
Pressure Overload and Right Ventricular Failure: From Pathophysiology to Treatment

 , and
, and
Abstract
:1. Introduction
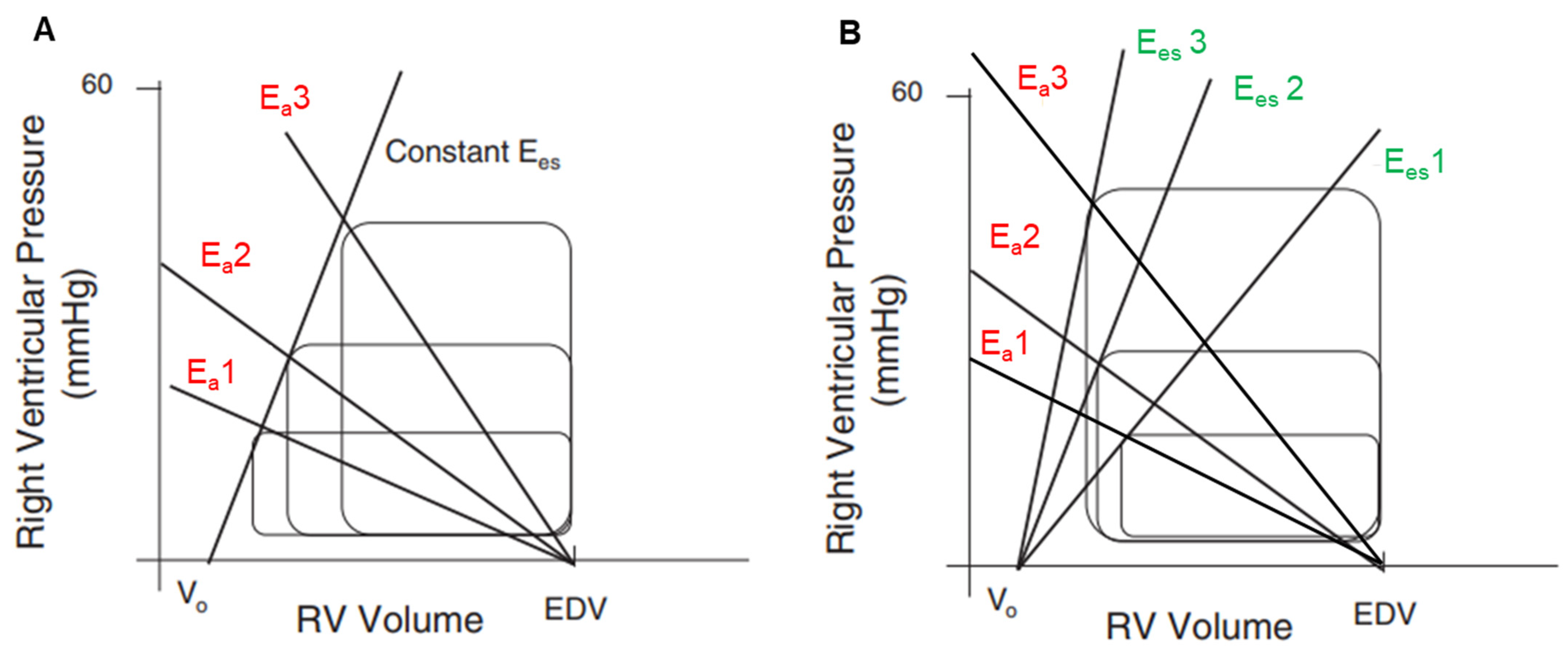
2. Determinants of RV Systolic Function and Coupling
3. RV under Pressure: Homeometric Adaptation
3.1. Adaptive Mechanisms to Increased Afterload
3.2. Transition from Adaptive to Maladaptive Hypertrophy
3.3. RV under Pressure: Heterometric Adaptation and RV Failure
4. Assessment of RV-PA Coupling at Bedside
4.1. Pressure–Volume Loops
4.2. Cardiac Magnetic Resonance Imaging
4.3. Echocardiography
5. Medical Management of Pressure-Overloaded RV Failure
5.1. Preload and Volume Management
5.2. Afterload Reduction
5.3. Vasopressor Therapy
5.4. Inotrope Therapy
5.5. Is There a Place for Mechanical Support in RV Failure?
6. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Starr, I.; Jeffers, W.A.; Meade, R.H. The Absence of Conspicuous Increments of Venous Pressure after Severe Damage to the Right Ventricle of the Dog, with a Discussion of the Relation between Clinical Congestive Failure and Heart Disease. Am. Heart J. 1943, 26, 291–301. [Google Scholar] [CrossRef]
- Downing, T.E.; Allen, K.Y.; Glatz, A.C.; Rogers, L.S.; Ravishankar, C.; Rychik, J.; Faerber, J.A.; Fuller, S.; Montenegro, L.M.; Steven, J.M.; et al. Long-Term Survival after the Fontan Operation: Twenty Years of Experience at a Single Center. J. Thorac. Cardiovasc. Surg. 2017, 154, 243–253.e2. [Google Scholar] [CrossRef] [PubMed]
- Miranda, W.R.; Jain, C.C.; Borlaug, B.A.; Jaffe, A.S.; Connolly, H.M.; Burchill, L.J.; Egbe, A.C. Exercise Capacity, NT-ProBNP, and Exercise Hemodynamics in Adults Post-Fontan. J. Am. Coll. Cardiol. 2023, 81, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- Brassard, P.; Bédard, É.; Jobin, J.; Rodés-Cabau, J.; Poirier, P. Exercise Capacity and Impact of Exercise Training in Patients after a Fontan Procedure: A Review. Can. J. Cardiol. 2006, 22, 489–495. [Google Scholar] [CrossRef] [Green Version]
- La Gerche, A.; MacIsaac, A.I.; Burns, A.T.; Mooney, D.J.; Inder, W.J.; Voigt, J.-U.; Heidbüchel, H.; Prior, D.L. Pulmonary Transit of Agitated Contrast Is Associated with Enhanced Pulmonary Vascular Reserve and Right Ventricular Function during Exercise. J. Appl. Physiol. 2010, 109, 1307–1317. [Google Scholar] [CrossRef] [Green Version]
- Lalande, S.; Yerly, P.; Faoro, V.; Naeije, R. Pulmonary Vascular Distensibility Predicts Aerobic Capacity in Healthy Individuals: Pulmonary Vascular Reserve and Aerobic Exercise Capacity. J. Physiol. 2012, 590, 4279–4288. [Google Scholar] [CrossRef] [Green Version]
- Pinsky, M.R. The Right Ventricle: Interaction with the Pulmonary Circulation. Crit. Care 2016, 20, 266. [Google Scholar] [CrossRef] [Green Version]
- Zeder, K.; Banfi, C.; Steinrisser-Allex, G.; Maron, B.A.; Humbert, M.; Lewis, G.D.; Berghold, A.; Olschewski, H.; Kovacs, G. Diagnostic, Prognostic and Differential-Diagnostic Relevance of Pulmonary Haemodynamic Parameters during Exercise: A Systematic Review. Eur. Respir. J. 2022, 60, 2103181. [Google Scholar] [CrossRef]
- Berlin, D.A.; Bakker, J. Understanding Venous Return. Intensive Care Med. 2014, 40, 1564–1566. [Google Scholar] [CrossRef]
- Markel, T.A.; Wairiuko, G.M.; Lahm, T.; Crisostomo, P.R.; Wang, M.; Herring, C.M.; Meldrum, D.R. The Right Heart and Its Distinct Mechanisms of Development, Function, and Failure. J. Surg. Res. 2008, 146, 304–313. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef]
- Harjola, V.-P.; Mebazaa, A.; Čelutkienė, J.; Bettex, D.; Bueno, H.; Chioncel, O.; Crespo-Leiro, M.G.; Falk, V.; Filippatos, G.; Gibbs, S.; et al. Contemporary Management of Acute Right Ventricular Failure: A Statement from the Heart Failure Association and the Working Group on Pulmonary Circulation and Right Ventricular Function of the European Society of Cardiology: Contemporary Management of Acute RV Failure. Eur. J. Heart Fail. 2016, 18, 226–241. [Google Scholar] [CrossRef] [PubMed]
- Vanderpool, R.R.; Pinsky, M.R.; Naeije, R.; Deible, C.; Kosaraju, V.; Bunner, C.; Mathier, M.A.; Lacomis, J.; Champion, H.C.; Simon, M.A. RV-Pulmonary Arterial Coupling Predicts Outcome in Patients Referred for Pulmonary Hypertension. Heart 2015, 101, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benza, R.L.; Miller, D.P.; Gomberg-Maitland, M.; Frantz, R.P.; Foreman, A.J.; Coffey, C.S.; Frost, A.; Barst, R.J.; Badesch, D.B.; Elliott, C.G.; et al. Predicting Survival in Pulmonary Arterial Hypertension: Insights From the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010, 122, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugger, N.; Lichtblau, M.; Maeder, M.T.; Müller, H.; Pellaton, C.; Yerly, P. Two-Dimensional Transthoracic Echocardiography at Rest for the Diagnosis, Screening and Management of Pulmonary Hypertension. Swiss Med. Wkly. 2021, 151, w20486. [Google Scholar] [CrossRef]
- Tello, K.; Seeger, W.; Naeije, R.; Vanderpool, R.; Ghofrani, H.A.; Richter, M.; Tedford, R.J.; Bogaard, H.J. Right Heart Failure in Pulmonary Hypertension: Diagnosis and New Perspectives on Vascular and Direct Right Ventricular Treatment. Br. J. Pharmacol. 2021, 178, 90–107. [Google Scholar] [CrossRef]
- Brimioulle, S.; Wauthy, P.; Ewalenko, P.; Rondelet, B.; Vermeulen, F.; Kerbaul, F.; Naeije, R. Single-Beat Estimation of Right Ventricular End-Systolic Pressure-Volume Relationship. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, H1625–H1630. [Google Scholar] [CrossRef]
