



Pulmonary Function Tests in the Evaluation of Early Lung Disease in Cystic Fibrosis
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Pulmonary Function Measurements
2.3. HRQoL Measurement
2.4. Statistical Analysis
3. Results
3.1. Study Group
3.2. Correlation of Spirometry with MBNW and IOS
3.3. Correlation of PFTs with a Respiratory Infection
3.4. Correlation of PFTs with Pulmonary Exacerbation
3.5. Correlation of PFT Results with HRQoL
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esther, C.R., Jr.; Muhlebach, M.S.; Ehre, C.; Hill, D.B.; Wolfgang, M.C.; Kesimer, M.; Ramsey, K.A.; Markovetz, M.R.; Garbarine, I.C.; Forest, M.G.; et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med. 2019, 11, eaav3488. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.C.; Hall, G.L.; Sly, P.D.; Stick, S.M.; Douglas, T.A.; AREST-CF. Early Lung Disease in Infants and Preschool Children with Cystic Fibrosis. What Have We Learned and What Should We Do about It? Am. J. Respir. Crit.Care Med. 2017, 195, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Thia, L.P.; Hoo, A.-F.; Bush, A.; Aurora, P.; Wade, A.; Chudleigh, J.; Lum, S.; Stocks, J. Evolution of lung function during the first year of life in newborn screened cystic fibrosis infants. Thorax 2014, 69, 910–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, A.; Sly, P.D. Evolution of cystic fibrosis lung function in the early years. Curr. Opin. Pulm. Med. 2015, 21, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Bayfield, K.J.; Douglas, T.A.; Rosenow, T.; Davies, J.C.; Elborn, S.J.; Mall, M.; Paproki, A.; Ratjen, F.; Sly, P.D.; Smyth, A.R.; et al. Time to get serious about the detection and monitoring of early lung disease in cystic fibrosis. Thorax 2021, 76, 1255–1265. [Google Scholar] [CrossRef]
- Kraemer, R.; Baldwin, D.N.; Ammann, R.A.; Frey, U.; Gallati, S. Progression of pulmonary hyperinflation and trapped gas associated with genetic and environmental factors in children with cystic fibrosis. Respir. Res. 2006, 7, 138. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.-R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Breuer, O.; Caudri, D.; Stick, S.; Turkovic, L. Predicting disease progression in cystic fibrosis. Expert Rev. Respir. Med. 2018, 12, 905–917. [Google Scholar] [CrossRef]
- Tiddens, H.A.; Donaldson, S.H.; Rosenfeld, M.; Paré, P.D. Cystic fibrosis lung disease starts in the small airways: Can we treat it more effectively? Pediatr. Pulmonol. 2010, 45, 107–117. [Google Scholar] [CrossRef]
- Aurora, P.; Bush, A.; Gustafsson, P.; Oliver, C.; Wallis, C.; Price, J.; Stroobant, J.; Carr, S.; Stocks, J. Multiple-breath washout as a marker of lung disease in preschool children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 249–256. [Google Scholar] [CrossRef]
