 294, 2353–2364. [Google Scholar] [CrossRef] [Green Version]
- Chappell, M.C. Biochemical evaluation of the renin-angiotensin system: The good, bad, and absolute? Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H137–H152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riordan, J.F. Angiotensin-I-converting enzyme and its relatives. Genome Biol. 2003, 4, 225. [Google Scholar] [CrossRef] [Green Version]
- Fountain, J.H.; Kaur, J.; Lappin, S.L. Physiology, Renin Angiotensin System. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ruiz-Ortega, M.; Ruperez, M.; Lorenzo, O.; Esteban, V.; Blanco, J.; Mezzano, S.; Egido, J. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int. Suppl. 2002, S12–S22. [Google Scholar] [CrossRef] [Green Version]
- Pastore, L.; Tessitore, A.; Martinotti, S.; Toniato, E.; Alesse, E.; Bravi, M.C.; Ferri, C.; Desideri, G.; Gulino, A.; Santucci, A. Angiotensin II stimulates intercellular adhesion molecule-1 (ICAM-1) expression by human vascular endothelial cells and increases soluble ICAM-1 release in vivo. Circulation 1999, 100, 1646–1652. [Google Scholar] [CrossRef] [Green Version]
- Wolf, G.; Wenzel, U.; Burns, K.D.; Harris, R.C.; Stahl, R.A.; Thaiss, F. Angiotensin II activates nuclear transcription factor-kappaB through AT1 and AT2 receptors. Kidney Int. 2002, 61, 1986–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Xu, D.; Deng, F.; Yan, Y.; Li, J.; Zhang, C.; Chu, J. Angiotensin (1–7) Attenuates Sepsis-Induced Acute Kidney Injury by Regulating the NF-kappaB Pathway. Front. Pharmacol. 2021, 12, 601909. [Google Scholar] [CrossRef] [PubMed]
- Sachse, A.; Wolf, G. Angiotensin II-induced reactive oxygen species and the kidney. J. Am. Soc. Nephrol. 2007, 18, 2439–2446. [Google Scholar] [CrossRef] [Green Version]
- Gromotowicz-Poplawska, A.; Stankiewicz, A.; Kramkowski, K.; Gradzka, A.; Wojewodzka-Zelezniakowicz, M.; Dzieciol, J.; Szemraj, J.; Chabielska, E. The acute prothrombotic effect of aldosterone in rats is partially mediated via angiotensin II receptor type 1. Thromb. Res. 2016, 138, 114–120. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Russell, J.; Almeida-Paula, L.D.; Harding, J.W.; Granger, D.N. Angiotensin II-mediated microvascular thrombosis. Hypertension 2010, 56, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Bavishi, C.; Bangalore, S.; Messerli, F.H. Renin Angiotensin Aldosterone System Inhibitors in Hypertension: Is There Evidence for Benefit Independent of Blood Pressure Reduction? Prog. Cardiovasc. Dis. 2016, 59, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, H.; Liu, D.; Liu, Y.; Li, H.; Tan, X.; Liu, F.; Peng, Y.; Zhang, H. Effects of RAAS Inhibitors in Patients with Kidney Disease. Curr. Hypertens. Rep. 2017, 19, 72. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Christie, G.A.; Waring, W.S. Rapid onset of haemodynamic effects after angiotensin converting enzyme-inhibitor overdose: Implications for initial patient triage. Emerg. Med. J. 2006, 23, 854–857. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.S.; How, C.K.; Hsieh, V.C.; Chen, P.C. Preadmission Antihypertensive Drug Use and Sepsis Outcome: Impact of Angiotensin-Converting Enzyme Inhibitors (ACEIs) and Angiotensin Receptor Blockers (ARBs). Shock 2020, 53, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.W.; Suh, J.K.; Jang, E.; Lee, S.M. Effect of angiotensin converting enzyme inhibitor and angiotensin II receptor blocker on the patients with sepsis. Korean J. Intern. Med. 2021, 36, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Hsu, W.T.; Galm, B.P.; Schrank, G.; Hsu, T.C.; Lee, S.H.; Park, J.Y.; Lee, C.C. Effect of Renin-Angiotensin-Aldosterone System Inhibitors on Short-Term Mortality After Sepsis: A Population-Based Cohort Study. Hypertension 2020, 75, 483–491. [Google Scholar] [CrossRef]
- de Roquetaillade, C.; Jamme, M.; Charpentier, J.; Chiche, J.D.; Cariou, A.; Mira, J.P.; Pene, F.; Llitjos, J.F. Hemodynamic Impact of Cardiovascular Antihypertensive Medications in Patients With Sepsis-Related Acute Circulatory Failure. Shock 2020, 54, 315–320. [Google Scholar] [CrossRef]
- Dial, S.; Nessim, S.J.; Kezouh, A.; Benisty, J.; Suissa, S. Antihypertensive agents acting on the renin-angiotensin system and the risk of sepsis. Br. J. Clin. Pharmacol. 2014, 78, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Feidakis, A.; Panagiotou, M.R.; Tsoukakis, E.; Bacharaki, D.; Gounari, P.; Nikolopoulos, P.; Marathias, K.P.; Lionaki, S.; Vlahakos, D. Impact of Angiotensin-Converting Enzyme Inhibitors or Angiotensin Receptor Blockers on Acute Kidney Injury in Emergency Medical Admissions. J. Clin. Med. 2021, 10, 412. [Google Scholar] [CrossRef]
- Liu, J.; Xie, H.; Ye, Z.; Li, F.; Wang, L. Rates, predictors, and mortality of sepsis-associated acute kidney injury: A systematic review and meta-analysis. BMC Nephrol. 2020, 21, 318. [Google Scholar] [CrossRef]
- Mansfield, K.E.; Nitsch, D.; Smeeth, L.; Bhaskaran, K.; Tomlinson, L.A. Prescription of renin-angiotensin system blockers and risk of acute kidney injury: A population-based cohort study. BMJ Open 2016, 6, e012690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suberviola, B.; Rodrigo, E.; Gonzalez-Castro, A.; Serrano, M.; Heras, M.; Castellanos-Ortega, A. Association between exposure to angiotensin-converting enzyme inhibitors and angiotensin receptor blockers prior to septic shock and acute kidney injury. Med. Intensiva 2017, 41, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Flannery, A.H.; Kiser, A.S.; Behal, M.L.; Li, X.; Neyra, J.A. RAS inhibition and sepsis-associated acute kidney injury. J. Crit. Care 2022, 69, 153986. [Google Scholar] [CrossRef]