- Suga, H.; Sagawa, K.; Shoukas, A.A. Load Independence of the Instantaneous Pressure-Volume Ratio of the Canine Left Ventricle and Effects of Epinephrine and Heart Rate on the Ratio. Circ. Res. 1973, 32, 314–322. [Google Scholar] [CrossRef]
- Tedford, R.J.; Mudd, J.O.; Girgis, R.E.; Mathai, S.C.; Zaiman, A.L.; Housten-Harris, T.; Boyce, D.; Kelemen, B.W.; Bacher, A.C.; Shah, A.A.; et al. Right Ventricular Dysfunction in Systemic Sclerosis–Associated Pulmonary Arterial Hypertension. Circ. Heart Fail. 2013, 6, 953–963. [Google Scholar] [CrossRef] [Green Version]
- Burkhoff, D.; Mirsky, I.; Suga, H. Assessment of Systolic and Diastolic Ventricular Properties via Pressure-Volume Analysis: A Guide for Clinical, Translational, and Basic Researchers. Am. J. Physiol.-Heart Circ. Physiol. 2005, 289, H501–H512. [Google Scholar] [CrossRef]
- Sunagawa, K.; Yamada, A.; Senda, Y.; Kikuchi, Y.; Nakamura, M.; Shibahara, T.; Nose, Y. Estimation of the Hydromotive Source Pressure from Ejecting Beats of the Left Ventricle. IEEE Trans. Biomed. Eng. 1980, BME-27, 299–305. [Google Scholar] [CrossRef]
- Morimont, P.; Lambermont, B.; Ghuysen, A.; Gerard, P.; Kolh, P.; Lancellotti, P.; Tchana-Sato, V.; Desaive, T.; D’Orio, V. Effective Arterial Elastance as an Index of Pulmonary Vascular Load. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H2736–H2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghuysen, A.; Lambermont, B.; Kolh, P.; Tchana-Sato, V.; Magis, D.; Gerard, P.; Mommens, V.; Janssen, N.; Desaive, T.; D’Orio, V. Alteration of Right Ventricular-Pulmonary Vascular Coupling in A Porcine Model of Progressive Pressure Overloading. Shock 2008, 29, 197–204. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W. Pathophysiology of Cor Pulmonale in Chronic Obstructive Pulmonary Disease. Part One. Am. J. Respir. Crit. Care Med. 1994, 150, 833–852. [Google Scholar] [CrossRef] [PubMed]
- Vonk Noordegraaf, A.; Westerhof, B.E.; Westerhof, N. The Relationship Between the Right Ventricle and Its Load in Pulmonary Hypertension. J. Am. Coll. Cardiol. 2017, 69, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Vieillard-Baron, A.; Naeije, R.; Haddad, F.; Bogaard, H.J.; Bull, T.M.; Fletcher, N.; Lahm, T.; Magder, S.; Orde, S.; Schmidt, G.; et al. Diagnostic Workup, Etiologies and Management of Acute Right Ventricle Failure: A State-of-the-Art Paper. Intensive Care Med. 2018, 44, 774–790. [Google Scholar] [CrossRef]
- Bogaard, H.J.; Abe, K.; Vonk Noordegraaf, A.; Voelkel, N.F. The Right Ventricle Under Pressure. Chest 2009, 135, 794–804. [Google Scholar] [CrossRef] [Green Version]
- Hess, O.M.; Villari, B.; Krayenbuehl, H.P. Diastolic Dysfunction in Aortic Stenosis. Circulation 1993, 87, IV73–IV76. [Google Scholar]
- Lyon, R.C.; Zanella, F.; Omens, J.H.; Sheikh, F. Mechanotransduction in Cardiac Hypertrophy and Failure. Circ. Res. 2015, 116, 1462–1476. [Google Scholar] [CrossRef]
- De Man, F.S.; Tu, L.; Handoko, M.L.; Rain, S.; Ruiter, G.; François, C.; Schalij, I.; Dorfmüller, P.; Simonneau, G.; Fadel, E.; et al. Dysregulated Renin–Angiotensin–Aldosterone System Contributes to Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 780–789. [Google Scholar] [CrossRef] [Green Version]
- van der Bruggen, C.E.E.; Tedford, R.J.; Handoko, M.L.; van der Velden, J.; de Man, F.S. RV Pressure Overload: From Hypertrophy to Failure. Cardiovasc. Res. 2017, 113, 1423–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, S.; Witte, K.K.; Al-Hesayen, A.; Granton, J.J.; Parker, J.D. Cardiac Sympathetic Activation in Patients with Pulmonary Arterial Hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R1153–R1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsumi, A.; Orr, A.W.; Tzima, E.; Schwartz, M.A. Integrins in Mechanotransduction. J. Biol. Chem. 2004, 279, 12001–12004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, M.H.; Peng, J.; Wu, Y.; McNabb, M.; Nelson, O.L.; Granzier, H.; Gotthardt, M. Targeted Deletion of Titin N2B Region Leads to Diastolic Dysfunction and Cardiac Atrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 3444–3449. [Google Scholar] [CrossRef] [PubMed]
- Velez-Roa, S.; Ciarka, A.; Najem, B.; Vachiery, J.-L.; Naeije, R.; Van De Borne, P. Increased Sympathetic Nerve Activity in Pulmonary Artery Hypertension. Circulation 2004, 110, 1308–1312. [Google Scholar] [CrossRef] [Green Version]
- Bhullar, S.K.; Dhalla, N.S. Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells 2022, 11, 3336. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, J.; Fontes, S.K.; Bautista, E.N.; Cheng, Z. Physiological and Pathological Roles of Protein Kinase A in the Heart. Cardiovasc. Res. 2022, 118, 386–398. [Google Scholar] [CrossRef]
- Fu, Y.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Basal and β-Adrenergic Regulation of the Cardiac Calcium Channel Ca V 1.2 Requires Phosphorylation of Serine 1700. Proc. Natl. Acad. Sci. USA 2014, 111, 16598–16603. [Google Scholar] [CrossRef]
- Bekedam, F.T.; Goumans, M.J.; Bogaard, H.J.; De Man, F.S.; Llucià-Valldeperas, A. Molecular Mechanisms and Targets of Right Ventricular Fibrosis in Pulmonary Hypertension. Pharmacol. Ther. 2023, 244, 108389. [Google Scholar] [CrossRef]
- Oka, T.; Akazawa, H.; Naito, A.T.; Komuro, I. Angiogenesis and Cardiac Hypertrophy: Maintenance of Cardiac Function and Causative Roles in Heart Failure. Circ. Res. 2014, 114, 565–571. [Google Scholar] [CrossRef]