- Kent, L.; Reix, P.; Innes, J.; Zielen, S.; Le Bourgeois, M.; Braggion, C.; Lever, S.; Arets, H.; Brownlee, K.; Bradley, J.; et al. Lung clearance index: Evidence for use in clinical trials in cystic fibrosis. J. Cyst. Fibros. 2014, 13, 123–138. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, K.A.; Rosenow, T.; Turkovic, L.; Skoric, B.; Banton, G.; Adams, A.-M.; Simpson, S.J.; Murray, C.; Ranganathan, S.C.; Stick, S.M.; et al. Lung Clearance Index and Structural Lung Disease on Computed Tomography in Early Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Horsley, A.R.; Belcher, J.; Bayfield, K.; Bianco, B.; Cunningham, S.; Fullwood, C.; Jones, A.; Shawcross, A.; Smith, J.A.; Maitra, A.; et al. Longitudinal assessment of lung clearance index to monitor disease progression in children and adults with cystic fibrosis. Thorax 2022, 77, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, K.; Agrawal, A. Impulse oscillometry: The state-of-art for lung function testing. Lung India 2016, 33, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Gangell, C.L.; Horak, F., Jr.; Patterson, H.J.; Sly, P.D.; Stick, S.M.; Hall, G.L. Respiratory impedance in children with cystic fibrosis using forced oscillations in clinic. Eur. Respir. J. 2007, 30, 892–897. [Google Scholar] [CrossRef]
- Evans, D.J.; Schultz, A.; Verheggen, M.; Hall, G.L.; Simpson, S.J. Identifying pediatric lung disease: A comparison of forced oscillation technique outcomes. Pediatr. Pulmonol. 2019, 54, 751–758. [Google Scholar] [CrossRef]
- Ramsey, K.A.; Ranganathan, S.C.; Gangell, C.L.; Turkovic, L.; Park, J.; Skoric, B.; Stick, S.M.; Sly, P.D.; Hall, G.L. Impact of lung disease on respiratory impedance in young children with cystic fibrosis. Eur. Respir. J. 2015, 46, 1672–1679. [Google Scholar] [CrossRef] [Green Version]
- Spano, J.; Milla, C.E. Defining the Clinical Utility of the Lung Clearance Index. Are We There Yet? Am. J. Respir. Crit. Care Med. 2021, 203, 937–939. [Google Scholar] [CrossRef]
- Castellani, C.; Duff, A.J.; Bell, S.C.; Heijerman, H.G.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.M.; Rosenstein, B.J.; White, T.B.; Accurso, F.J.; Castellani, C.; Cutting, G.R.; Durie, P.R.; LeGrys, V.A.; Massie, J.; Parad, R.B.; et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic fibrosis foundation consensus report. J. Pediatr. 2008, 153, S4–S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boeck, K.; Wilschanski, M.; Castellani, C.; Taylor, C.; Cuppens, H.; Dodge, J.; Sinaasappel, M. Cystic fibrosis: Terminology and diagnostic algorithms. Thorax 2006, 61, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Beydon, N.; Davis, S.D.; Lombardi, E.; Allen, J.L.; Arets, H.G.M.; Aurora, P.; Bisgaard, H.; Davis, G.M.; Ducharme, F.M.; Eigen, H.; et al. An official American Thoracic Society/European Respiratory Society statement: Pulmonary function testing in preschool children. Am. J. Respir. Crit. Care Med. 2007, 175, 1304–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; Van Der Grinten, C.P.M.