- Hasegawa, D.; Lee, Y.I.; Prasitlumkum, N.; Chopra, L.; Nishida, K.; Smith, R.L.; Sato, R. Premorbid angiotensin converting enzyme inhibitors or angiotensin II receptor blockers in patients with sepsis. Am. J. Emerg. Med. 2022, 62, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Demiselle, J.; Seegers, V.; Lemerle, M.; Meziani, F.; Grelon, F.; Megarbane, B.; Anguel, N.; Mira, J.P.; Dequin, P.F.; Gergaud, S.; et al. Prior Exposure to Angiotensin II Receptor Blockers in Patients With Septic Shock to Individualize Mean Arterial Pressure Target? A Post Hoc Analysis of the Sepsis and Mean Arterial Pressure (SEPSISPAM) Trial. Crit. Care Med. 2021, 49, e412–e422. [Google Scholar] [CrossRef]
- Salgado, D.R.; Rocco, J.R.; Silva, E.; Vincent, J.L. Modulation of the renin-angiotensin-aldosterone system in sepsis: A new therapeutic approach? Expert. Opin. Ther. Targets 2010, 14, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.; Tao, X.; Lin, Y.; Zheng, H.; Ning, L.; Lu, H.S.; Daugherty, A.; Shi, P.; Mullick, A.E.; Chen, S.; et al. Loss of Hepatic Angiotensinogen Attenuates Sepsis-Induced Myocardial Dysfunction. Circ. Res. 2021, 129, 547–564. [Google Scholar] [CrossRef]
- Chen, X.S.; Wang, S.H.; Liu, C.Y.; Gao, Y.L.; Meng, X.L.; Wei, W.; Shou, S.T.; Liu, Y.C.; Chai, Y.F. Losartan attenuates sepsis-induced cardiomyopathy by regulating macrophage polarization via TLR4-mediated NF-kappaB and MAPK signaling. Pharmacol. Res. 2022, 185, 106473. [Google Scholar] [CrossRef]
- Chen, X.S.; Cui, J.R.; Meng, X.L.; Wang, S.H.; Wei, W.; Gao, Y.L.; Shou, S.T.; Liu, Y.C.; Chai, Y.F. Angiotensin-(1-7) ameliorates sepsis-induced cardiomyopathy by alleviating inflammatory response and mitochondrial damage through the NF-kappaB and MAPK pathways. J. Transl. Med. 2023, 21, 2. [Google Scholar] [CrossRef]
- Akpinar, E.; Halici, Z.; Cadirci, E.; Bayir, Y.; Karakus, E.; Calik, M.; Topcu, A.; Polat, B. What is the role of renin inhibition during rat septic conditions: Preventive effect of aliskiren on sepsis-induced lung injury. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 969–978. [Google Scholar] [CrossRef]
- Ugan, R.A.; Un, H.; Gurbuz, M.A.; Kaya, G.; Kahramanlar, A.; Aksakalli-Magden, Z.B.; Halici, Z.; Cadirci, E. Possible contribution of the neprilysin/ACE pathway to sepsis in mice. Life Sci. 2020, 258, 118177. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Lou, A.; Lin, Y.; Xu, Q.; Cui, W.; Huang, W.; Wang, G.; Li, Y.; Sun, J.; et al. Sacubitril/valsartan alleviates sepsis-induced acute lung injury via inhibiting GSDMD-dependent macrophage pyroptosis in mice. FEBS J. 2023, 290, 2180–2198. [Google Scholar] [CrossRef] [PubMed]
- Al-Kadi, A.; El-Daly, M.; El-Tahawy, N.F.G.; Khalifa, M.M.A.; Ahmed, A.F. Angiotensin aldosterone inhibitors improve survival and ameliorate kidney injury induced by sepsis through suppression of inflammation and apoptosis. Fundam. Clin. Pharmacol. 2022, 36, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Azizi-Malekabadi, H.; Beheshti, F.; Abareshi, A.; Norouzi, F.; Khazaei, M.; Soukhtanloo, M.; Hosseini, M. Angiotensin-Converting Enzyme Inhibitor Captopril: Does it Improve Renal Function in Lipopolysaccharide-induced Inflammation Model in Rats. Saudi J. Kidney Dis. Transpl. 2020, 31, 727–738. [Google Scholar] [CrossRef]