- Vonk Noordegraaf, A.; Galie, N. The Role of the Right Ventricle in Pulmonary Arterial Hypertension. Eur. Respir. Rev. 2011, 20, 243–253. [Google Scholar] [CrossRef]
- Van Der Bruggen, C.E.; Happé, C.M.; Dorfmüller, P.; Trip, P.; Spruijt, O.A.; Rol, N.; Hoevenaars, F.P.; Houweling, A.C.; Girerd, B.; Marcus, J.T.; et al. Bone Morphogenetic Protein Receptor Type 2 Mutation in Pulmonary Arterial Hypertension: A View on the Right Ventricle. Circulation 2016, 133, 1747–1760. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.; Kokkonen-Simon, K.M.; Kirk, J.A.; Kolb, T.M.; Damico, R.L.; Mathai, S.C.; Mukherjee, M.; Shah, A.A.; Wigley, F.M.; Margulies, K.B.; et al. Right Ventricular Myofilament Functional Differences in Humans With Systemic Sclerosis–Associated Versus Idiopathic Pulmonary Arterial Hypertension. Circulation 2018, 137, 2360–2370. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arroyo, J.; Santos-Martinez, L.E.; Aranda, A.; Pulido, T.; Beltran, M.; Muñoz-Castellanos, L.; Dominguez-Cano, E.; Sonnino, C.; Voelkel, N.F.; Sandoval, J. Differences in Right Ventricular Remodeling Secondary to Pressure Overload in Patients with Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Hurdman, J.; Condliffe, R.; Elliot, C.A.; Davies, C.; Hill, C.; Wild, J.M.; Capener, D.; Sephton, P.; Hamilton, N.; Armstrong, I.J.; et al. ASPIRE Registry: Assessing the Spectrum of Pulmonary Hypertension Identified at a REferral Centre. Eur. Respir. J. 2012, 39, 945–955. [Google Scholar] [CrossRef] [Green Version]
- Ohira, H.; deKemp, R.; Pena, E.; Davies, R.A.; Stewart, D.J.; Chandy, G.; Contreras-Dominguez, V.; Dennie, C.; Mc Ardle, B.; Mc Klein, R.; et al. Shifts in Myocardial Fatty Acid and Glucose Metabolism in Pulmonary Arterial Hypertension: A Potential Mechanism for a Maladaptive Right Ventricular Response. Eur. Heart J. Cardiovasc. Imaging 2016, 17, 1424–1431. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, W.; Yang, Y.; Wu, W.; Cai, Q.; Ma, X.; Xiong, C.; He, J.; Fang, W. Quantitative Assessment of Right Ventricular Glucose Metabolism in Idiopathic Pulmonary Arterial Hypertension Patients: A Longitudinal Study. Eur. Heart J.-Cardiovasc. Imaging 2016, 17, 1161–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L.; Fang, Y.; Ryan, J.J.; Piao, L. Metabolism and Bioenergetics in the Right Ventricle and Pulmonary Vasculature in Pulmonary Hypertension. Pulm. Circ. 2013, 3, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.J.; Huston, J.; Kutty, S.; Hatton, N.D.; Bowman, L.; Tian, L.; Herr, J.E.; Johri, A.M.; Archer, S.L. Right Ventricular Adaptation and Failure in Pulmonary Arterial Hypertension. Can. J. Cardiol. 2015, 31, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Rain, S.; Handoko, M.L.; Trip, P.; Gan, C.T.-J.; Westerhof, N.; Stienen, G.J.; Paulus, W.J.; Ottenheijm, C.A.C.; Marcus, J.T.; Dorfmüller, P.; et al. Right Ventricular Diastolic Impairment in Patients With Pulmonary Arterial Hypertension. Circulation 2013, 128, 2016–2025. [Google Scholar] [CrossRef] [Green Version]
- Kusakari, Y.; Urashima, T.; Shimura, D.; Amemiya, E.; Miyasaka, G.; Yokota, S.; Fujimoto, Y.; Akaike, T.; Inoue, T.; Minamisawa, S. Impairment of Excitation-Contraction Coupling in Right Ventricular Hypertrophied Muscle with Fibrosis Induced by Pulmonary Artery Banding. PLoS ONE 2017, 12, e0169564. [Google Scholar] [CrossRef] [Green Version]
- Rain, S.; Andersen, S.; Najafi, A.; Gammelgaard Schultz, J.; Da Silva Gonçalves Bós, D.; Handoko, M.L.; Bogaard, H.-J.; Vonk-Noordegraaf, A.; Andersen, A.; Van Der Velden, J.; et al. Right Ventricular Myocardial Stiffness in Experimental Pulmonary Arterial Hypertension: Relative Contribution of Fibrosis and Myofibril Stiffness. Circ. Heart Fail. 2016, 9, e002636. [Google Scholar] [CrossRef] [PubMed]
- Van Wolferen, S.A.; Marcus, J.T.; Westerhof, N.; Spreeuwenberg, M.D.; Marques, K.M.J.; Bronzwaer, J.G.F.; Henkens, I.R.; Gan, C.T.-J.; Boonstra, A.; Postmus, P.E.; et al. Right Coronary Artery Flow Impairment in Patients with Pulmonary Hypertension. Eur. Heart J. 2007, 29, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiter, G.; Ying Wong, Y.; De Man, F.S.; Louis Handoko, M.; Jaspers, R.T.; Postmus, P.E.; Westerhof, N.; Niessen, H.W.M.; Van Der Laarse, W.J.; Vonk-Noordegraaf, A. Right Ventricular Oxygen Supply Parameters Are Decreased in Human and Experimental Pulmonary Hypertension. J. Heart Lung Transplant. 2013, 32, 231–240. [Google Scholar] [CrossRef]
- Nootens, M.; Kaufmann, E.; Rector, T.; Toher, C.; Judd, D.; Francis, G.S.; Rich, S. Neurohormonal Activation in Patients with Right Ventricular Failure from Pulmonary Hypertension: Relation to Hemodynamic Variables and Endothelin Levels. J. Am. Coll. Cardiol. 1995, 26, 1581–1585. [Google Scholar] [CrossRef] [Green Version]
- Campo, A.; Mathai, S.C.; Le Pavec, J.; Zaiman, A.L.; Hummers, L.K.; Boyce, D.; Housten, T.; Lechtzin, N.; Chami, H.; Girgis, R.E.; et al. Outcomes of Hospitalisation for Right Heart Failure in Pulmonary Arterial Hypertension. Eur. Respir. J. 2011, 38, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Schmeißer, A.; Rauwolf, T.; Groscheck, T.; Fischbach, K.; Kropf, S.; Luani, B.; Tanev, I.; Hansen, M.; Meißler, S.; Schäfer, K.; et al. Predictors and Prognosis of Right Ventricular Function in Pulmonary Hypertension Due to Heart Failure with Reduced Ejection Fraction. ESC Heart Fail. 2021, 8, 2968–2981. [Google Scholar] [CrossRef]