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, P.D.; Latzin, P.; Verbanck, S.; Hall, G.L.; Horsley, A.; Gappa, M.; Thamrin, C.; Arets, H.G.M.; Aurora, P.; Fuchs, S.I.; et al. Consensus statement for inert gas washout measurement using multiple- and single-breath tests. Eur. Respir. J. 2013, 41, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, G.G.; Bates, J.; Berger, K.I.; Calverley, P.; De Melo, P.L.; Dellacà, R.L.; Farre, R.; Hall, G.; Ioan, I.; Irvin, C.G.; et al. Technical standards for respiratory oscillometry. Eur. Respir. J. 2020, 55, 1900753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quittner, A.L.; Buu, A.; Messer, M.A.; Modi, A.C.; Watrous, M. Development and validation of The Cystic Fibrosis Questionnaire in the United States: A health-related quality-of-life measure for cystic fibrosis. Chest 2005, 128, 2347–2354. [Google Scholar] [CrossRef]
- Sands, D.; Borawska-Kowalczyk, U. Polska adaptacja Kwestionariusza Jakości Życia przeznaczonego dla dzieci i dorosłych chorych na mukowiscydozę oraz ich rodziców (CFQ-R). Pediatr. Polska. 2009, 84, 165–172. [Google Scholar] [CrossRef]
- Quittner, A.L.; Modi, A.C.; Wainwright, C.; Otto, K.; Kirihara, J.; Montgomery, A.B. Determination of the Minimal Clinically Important Difference Scores for the Cystic Fibrosis Questionnaire-Revised Respiratory Symptom Scale in Two Populations of Patients with Cystic Fibrosis and Chronic Pseudomonas aeruginosa Airway Infection. Chest 2009, 135, 1610–1618. [Google Scholar] [CrossRef] [Green Version]
- Walicka-Serzysko, K.; Postek, M.; Sands, D. Silent lung zone—Application of multi-breath nitrogen washout test (MBNW) in the diagnosis of small airways diseases in children—Preliminary report based on literature and own experience. Dev. Period Med. 2017, 21, 369–379. [Google Scholar]
- Aurora, P.; Gustafsson, P.; Bush, A.; Lindblad, A.; Oliver, C.; Wallis, C.E.; Stocks, J. Multiple breath inert gas washout as a measure of ventilation distribution in children with cystic fibrosis. Thorax 2004, 59, 1068–1073. [Google Scholar] [CrossRef] [Green Version]
- Stanojevic, S.; Davis, S.D.; Retsch-Bogart, G.; Webster, H.; Davis, M.; Johnson, R.C.; Jensen, R.; Pizarro, M.E.; Kane, M.; Clem, C.C.; et al. Progression of Lung Disease in Preschool Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, 1216–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayment, J.H.; Stanojevic, S.; Davis, S.D.; Retsch-Bogart, G.; Ratjen, F. Lung clearance index to monitor treatment response in pulmonary exacerbations in preschool children with cystic fibrosis. Thorax 2018, 73, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Walicka-Serzysko, K.; Postek, M.; Milczewska, J.; Sands, D. Change in lung clearance index with microbiological status in children with cystic fibrosis. Pediatr. Pulmonol. 2019, 54, 729–736. [Google Scholar] [CrossRef]