- Kostakoglu, U.; Mercantepe, T.; Yilmaz, H.K.; Tumkaya, L.; Batcik, S.; Pinarbas, E.; Uydu, H.A. The Protective Effects of Perindopril Against Acute Kidney Damage Caused by Septic Shock. Inflammation 2021, 44, 148–159. [Google Scholar] [CrossRef]
- Leisman, D.E.; Fernandes, T.D.; Bijol, V.; Abraham, M.N.; Lehman, J.R.; Taylor, M.D.; Capone, C.; Yaipan, O.; Bellomo, R.; Deutschman, C.S. Impaired angiotensin II type 1 receptor signaling contributes to sepsis-induced acute kidney injury. Kidney Int. 2021, 99, 148–160. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
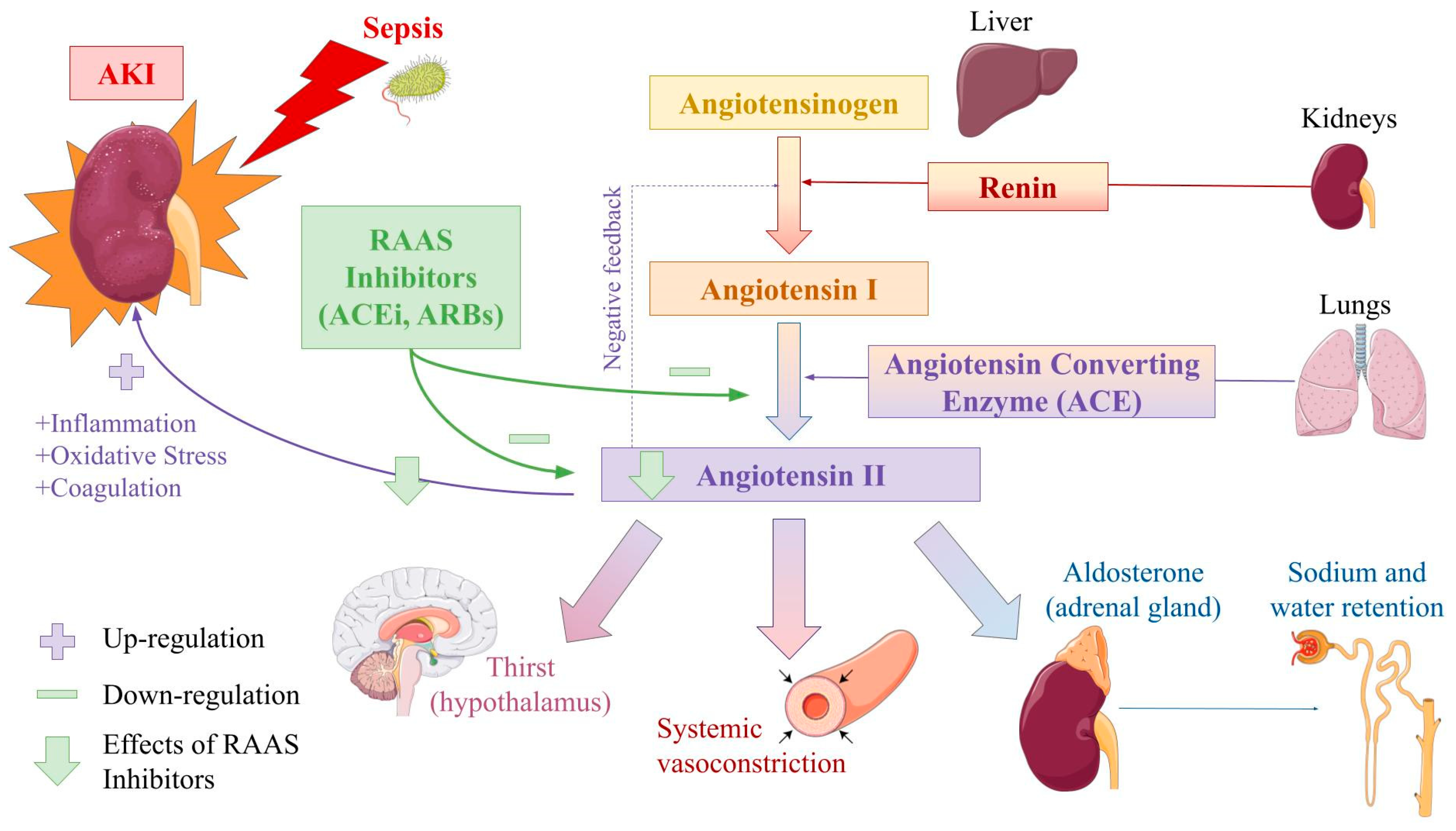{kind=link}
| Study | AKI * Status of Septic Patient | Number of Patients | Median Age (in years) | Male n (%) | Overall Rate of S-AKI * in Septic Patients | Overall Mortality Rate n (%) |
|---|---|---|---|---|---|---|
| Suh 2013 [7] | non-AKI | 419 | 58.7 ± 18.3 | 195 (46.5) | 57.70% | 12 (2.9) |
| AKI | 573 | 66.8 ± 14.3 | 310 (54.1) | 94 (16.4) | ||
| Hsu 2019 [8] | non-AKI | 597 | 68.3 ± 15.5 | 366 (61.3) | 14.22% | 127 (21.3) |
| AKI | 99 | 68.8 ± 16.1 | 63 (63.6) | 71 (71.7) | ||
| Hoste 2003 [11] | non-AKI | 155 | 53 | 106 (68.4) | 16.20% | 44 (28.4) |
| AKI | 30 | 62 | 20 (66.6) | 17 (56.7) | ||
| Murugan 2010 [12] | non-AKI | 1205 | 65.2 ± 17.1 | 634 (52.6) | 34.37% | 16 (1.3) |
| AKI | 631 | 73.4 ± 14.5 | 320 (50.7) | 70 (11.1) | ||
| Poukkanen 2013 [13] | non-AKI | 430 | 63.0 ± 11.0 | 285 (66.3) | 53.15% | 18.1 (78) |
| AKI | 488 | 66.0 ± 10.0 | 305 (62.1) | 29.3 (143) | ||
| Bu 2019 [14] | non-AKI | 90 | 63.54 ± 17.46 | 49 (54.4) | 59.46% | 9 (10.00) |
| AKI | 132 | 65.70 ± 16.92 | 74 (56.1) | 55 (41.67) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tibi, S.; Zeynalvand, G.; Mohsin, H. Role of the Renin Angiotensin Aldosterone System in the Pathogenesis of Sepsis-Induced Acute Kidney Injury: A Systematic Review. J. Clin. Med. 2023, 12, 4566. https://doi.org/10.3390/jcm12144566
Tibi S, Zeynalvand G, Mohsin H. Role of the Renin Angiotensin Aldosterone System in the Pathogenesis of Sepsis-Induced Acute Kidney Injury: A Systematic Review. Journal of Clinical Medicine. 2023; 12(14):4566. https://doi.org/10.3390/jcm12144566
Chicago/Turabian StyleTibi, Sedra, Garbel Zeynalvand, and Hina Mohsin. 2023. "Role of the Renin Angiotensin Aldosterone System in the Pathogenesis of Sepsis-Induced Acute Kidney Injury: A Systematic Review" Journal of Clinical Medicine 12, no. 14: 4566. https://doi.org/10.3390/jcm12144566
APA StyleTibi, S., Zeynalvand, G., & Mohsin, H. (2023). Role of the Renin Angiotensin Aldosterone System in the Pathogenesis of Sepsis-Induced Acute Kidney Injury: A Systematic Review. Journal of Clinical Medicine, 12(14), 4566. https://doi.org/10.3390/jcm12144566