- Richter, M.J.; Peters, D.; Ghofrani, H.A.; Naeije, R.; Roller, F.; Sommer, N.; Gall, H.; Grimminger, F.; Seeger, W.; Tello, K. Evaluation and Prognostic Relevance of Right Ventricular–Arterial Coupling in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Simpson, C.E.; Houston, B.A.; Wand, A.; Sato, T.; Kolb, T.M.; Mathai, S.C.; Kass, D.A.; Hassoun, P.M.; Damico, R.L.; et al. Multi-Beat Right Ventricular-Arterial Coupling Predicts Clinical Worsening in Pulmonary Arterial Hypertension. J. Am. Heart Assoc. 2020, 9, e016031. [Google Scholar] [CrossRef]
- Rako, Z.A.; Kremer, N.; Yogeswaran, A.; Richter, M.J.; Tello, K. Adaptive versus Maladaptive Right Ventricular Remodelling. ESC Heart Fail. 2023, 10, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Vonk-Noordegraaf, A.; Haddad, F.; Chin, K.M.; Forfia, P.R.; Kawut, S.M.; Lumens, J.; Naeije, R.; Newman, J.; Oudiz, R.J.; Provencher, S.; et al. Right Heart Adaptation to Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2013, 62, D22–D33. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Sokos, G.; Taylor, D.O.; Starling, R.C.; Young, J.B.; Tang, W.H.W. Importance of Venous Congestion for Worsening of Renal Function in Advanced Decompensated Heart Failure. J. Am. Coll. Cardiol. 2009, 53, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitker, L.; Sens, F.; Payet, C.; Turquier, S.; Duclos, A.; Cottin, V.; Juillard, L. Presence of Kidney Disease as an Outcome Predictor in Patients with Pulmonary Arterial Hypertension. Am. J. Nephrol. 2018, 47, 134–143. [Google Scholar] [CrossRef]
- Rosenkranz, S.; Howard, L.S.; Gomberg-Maitland, M.; Hoeper, M.M. Systemic Consequences of Pulmonary Hypertension and Right-Sided Heart Failure. Circulation 2020, 141, 678–693. [Google Scholar] [CrossRef]
- Schefold, J.C.; Filippatos, G.; Hasenfuss, G.; Anker, S.D.; Von Haehling, S. Heart Failure and Kidney Dysfunction: Epidemiology, Mechanisms and Management. Nat. Rev. Nephrol. 2016, 12, 610–623. [Google Scholar] [CrossRef]
- Rommel, K.-P.; Besler, C.; Noack, T.; Blazek, S.; Von Roeder, M.; Fengler, K.; Ender, J.; Gutberlet, M.; Desch, S.; Borger, M.A.; et al. Physiological and Clinical Consequences of Right Ventricular Volume Overload Reduction After Transcatheter Treatment for Tricuspid Regurgitation. JACC Cardiovasc. Interv. 2019, 12, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhai, Z.; Huang, K.; Xie, W.; Wan, J.; Wang, C. Bosentan Therapy for Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension: A Systemic Review and Meta-Analysis. Clin. Respir. J. 2018, 12, 2065–2074. [Google Scholar] [CrossRef]
- Medvedofsky, D.; Mor-Avi, V.; Amzulescu, M.; Fernández-Golfín, C.; Hinojar, R.; Monaghan, M.J.; Otani, K.; Reiken, J.; Takeuchi, M.; Tsang, W.; et al. Three-Dimensional Echocardiographic Quantification of the Left-Heart Chambers Using an Automated Adaptive Analytics Algorithm: Multicentre Validation Study. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Tji-Joong Gan, C.; Lankhaar, J.-W.; Marcus, J.T.; Westerhof, N.; Marques, K.M.; Bronzwaer, J.G.F.; Boonstra, A.; Postmus, P.E.; Vonk-Noordegraaf, A. Impaired Left Ventricular Filling Due to Right-to-Left Ventricular Interaction in Patients with Pulmonary Arterial Hypertension. Am. J. Physiol.-Heart Circ. Physiol. 2006, 290, H1528–H1533. [Google Scholar] [CrossRef]
- Greiner, S.; Jud, A.; Aurich, M.; Hess, A.; Hilbel, T.; Hardt, S.; Katus, H.A.; Mereles, D. Reliability of Noninvasive Assessment of Systolic Pulmonary Artery Pressure by Doppler Echocardiography Compared to Right Heart Catheterization: Analysis in a Large Patient Population. J. Am. Heart Assoc. 2014, 3, e001103. [Google Scholar] [CrossRef] [Green Version]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for Cardiac Chamber Quantification by Echocardiography in Adults: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weatherald, J.; Boucly, A.; Chemla, D.; Savale, L.; Peng, M.; Jevnikar, M.; Jaïs, X.; Taniguchi, Y.; O’Connell, C.; Parent, F.; et al. Prognostic Value of Follow-Up Hemodynamic Variables After Initial Management in Pulmonary Arterial Hypertension. Circulation 2018, 137, 693–704. [Google Scholar] [CrossRef]
- Nickel, N.; Golpon, H.; Greer, M.; Knudsen, L.; Olsson, K.; Westerkamp, V.; Welte, T.; Hoeper, M.M. The Prognostic Impact of Follow-up Assessments in Patients with Idiopathic Pulmonary Arterial Hypertension. Eur. Respir. J. 2012, 39, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Diepen, S.; Katz, J.N.; Albert, N.M.; Henry, T.D.; Jacobs, A.K.; Kapur, N.K.; Kilic, A.; Menon, V.; Ohman, E.M.; Sweitzer, N.K.; et al. Contemporary Management of Cardiogenic Shock: A Scientific Statement From the American Heart Association. Circulation 2017, 136, e232–e268. [Google Scholar] [CrossRef]
- Tello, K.; Richter, M.J.; Yogeswaran, A.; Ghofrani, H.A.; Naeije, R.; Vanderpool, R.; Gall, H.; Tedford, R.J.; Seeger, W.; Lahm, T. Sex Differences in Right Ventricular–Pulmonary Arterial Coupling in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020, 202, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Sanz, J.; Conroy, J.; Narula, J. Imaging of the Right Ventricle. Cardiol. Clin. 2012, 30, 189–203. [Google Scholar] [CrossRef]