- Walicka-Serzysko, K.; Postek, M.; Milczewska, J.; Sands, D. Lung function deterioration in school children with cystic fibrosis. Pediatr. Pulmonol. 2020, 55, 3030–3038. [Google Scholar] [CrossRef] [PubMed]
- De Boer, K.; Vandemheen, K.L.; Tullis, E.; Doucette, S.; Fergusson, D.; Freitag, A.; Paterson, N.; Jackson, M.; Lougheed, M.D.; Kumar, V.; et al. Exacerbation frequency and clinical outcomes in adult patients with cystic fibrosis. Thorax 2011, 66, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Waters, V.; Ratjen, F. Pulmonary Exacerbations in Children with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S2), S200–S2006. [Google Scholar] [CrossRef]
- Sanders, D.B.; Bittner, R.C.L.; Rosenfeld, M.; Hoffman, L.R.; Redding, G.J.; Goss, C.H. Failure to recover to baseline pulmonary function after cystic fibrosis pulmonary exacerbation. Am. J. Respir. Crit. Care Med. 2010, 182, 627–632. [Google Scholar] [CrossRef] [Green Version]
- Sanders, D.B.; Bittner, R.C.; Rosenfeld, M.; Redding, G.J.; Goss, C.H. Pulmonary exacerbations are associated with subsequent FEV1 decline in both adults and children with cystic fibrosis. Pediatr. Pulmonol. 2011, 46, 393–400. [Google Scholar] [CrossRef]
- Waters, V.; Stanojevic, S.; Atenafu, E.G.; Lu, A.; Yau, Y.; Tullis, E.; Ratjen, F. Effect of pulmonary exacerbations on long-term lung function decline in cystic fibrosis. Eur. Respir. J. 2012, 40, 61–66. [Google Scholar] [CrossRef]
- Heltshe, S.L.; Goss, C.H.; Thompson, V.; Sagel, S.D.; Sanders, D.B.; Marshall, B.C.; Quon, B. Short-term and long-term response to pulmonary exacerbation treatment in cystic fibrosis. Thorax 2016, 71, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Sanders, D.B.; Hoffman, L.R.; Emerson, J.; Gibson, R.L.; Rosenfeld, M.; Redding, G.J.; Goss, C.H. Return of FEV1 after pulmonary exacerbation in children with cystic fibrosis. Pediatr. Pulmonol. 2010, 45, 127–134. [Google Scholar] [CrossRef]
- Perrem, L.; Stanojevic, S.; Shaw, M.; Jensen, R.; McDonald, N.; Isaac, S.M.; Davis, M.; Clem, C.; Guido, J.; Jara, S.; et al. Lung Clearance Index to Track Acute Respiratory Events in School-Age Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 203, 977–986. [Google Scholar] [CrossRef]
- VanDevanter, E.J.; Heltshe, S.L.; Skalland, M.; Lechtzin, N.; Nichols, D.; Goss, C.H. The effect of oral and intravenous antimicrobials on pulmonary exacerbation recovery in cystic fibrosis. J. Cyst. Fibros. 2021, 20, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Lucy, P.; Sanja, S.; Melinda, S.; Hartmut, G.; Neil, S.; Valerie, W.; Sanders, D.B.; Davis, S.D.; Ratjen, F. Evaluation of clinically relevant changes in the lung clearance index in children with cystic fibrosis and healthy controls. Thorax 2023, 78, 362. [Google Scholar]