- Trip, P.; Kind, T.; van de Veerdonk, M.C.; Marcus, J.T.; de Man, F.S.; Westerhof, N.; Vonk-Noordegraaf, A. Accurate Assessment of Load-Independent Right Ventricular Systolic Function in Patients with Pulmonary Hypertension. J. Heart Lung Transplant. 2013, 32, 50–55. [Google Scholar] [CrossRef]
- Brewis, M.J.; Bellofiore, A.; Vanderpool, R.R.; Chesler, N.C.; Johnson, M.K.; Naeije, R.; Peacock, A.J. Imaging Right Ventricular Function to Predict Outcome in Pulmonary Arterial Hypertension. Int. J. Cardiol. 2016, 218, 206–211. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Lin, Y.; Zhu, Y.; Gao, L.; Ji, M.; Zhang, L.; Xie, M.; Li, Y. Clinical Usefulness of Right Ventricle–Pulmonary Artery Coupling in Cardiovascular Disease. J. Clin. Med. 2023, 12, 2526. [Google Scholar] [CrossRef]
- Guazzi, M.; Bandera, F.; Pelissero, G.; Castelvecchio, S.; Menicanti, L.; Ghio, S.; Temporelli, P.L.; Arena, R. Tricuspid Annular Plane Systolic Excursion and Pulmonary Arterial Systolic Pressure Relationship in Heart Failure: An Index of Right Ventricular Contractile Function and Prognosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1373–H1381. [Google Scholar] [CrossRef]
- Guazzi, M.; Dixon, D.; Labate, V.; Beussink-Nelson, L.; Bandera, F.; Cuttica, M.J.; Shah, S.J. RV Contractile Function and Its Coupling to Pulmonary Circulation in Heart Failure With Preserved Ejection Fraction. JACC Cardiovasc. Imaging 2017, 10, 1211–1221. [Google Scholar] [CrossRef]
- Guazzi, M.; Naeije, R.; Arena, R.; Corrà, U.; Ghio, S.; Forfia, P.; Rossi, A.; Cahalin, L.P.; Bandera, F.; Temporelli, P. Echocardiography of Right Ventriculoarterial Coupling Combined With Cardiopulmonary Exercise Testing to Predict Outcome in Heart Failure. Chest 2015, 148, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Tello, K.; Axmann, J.; Ghofrani, H.A.; Naeije, R.; Narcin, N.; Rieth, A.; Seeger, W.; Gall, H.; Richter, M.J. Relevance of the TAPSE/PASP Ratio in Pulmonary Arterial Hypertension. Int. J. Cardiol. 2018, 266, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Tello, K.; Wan, J.; Dalmer, A.; Vanderpool, R.; Ghofrani, H.A.; Naeije, R.; Roller, F.; Mohajerani, E.; Seeger, W.; Herberg, U.; et al. Validation of the Tricuspid Annular Plane Systolic Excursion/Systolic Pulmonary Artery Pressure Ratio for the Assessment of Right Ventricular-Arterial Coupling in Severe Pulmonary Hypertension. Circ. Cardiovasc. Imaging 2019, 12, e009047. [Google Scholar] [CrossRef]
- Kim, D.; Jang, W.J.; Park, T.K.; Cho, Y.H.; Choi, J.-O.; Jeon, E.-S.; Yang, J.H. Echocardiographic Predictors of Successful Extracorporeal Membrane Oxygenation Weaning After Refractory Cardiogenic Shock. J. Am. Soc. Echocardiogr. 2021, 34, 414–422.e4. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kiernan, M.S.; Bernstein, D.; Bozkurt, B.; Jacob, M.; Kapur, N.K.; Kociol, R.D.; Lewis, E.F.; Mehra, M.R.; Pagani, F.D.; et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2018, 137, e578–e622. [Google Scholar] [CrossRef]
- Campbell, P.; Drazner, M.H.; Kato, M.; Lakdawala, N.; Palardy, M.; Nohria, A.; Stevenson, L.W. Mismatch of Right- and Left-Sided Filling Pressures in Chronic Heart Failure. J. Card. Fail. 2011, 17, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Ltaief, Z.; Yerly, P.; Liaudet, L. Pulmonary Hypertension in Left Heart Diseases: Pathophysiology, Hemodynamic Assessment and Therapeutic Management. Int. J. Mol. Sci. 2023, 24, 9971. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, M.F.; Cancelli, E.; Rusca, M.; Kirsch, M.; Yerly, P.; Liaudet, L. The Prognostic Value of Pulmonary Artery Compliance in Cardiogenic Shock. Pulm. Circ. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Marenzi, G.; Lauri, G.; Grazi, M.; Assanelli, E.; Campodonico, J.; Agostoni, P. Circulatory Response to Fluid Overload Removal by Extracorporeal Ultrafiltration in Refractory Congestive Heart Failure. J. Am. Coll. Cardiol. 2001, 38, 963–968. [Google Scholar] [CrossRef] [Green Version]
- Mullens, W.; Damman, K.; Harjola, V.-P.; Mebazaa, A.; Brunner-La Rocca, H.-P.; Martens, P.; Testani, J.M.; Tang, W.H.W.; Orso, F.; Rossignol, P.; et al. The Use of Diuretics in Heart Failure with Congestion—A Position Statement from the Heart Failure Association of the European Society of Cardiology: Diuretics in Heart Failure. Eur. J. Heart Fail. 2019, 21, 137–155. [Google Scholar] [CrossRef] [Green Version]
- Mullens, W.; Dauw, J.; Martens, P.; Verbrugge, F.H.; Nijst, P.; Meekers, E.; Tartaglia, K.; Chenot, F.; Moubayed, S.; Dierckx, R.; et al. Acetazolamide in Acute Decompensated Heart Failure with Volume Overload. N. Engl. J. Med. 2022, 387, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.R. Ultrafiltration in Acute Heart Failure. Card. Fail. Rev. 2019, 5, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Konstantinides, S.V.; Meyer, G.; Becattini, C.; Bueno, H.; Geersing, G.-J.; Harjola, V.-P.; Huisman, M.V.; Humbert, M.; Jennings, C.S.; Jiménez, D.; et al. 2019 ESC Guidelines for the Diagnosis and Management of Acute Pulmonary Embolism Developed in Collaboration with the European Respiratory Society (ERS). Eur. Heart J. 2020, 41, 543–603. [Google Scholar] [CrossRef] [PubMed]
- Madani, M.M. Surgical Treatment of Chronic Thromboembolic Pulmonary Hypertension: Pulmonary Thromboendarterectomy. Methodist DeBakey Cardiovasc. J. 2016, 12, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbelen, T.; Godinas, L.; Maleux, G.; Coolen, J.; Claessen, G.; Belge, C.; Meyns, B.; Delcroix, M. Chronic Thromboembolic Pulmonary Hypertension: Diagnosis, Operability Assessment and Patient Selection for Pulmonary Endarterectomy. Ann. Cardiothorac. Surg. 2022, 11, 82–97. [Google Scholar] [CrossRef]
- Vieillard-Baron, A.; Price, L.C.; Matthay, M.A. Acute Cor Pulmonale in ARDS. Intensive Care Med. 2013, 39, 1836–1838. [Google Scholar] [CrossRef] [Green Version]
- Vieillard-Baron, A.; Schmitt, J.-M.; Augarde, R.; Fellahi, J.L.; Prin, S.; Page, B.; Beauchet, A.; Jardin, F. Acute Cor Pulmonale in Acute Respiratory Distress Syndrome Submitted to Protective Ventilation: Incidence, Clinical Implications, and Prognosis. Crit. Care Med. 2001, 29, 1551–1555. [Google Scholar] [CrossRef]
- Bull, T.M.; Clark, B.; McFann, K.; Moss, M. National Institutes of Health/National Heart, Lung, and Blood Institute ARDS Network Pulmonary Vascular Dysfunction Is Associated with Poor Outcomes in Patients with Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2010, 182, 1123–1128. [Google Scholar] [CrossRef] [Green Version]
- Lhéritier, G.; Legras, A.; Caille, A.; Lherm, T.; Mathonnet, A.; Frat, J.-P.; Courte, A.; Martin-Lefèvre, L.; Gouëllo, J.-P.; Amiel, J.-B.; et al. Prevalence and Prognostic Value of Acute Cor Pulmonale and Patent Foramen Ovale in Ventilated Patients with Early Acute Respiratory Distress Syndrome: A Multicenter Study. Intensive Care Med. 2013, 39, 1734–1742. [Google Scholar] [CrossRef]
- L’Heureux, M.; Sternberg, M.; Brath, L.; Turlington, J.; Kashiouris, M.G. Sepsis-Induced Cardiomyopathy: A Comprehensive Review. Curr. Cardiol. Rep. 2020, 22, 35. [Google Scholar] [CrossRef] [PubMed]
- Ventetuolo, C.E.; Klinger, J.R. Management of Acute Right Ventricular Failure in the Intensive Care Unit. Ann. Am. Thorac. Soc. 2014, 11, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, A.M.; Yuan, S. Response of the Pulmonary Vasculature to Hypoxia and H+ Ion Concentration Changes. J. Clin. Investig. 1966, 45, 399–411. [Google Scholar] [CrossRef] [PubMed]
- The Acute Respiratory Distress Syndrome Network. Ventilation with Lower Tidal Volumes as Compared with Traditional Tidal Volumes for Acute Lung Injury and the Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef] [Green Version]
- Bhorade, S.; Christenson, J.; O’connor, M.; Lavoie, A.; Pohlman, A.; Hall, J.B. Response to Inhaled Nitric Oxide in Patients with Acute Right Heart Syndrome. Am. J. Respir. Crit. Care Med. 1999, 159, 571–579. [Google Scholar] [CrossRef]
- Rossaint, R.; Slama, K.; Steudel, W.; Gerlach, H.; Pappert, D.; Veit, S.; Falke, K. Effects of Inhaled Nitric Oxide on Right Ventricular Function in Severe Acute Respiratory Distress Syndrome. Intensive Care Med. 1995, 21, 197–203. [Google Scholar] [CrossRef]
- Muzaffar, S.; Shukla, N.; Angelini, G.D.; Jeremy, J.Y. Inhaled Prostacyclin Is Safe, Effective, and Affordable in Patients with Pulmonary Hypertension, Right-Heart Dysfunction, and Refractory Hypoxemia after Cardiothoracic Surgery. J. Thorac. Cardiovasc. Surg. 2004, 128, 949–950. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Hollenberg, S.M. Vasoactive Drugs in Circulatory Shock. Am. J. Respir. Crit. Care Med. 2011, 183, 847–855. [Google Scholar] [CrossRef]
- Ghignone, M.; Girling, L.; Prewitt, R.M. Volume Expansion versus Norepinephrine in Treatment of a Low Cardiac Output Complicating an Acute Increase in Right Ventricular Afterload in Dogs. Anesthesiology 1984, 60, 132–135. [Google Scholar] [CrossRef]
- Schreuder, W.O.; Schneider, A.J.; Groeneveld, A.B.J.; Thijs, L.G. The Influence of Catecholamines on Right Ventricular Function in Septic Shock. Intensive Care Med. 1988, 14, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Evora, P.R.; Pearson, P.J.; Schaff, H.V. Arginine Vasopressin Induces Endothelium-Dependent Vasodilatation of the Pulmonary Artery. V1-Receptor-Mediated Production of Nitric Oxide. Chest 1993, 103, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, J.; Golden, P.J.; Kajiura, L.N.; Murata, L.-A.M.; Uyehara, C.F.T. Vasopressin Decreases Pulmonary-to-Systemic Vascular Resistance Ratio in a Porcine Model of Severe Hemorrhagic Shock. Shock Augusta Ga 2015, 43, 475–482. [Google Scholar] [CrossRef]
- Leather, H.A.; Segers, P.; Berends, N.; Vandermeersch, E.; Wouters, P.F. Effects of Vasopressin on Right Ventricular Function in an Experimental Model of Acute Pulmonary Hypertension*. Crit. Care Med. 2002, 30, 2548. [Google Scholar] [CrossRef]
- Joshi, S.; Quinones Cardona, V.; Menkiti, O.R. Use of Vasopressin in Persistent Pulmonary Hypertension of the Newborn: A Case Series. SAGE Open Med. Case Rep. 2022, 10, 2050313X221102289. [Google Scholar] [CrossRef]
- Ryan, J.J.; Archer, S.L. The Right Ventricle in Pulmonary Arterial Hypertension: Disorders of Metabolism, Angiogenesis and Adrenergic Signaling in Right Ventricular Failure. Circ. Res. 2014, 115, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Kerbaul, F.; Rondelet, B.; Motte, S.; Fesler, P.; Hubloue, I.; Ewalenko, P.; Naeije, R.; Brimioulle, S. Effects of Norepinephrine and Dobutamine on Pressure Load-Induced Right Ventricular Failure*. Crit. Care Med. 2004, 32, 1035. [Google Scholar] [CrossRef]
- Piao, L.; Fang, Y.-H.; Parikh, K.S.; Ryan, J.J.; D’Souza, K.M.; Theccanat, T.; Toth, P.T.; Pogoriler, J.; Paul, J.; Blaxall, B.C.; et al. GRK2-Mediated Inhibition of Adrenergic and Dopaminergic Signaling in Right Ventricular Hypertrophy: Therapeutic Implications in Pulmonary Hypertension. Circulation 2012, 126, 2859–2869. [Google Scholar] [CrossRef] [Green Version]