- Stanojevic, S.; Davis, S.D.; Perrem, L.; Shaw, M.; Retsch-Bogart, G.; Davis, M.; Jensen, R.; Clem, C.C.; Isaac, S.M.; Guido, J.; et al. Determinants of lung disease progression measured by lung clearance index in children with cystic fibrosis. Eur. Respir. J. 2021, 58, 2003380. [Google Scholar] [CrossRef]
- Wojsyk-Banaszak, I.; Więckowska, B.; Stachowiak, Z.; Kycler, M.; Szczepankiewicz, A. Predictive value of impulse oscillometry and multiple breath washout parameters in pediatric patients with cystic fibrosis pulmonary exacerbation. Pediatr. Pulmonol. 2022, 57, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Sakarya, A.; Uyan, Z.S.; Baydemir, C.; Anık, Y.; Erdem, E.; Gokdemir, Y.; Karadag, B.; Karakoc, F.; Ersu, R. Evaluation of children with cystic fibrosis by impulse oscillometry when stable and at exacerbation. Pediatr. Pulmonol. 2016, 51, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Cheney, J.; Vidmar, S.; Gailer, N.; Wainwright, C.; Douglas, T.A.; group ACFBLAs. Health-related quality-of-life in children with cystic fibrosis aged 5-years and associations with health outcomes. J. Cyst. Fibros. 2020, 19, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quittner, A.L.; Sawicki, G.S.; McMullen, A.; Rasouliyan, L.; Pasta, D.J.; Yegin, A.; Konstan, M.W. Psychometric evaluation of the Cystic Fibrosis Questionnaire-Revised in a national sample. Qual. Life Res. 2012, 21, 1267–1278. [Google Scholar] [CrossRef]
- Sawicki, G.S.; Rasouliyan, L.; McMullen, A.H.; Wagener, J.S.; McColley, S.A.; Pasta, D.J.; Quittner, A.L. Longitudinal assessment of health-related quality of life in an observational cohort of patients with cystic fibrosis. Pediatr. Pulmonol. 2010, 46, 36–44. [Google Scholar] [CrossRef]
- Van Horck, M.; Winkens, B.; Wesseling, G.; de Winter-de Groot, K.; de Vreede, I.; Jöbsis, Q.; Dompeling, E. Factors associated with changes in health-related quality of life in children with cystic fibrosis during 1-year follow-up. Eur. J. Pediatr. 2017, 176, 1047–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, F.; Proesmans, M.; Boon, M.; Havermans, T.; De Boeck, K. Lung clearance index predicts pulmonary exacerbations in young patients with cystic fibrosis. Thorax 2014, 69, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’neill, K.; Tunney, M.M.; Johnston, E.; Rowan, S.; Downey, D.G.; Rendall, J.; Reid, A.; Bradbury, I.; Elborn, J.S.; Bradley, J.M. Lung Clearance Index in Adults and Children with Cystic Fibrosis. Chest 2016, 150, 1323–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattie, P.; Ranganathan, S.; Harrison, J.; Vidmar, S.; Hall, G.L.; Foong, R.E.; Harper, A.; Ramsey, K.; Wurzel, D. Quality of life is poorly correlated to lung disease severity in school-aged children with cystic fibrosis. J. Cyst. Fibros. 2022, 21, e188–e203. [Google Scholar] [CrossRef] [PubMed]