- Vizza, C.D.; Rocca, G.D.; Roma, A.D.; Iacoboni, C.; Pierconti, F.; Venuta, F.; Rendina, E.; Schmid, G.; Pietropaoli, P.; Fedele, F. Acute Hemodynamic Effects of Inhaled Nitric Oxide, Dobutamine and a Combination of the Two in Patients with Mild to Moderate Secondary Pulmonary Hypertension. Crit. Care Lond. Engl. 2001, 5, 355–361. [Google Scholar] [CrossRef]
- Acosta, F.; Sansano, T.; Palenciano, C.G.; Falcon, L.; Domenech, P.; Robles, R.; Bueno, F.S.; Ramirez, P.; Parrilla, P. Effects of Dobutamine on Right Ventricular Function and Pulmonary Circulation in Pulmonary Hypertension During Liver Transplantation. Transplant. Proc. 2005, 37, 3869–3870. [Google Scholar] [CrossRef]
- Farah, A.E.; Frangakis, C.J. Studies on the Mechanism of Action of the Bipyridine Milrinone on the Heart. Basic Res. Cardiol. 1989, 84 (Suppl. 1), 85–103. [Google Scholar] [CrossRef] [PubMed]
- Honerjäger, P. Pharmacology of Bipyridine Phosphodiesterase III Inhibitors. Am. Heart J. 1991, 121, 1939–1944. [Google Scholar] [CrossRef] [PubMed]
- Alousi, A.A.; Johnson, D.C. Pharmacology of the Bipyridines: Amrinone and Milrinone. Circulation 1986, 73, III10-24. [Google Scholar]
- Alfranca, A.; Iñiguez, M.A.; Fresno, M.; Redondo, J.M. Prostanoid Signal Transduction and Gene Expression in the Endothelium: Role in Cardiovascular Diseases. Cardiovasc. Res. 2006, 70, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Rabe, K.F.; Tenor, H.; Dent, G.; Schudt, C.; Nakashima, M.; Magnussen, H. Identification of PDE Isozymes in Human Pulmonary Artery and Effect of Selective PDE Inhibitors. Am. J. Physiol.-Lung Cell Mol. Physiol. 1994, 266, L536–L543. [Google Scholar] [CrossRef]
- Eichhorn, E.J.; Konstam, M.A.; Weiland, D.S.; Roberts, D.J.; Martin, T.T.; Stransky, N.B.; Salem, D.N. Differential Effects of Milrinone and Dobutamine on Right Ventricular Preload, Afterload and Systolic Performance in Congestive Heart Failure Secondary to Ischemic or Idiopathic Dilated Cardiomyopathy. Am. J. Cardiol. 1987, 60, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.P.; Bittner, H.B.; Davis, R.D.; Van Trigt, P. Milrinone Improves Pulmonary Hemodynamics and Right Ventricular Function in Chronic Pulmonary Hypertension. Ann. Thorac. Surg. 1997, 63, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Di Santo, P.; Jung, R.G.; Marbach, J.A.; Hutson, J.; Simard, T.; Ramirez, F.D.; Harnett, D.T.; Merdad, A.; Almufleh, A.; et al. Milrinone as Compared with Dobutamine in the Treatment of Cardiogenic Shock. N. Engl. J. Med. 2021, 385, 516–525. [Google Scholar] [CrossRef]
- Toller, W.G.; Stranz, C.; Warltier, D.C. Levosimendan, a New Inotropic and Vasodilator Agent. Anesthesiology 2006, 104, 556–569. [Google Scholar] [CrossRef] [Green Version]
- Vildbrad, M.D.; Andersen, A.; Holmboe, S.; Ringgaard, S.; Nielsen, J.M.; Nielsen-Kudsk, J.E. Acute Effects of Levosimendan in Experimental Models of Right Ventricular Hypertrophy and Failure. Pulm. Circ. 2014, 4, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Kerbaul, F.; Rondelet, B.; Demester, J.-P.; Fesler, P.; Huez, S.; Naeije, R.; Brimioulle, S. Effects of Levosimendan versus Dobutamine on Pressure Load-Induced Right Ventricular Failure. Crit. Care Med. 2006, 34, 2814–2819. [Google Scholar] [CrossRef] [PubMed]
- Kerbaul, F.; Gariboldi, V.; Giorgi, R.; Mekkaoui, C.; Guieu, R.; Fesler, P.; Gouin, F.; Brimioulle, S.; Collart, F. Effects of Levosimendan on Acute Pulmonary Embolism-Induced Right Ventricular Failure. Crit. Care Med. 2007, 35, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Schwarte, L.A.; Schwartges, I.; Thomas, K.; Schober, P.; Picker, O. The Effects of Levosimendan and Glibenclamide on Circulatory and Metabolic Variables in a Canine Model of Acute Hypoxia. Intensive Care Med. 2011, 37, 701–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiklund, A.; Kylhammar, D.; Rådegran, G. Levosimendan Attenuates Hypoxia-Induced Pulmonary Hypertension in a Porcine Model. J. Cardiovasc. Pharmacol. 2012, 59, 441–449. [Google Scholar] [CrossRef]
- Revermann, M.; Schloss, M.; Mieth, A.; Babelova, A.; Schröder, K.; Neofitidou, S.; Buerkl, J.; Kirschning, T.; Schermuly, R.T.; Hofstetter, C.; et al. Levosimendan Attenuates Pulmonary Vascular Remodeling. Intensive Care Med. 2011, 37, 1368–1377. [Google Scholar] [CrossRef]
- Antila, S.; Sundberg, S.; Lehtonen, L.A. Clinical Pharmacology of Levosimendan. Clin. Pharmacokinet. 2007, 46, 535–552. [Google Scholar] [CrossRef]
- Masarone, D.; Melillo, E.; Errigo, V.; Martucci, M.L.; Pacileo, R.; Pollesello, P.; Petraio, A.; Pacileo, G. Hemodynamic Effects of Levosimendan in Outpatients With Advanced Heart Failure: An Echocardiographic Pilot Study. J. Cardiovasc. Pharmacol. 2022, 79, e36–e40. [Google Scholar] [CrossRef]
- Slawsky, M.T.; Colucci, W.S.; Gottlieb, S.S.; Greenberg, B.H.; Haeusslein, E.; Hare, J.; Hutchins, S.; Leier, C.V.; LeJemtel, T.H.; Loh, E.; et al. Acute Hemodynamic and Clinical Effects of Levosimendan in Patients With Severe Heart Failure. Circulation 2000, 102, 2222–2227. [Google Scholar] [CrossRef] [Green Version]