- Perrem, L.; Stanojevic, S.; Shaw, M.; Davis, S.; Retsch-Bogart, G.; Ratjen, F. Changes in the parent cystic fibrosis questionnaire-revised (CFQ-R) with respiratory symptoms in preschool children with cystic fibrosis. J. Cyst. Fibros. 2020, 19, 492–498. [Google Scholar] [CrossRef]


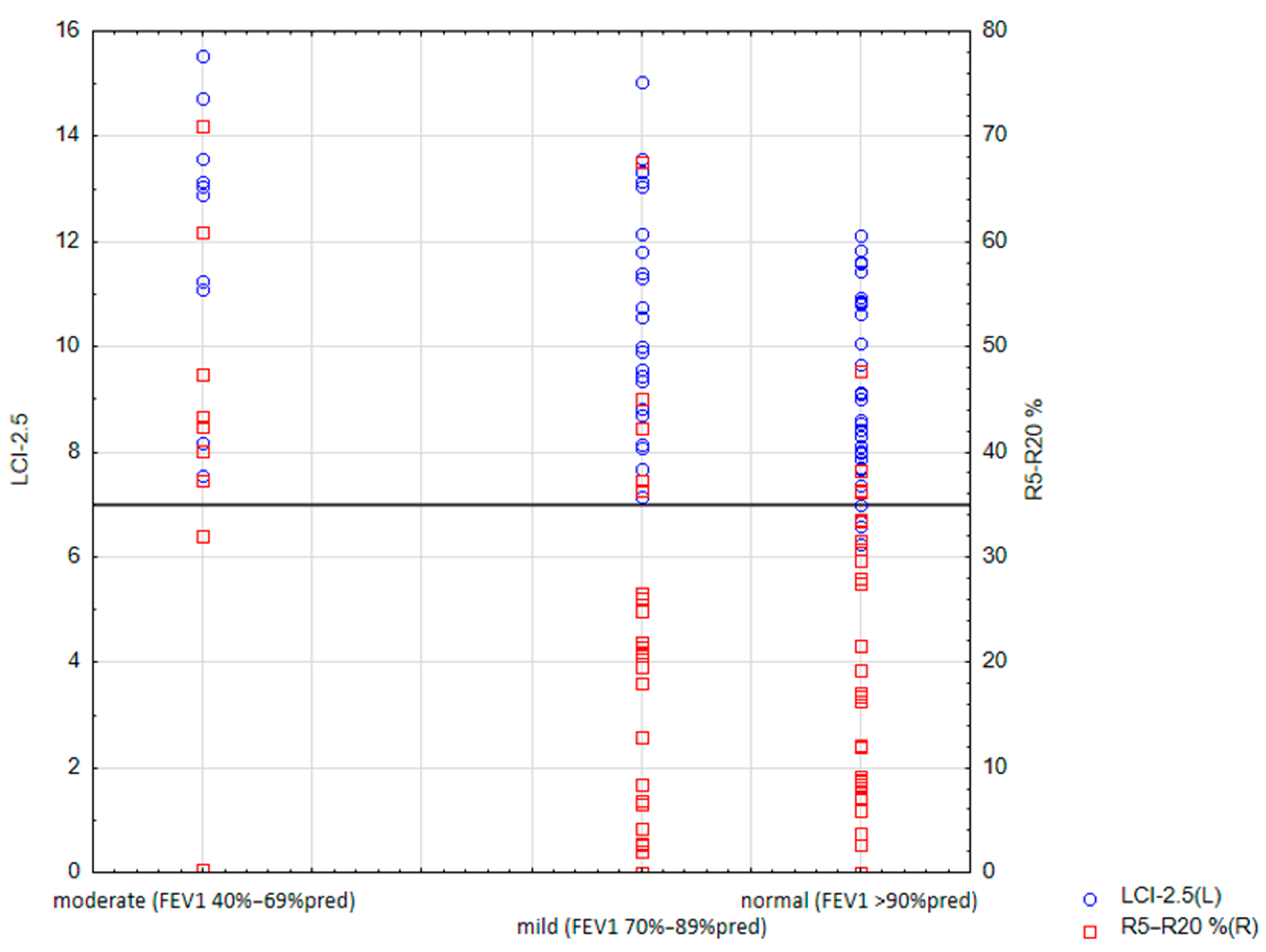{kind=link}
| Number of Patients | 69 |
| Age in years, mean ± SD | 14.09 ± 3.26 |
| Male/female | 32/37 |
| Weight z-score | −0.09 ± 0.82 |
| Height z-score | 0.25 ± 0.88 |
| BMI, mean ± SD | 18.56 ± 2.99 |
| BMI z-score mean ± SD | −0.31 ± 0.80 |
| F508del homozygous, n (%) | 26 (38%) |
| F508del heterozygous | 34 (49%) |
| Other/other | 9 (13%) |
| MSSA infection | 53 (77%) |
| Pseudomonas aeruginosa infection | 16 (23%) |
| Aspergillus fumigatus infection | 8 (12%) |
| Scedosporium apiospermum infection | 5 (7%) |
| FEV1 z-score < −1.64 | 23 (33%) |
| FEV1 z-score, mean ± SD | −1.15 ± 1.5 |
| FEV1 %predicted, mean ± SD | 85.72 ± 17.66 |
| LCI 2.5%, mean ± SD | 9.86 ± 2.50 |
| MBNW Parameters | IOS Parameters | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FEV1 | N (%) | LCI > 6.98 N (%) | LCI Mean [Range] | R5Hz%Pred Mean ± SD | R5Hz-20Hz%Pred Mean ± SD | AX Mean ± SD | Fres Mean ± SD | X20Hz Mean ± SD | X20Hz%Pred Mean ± SD | X5Hz Mean ± SD | X5Hz%Pred Mean ± SD |
| FEV1 ≥ 90%pred | 34 (49.3) | 29 (85) | 8.97; [6.25–12.11] | 92.57 ± 36.11 | 17.32 ± 13.68 | 0.64 ± 0.52 | 13.75 ± 4.58 | 0.07 ± 0.05 | 111.33 ± 91.89 | −0.15 ± 0.076 | 115.33 ± 50.98 |
| FEV1 70–89%pred | 25 (36.2) | 25 (100) | 10.53; [7.14–15.04] | 110.17 ± 44.59 | 20.66 ± 16.75 | 0.82 ± 0.97 | 14.67 ± 5.30 | 0.05 ± 0.06 | 66.74 ± 96.52 | −0.15 ± 0.11 | 150.65 ± 91.61 |
| FEV1 40–69%pred | 10 (14.5) | 10 (100) | 12.10; [7.54–15.54] | 129.33 ± 49.16 | 41.65 ± 19.66 | 1.71 ± 0.94 | 19.58 ± 4.70 | 0.001 ± 0.06 | −12.56 ± 118.43 | −0.28 ± 0.10 | 240.11 ± 128.29 |
| FEV1 < 40%pred | - | - | - | - | - | - | - | - | - | - | - |
| FEV1/FVC z-score < −1.96 | 19 (28) | - | - | - | - | - | - | - | - | - | - |
| p FEV1 ≥ 90%pred vs. FEV1 40–69%pred | - | - | 0.003 | 0.016 | 0.003 | 0.004 | 0.011 | 0.02 | 0.02 | 0.004 | < 0.001 |
| p FEV1 ≥ 90%pred vs FEV1 70–89%pred | - | - | 0.033 | - | - | - | - | - | - | - | - |
| LCI > 6.98 | 63 (92.6) | - | - | - | - | - | - | - | - | - | - |
| Parameter | RSpearmana (p < 0.05) |
|---|---|
| FEV1/FVC %pred | 0.040 |
| FEV1/FVC z-score | 0.037 |
| MEF25% | 0.021 |
| MEF25 z-score | 0.020 |
| R at 5 Hz %pred | 0.039 |
| LCI-2.5 | 0.001 |
| 0 PEx | 1–3 PExs | >3 PExs | |
|---|---|---|---|
| PEx oral | 2 | 45 | 22 |
| PEx iv | 35 | 26 | 8 |
| PEx oral + iv | 2 | 33 | 34 |