- Duygu, H.; Ozerkan, F.; Zoghi, M.; Nalbantgil, S.; Yildiz, A.; Akilli, A.; Akin, M.; Nazli, C.; Ergene, O. Effect of Levosimendan on Right Ventricular Systolic and Diastolic Functions in Patients with Ischaemic Heart Failure: Effect of Levosimendan on Right Ventricular Functions. Int. J. Clin. Pract. 2007, 62, 228–233. [Google Scholar] [CrossRef]
- Jiang, R.; Zhao, Q.; Wu, W.; Zhang, R.; Yuan, P.; Gong, S.; He, J.; Luo, C.; Qiu, H.; Wang, L.; et al. Efficacy and Safety of a Calcium Sensitizer, Levosimendan, in Patients with Right Heart Failure Due to Pulmonary Hypertension. Clin. Respir. J. 2018, 12, 1518–1525. [Google Scholar] [CrossRef]
- Packer, M.; Colucci, W.; Fisher, L.; Massie, B.M.; Teerlink, J.R.; Young, J.; Padley, R.J.; Thakkar, R.; Delgado-Herrera, L.; Salon, J.; et al. Effect of Levosimendan on the Short-Term Clinical Course of Patients With Acutely Decompensated Heart Failure. JACC Heart Fail. 2013, 1, 103–111. [Google Scholar] [CrossRef]
- Mebazaa, A.; Nieminen, M.S.; Packer, M.; Cohen-Solal, A.; Kleber, F.X.; Pocock, S.J.; Thakkar, R.; Padley, R.J.; Põder, P.; Kivikko, M.; et al. Levosimendan vs Dobutamine for Patients With Acute Decompensated Heart Failure: The SURVIVE Randomized Trial. JAMA 2007, 297, 1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husebye, T.; Eritsland, J.; Müller, C.; Sandvik, L.; Arnesen, H.; Seljeflot, I.; Mangschau, A.; Bjørnerheim, R.; Andersen, G.Ø. Levosimendan in Acute Heart Failure Following Primary Percutaneous Coronary Intervention-treated Acute ST-elevation Myocardial Infarction. Results from the LEAF Trial: A Randomized, Placebo-controlled Study. Eur. J. Heart Fail. 2013, 15, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Comín-Colet, J.; Manito, N.; Segovia-Cubero, J.; Delgado, J.; García Pinilla, J.M.; Almenar, L.; Crespo-Leiro, M.G.; Sionis, A.; Blasco, T.; Pascual-Figal, D.; et al. Efficacy and Safety of Intermittent Intravenous Outpatient Administration of Levosimendan in Patients with Advanced Heart Failure: The LION-HEART Multicentre Randomised Trial. Eur. J. Heart Fail. 2018, 20, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Altenberger, J.; Parissis, J.T.; Costard-Jaeckle, A.; Winter, A.; Ebner, C.; Karavidas, A.; Sihorsch, K.; Avgeropoulou, E.; Weber, T.; Dimopoulos, L.; et al. Efficacy and Safety of the Pulsed Infusions of Levosimendan in Outpatients with Advanced Heart Failure (LevoRep) Study: A Multicentre Randomized Trial. Eur. J. Heart Fail. 2014, 16, 898–906. [Google Scholar] [CrossRef]
- Houston, B.A.; Brittain, E.L.; Tedford, R.J. Right Ventricular Failure. N. Engl. J. Med. 2023, 388, 1111–1125. [Google Scholar] [CrossRef]
- Grignola, J.C.; Domingo, E. Acute Right Ventricular Dysfunction in Intensive Care Unit. BioMed Res. Int. 2017, 2017, 8217105. [Google Scholar] [CrossRef] [Green Version]
- Becher, P.M.; Goßling, A.; Schrage, B.; Twerenbold, R.; Fluschnik, N.; Seiffert, M.; Bernhardt, A.M.; Reichenspurner, H.; Blankenberg, S.; Westermann, D. Procedural Volume and Outcomes in Patients Undergoing VA-ECMO Support. Crit. Care 2020, 24, 291. [Google Scholar] [CrossRef] [PubMed]
- Klinke, A.; Schubert, T.; Müller, M.; Legchenko, E.; Zelt, J.G.E.; Shimauchi, T.; Napp, L.C.; Rothman, A.M.K.; Bonnet, S.; Stewart, D.J.; et al. Emerging Therapies for Right Ventricular Dysfunction and Failure. Cardiovasc. Diagn. Ther. 2020, 10, 1735–1767. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shah, R.; Dimmeler, S.; Freedman, J.E.; Holley, C.; Lee, J.-M.; Moore, K.; Musunuru, K.; Wang, D.-Z.; Xiao, J.; et al. Noncoding RNAs in Cardiovascular Disease: Current Knowledge, Tools and Technologies for Investigation, and Future Directions: A Scientific Statement From the American Heart Association. Circ. Genomic Precis. Med. 2020, 13, e000062. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cardiac Index | PVR | SVR | RV/PA Coupling | RV Ischemia | Remarks | |
|---|---|---|---|---|---|---|
| Volume Depletion | (↑) | - | - | (↑) | (↓) | Improves end-organ function, RV/LV interference and tricuspid regurgitation Target RAP 8–12 mmHg |
| Vasopressors | ||||||
| Norepinephrine | ↑ | ↑ | ↑↑ | ↑ | ↓ | Often used as first-line therapy |
| Low-Dose Vasopressin | ↑ | ↓ | ↑↑ | ↑ | ↓ | PA vasodilator at low dose |
| Inotropes | ||||||
| Dobutamine | (↑) | ↓ | ↓ | ↑ | ↑ | Triggers arrythmias and favors hypotension |
| PDE3 Inhibitors | ↑ | ↓ | ↓ | ↑ | ↑ | Hypotension |
| Levosimendan | ↑ | ↓ | ↓ | ↑ | ↓ | Hypotension |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dayer, N.; Ltaief, Z.; Liaudet, L.; Lechartier, B.; Aubert, J.-D.; Yerly, P. Pressure Overload and Right Ventricular Failure: From Pathophysiology to Treatment. J. Clin. Med. 2023, 12, 4722. https://doi.org/10.3390/jcm12144722
Dayer N, Ltaief Z, Liaudet L, Lechartier B, Aubert J-D, Yerly P. Pressure Overload and Right Ventricular Failure: From Pathophysiology to Treatment. Journal of Clinical Medicine. 2023; 12(14):4722. https://doi.org/10.3390/jcm12144722
Chicago/Turabian StyleDayer, Nicolas, Zied Ltaief, Lucas Liaudet, Benoit Lechartier, John-David Aubert, and Patrick Yerly. 2023. "Pressure Overload and Right Ventricular Failure: From Pathophysiology to Treatment" Journal of Clinical Medicine 12, no. 14: 4722. https://doi.org/10.3390/jcm12144722
APA StyleDayer, N., Ltaief, Z., Liaudet, L., Lechartier, B., Aubert, J. -D., & Yerly, P. (2023). Pressure Overload and Right Ventricular Failure: From Pathophysiology to Treatment. Journal of Clinical Medicine, 12(14), 4722. https://doi.org/10.3390/jcm12144722