| Parameter | FEV1 %pred | FEV1 z-Score | FVC %pred | FEV1/FVC %pred | FEV1/FVC z-Score | MEF25 %pred | MEF25 z-Score | MEF50 %pred | MEF50 z-Score | X at 20 Hz %pred | X at 5 Hz %pred | LCI-2.5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rspearmana (p < 0.05) | −0.38 | −0.39 | −0.28 | −0.31 | −0.32 | −0.42 | −0.42 | −0.38 | −0.37 | −0.26 | 0.34 | 0.57 |
| Group/RSpearman | Rtot | Rtot z-Score | sReff %pred | sReff z-Score | Reff | Reff %pred | Reff z-Score | FRC %pred | FRC z-Score | TLC %pred | TLC z-Score | RV%TLC | RV/TLC %pred | RV/TLC z-Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PEx oral | −0.03 | 0.03 | 0.14 | 0.17 | 0.04 | 0.10 | 0.12 | 0.23 | 0.24 | 0.17 | 0.16 | 0.22 | 0.21 | 0.20 |
| PEx iv | 0.27 * | 0.26 * | 0.36 * | 0.41 * | 0.32 * | 0.27 * | 0.31 * | 0.24 | 0.21 | 0.22 | 0.21 | 0.38 * | 0.42 * | 0.34 * |
| Parameter | p 0–3 PEx iv vs. >3 PEx iv |
|---|---|
| FEV1 z-score | 0.044 |
| FVC %pred | 0.049 |
| MEF25 %pred | 0.031 |
| MEF25 z-score | 0.030 |
| X at 5 Hz %pred | 0.047 |
| LCI-2.5 | <0.001 |
| Parameter | p 0–3 PExs vs. >3 PExs |
|---|---|
| sReff % | 0.020 |
| sReff z-score | 0.011 |
| X at 20 Hz %pred | 0.040 |
| LCI-2.5 | 0.001 |
| CFQ-R | ||||||||
|---|---|---|---|---|---|---|---|---|
| Physical | Emotional State | Social | Body Imagine | Eating | Treatment Burden | Respiratory | Digestion | |
| M ± SD | 85.31 ± 16.4 | 74.14 ± 15.5 | 67.82 ± 18.2 | 74.88 ± 24.1 | 79.39 ± 22.9 | 64.4 ± 16.1 | 77.86 ± 16.4 | 83.09 ± 17.4 |
| MIN | 38.89 | 26.67 | 19.05 | 0 | 0 | 33.33 | 22.22 | 33.33 |
| MAX | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| CFQ-R | Parameter | RSpearmana (p < 0.05) |
|---|---|---|
| Physical | FEV1/FVC %pred | 0.28 |
| FEV1/FVC z-score | 0.28 | |
| MEF25%pred | 0.24 | |
| MEF25 z-score | 0.24 | |
| Respiratory | R at 20 Hz | −0.25 |
| Treatment burden | R at 20 Hz | 0.32 |
| R at 5 Hz | 0.32 | |
| X at 5 Hz | −0.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walicka-Serzysko, K.; Postek, M.; Borawska-Kowalczyk, U.; Milczewska, J.; Sands, D. Pulmonary Function Tests in the Evaluation of Early Lung Disease in Cystic Fibrosis. J. Clin. Med. 2023, 12, 4735. https://doi.org/10.3390/jcm12144735
Walicka-Serzysko K, Postek M, Borawska-Kowalczyk U, Milczewska J, Sands D. Pulmonary Function Tests in the Evaluation of Early Lung Disease in Cystic Fibrosis. Journal of Clinical Medicine. 2023; 12(14):4735. https://doi.org/10.3390/jcm12144735
Chicago/Turabian StyleWalicka-Serzysko, Katarzyna, Magdalena Postek, Urszula Borawska-Kowalczyk, Justyna Milczewska, and Dorota Sands. 2023. "Pulmonary Function Tests in the Evaluation of Early Lung Disease in Cystic Fibrosis" Journal of Clinical Medicine 12, no. 14: 4735. https://doi.org/10.3390/jcm12144735
APA StyleWalicka-Serzysko, K., Postek, M., Borawska-Kowalczyk, U., Milczewska, J., & Sands, D. (2023). Pulmonary Function Tests in the Evaluation of Early Lung Disease in Cystic Fibrosis. Journal of Clinical Medicine, 12(14), 4735. https://doi.org/10.3390/jcm12144735