
Targeting IRAK4 with Emavusertib in Lymphoma Models with Secondary Resistance to PI3K and BTK Inhibitors

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Lines
2.2. Treatments
2.3. Flow Cytometry
2.4. Immunofluorescence and Confocal Microscopy
3. Results
3.1. The IRAK4 Inhibitor Emavusertib Is Beneficial in MYD88 Mutated Lymphoma Cells
3.2. Emavusertib Increases Sensitivity to PI3K and BTK Inhibitors in Resistant Models of MZL
3.3. Emavusertib Affects Proliferation and Induces Apoptosis in Both Sensitive and Resistant Marginal Zone Lymphoma Models
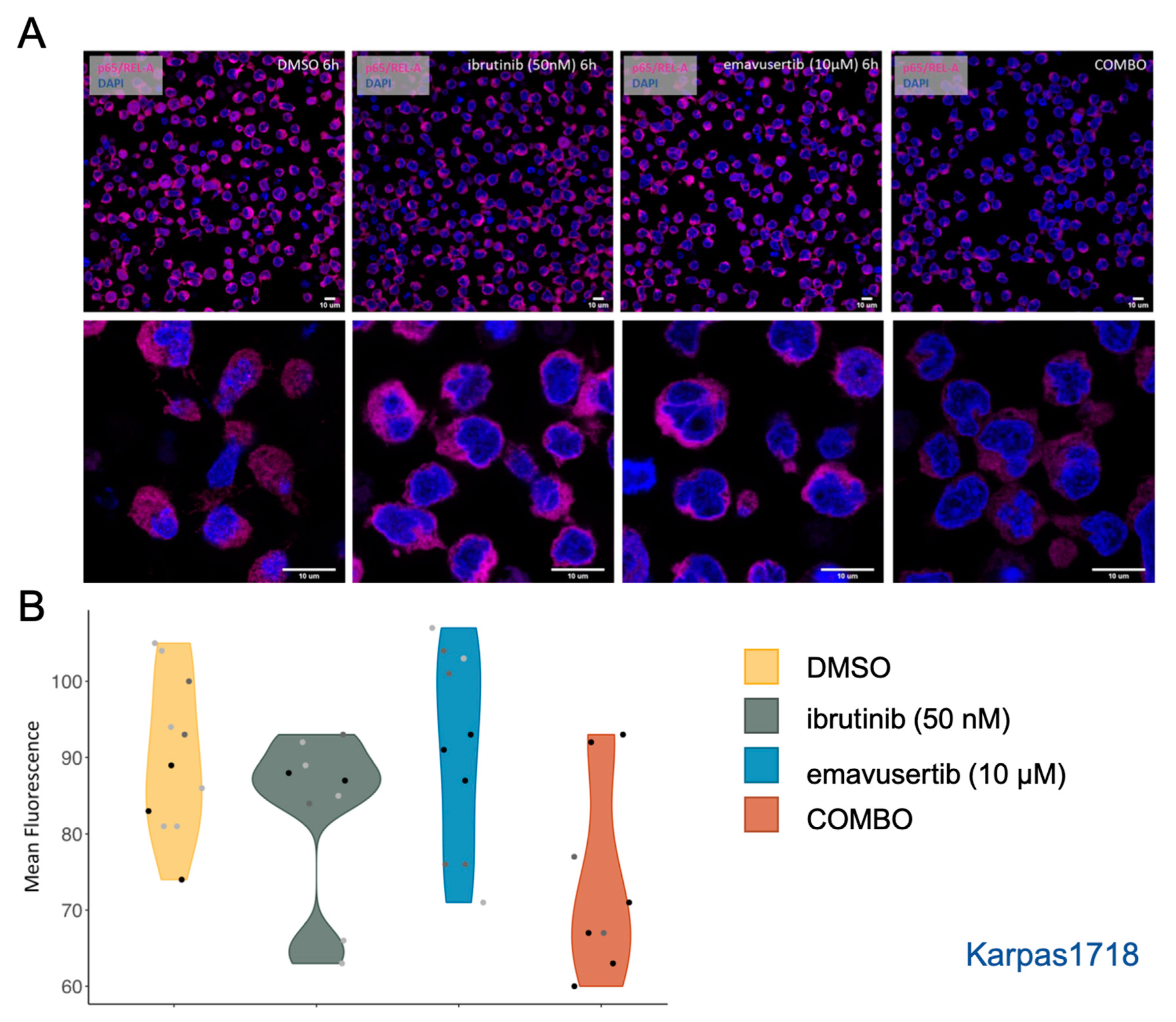
3.4. Emavusertib Reduces Total and Nuclear REL-A in MZL Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dreyling, M.; Santoro, A.; Santoro, A.; Mollica, L.; Leppa, S.; Follows, G.; Lenz, G.; Kim, W.S.; Nagler, A.; Dimou, M.; et al. Long-term safety and efficacy of the PI3K inhibitor copanlisib in patients with relapsed or refractory indolent lymphoma: 2-year follow-up of the CHRONOS-1 study. Am. J. Hematol. 2020, 95, 362–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, A.; de Vos, S.; Coleman, M.; Martin, P.; Flowers, C.R.; Thieblemont, C.; Morschhauser, F.; Collins, G.P.; Ma, S.; Peles, S.; et al. Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: Long-term follow-up and biomarker analysis. Blood Adv. 2020, 4, 5773–5784. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Sanz, R.G.; et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: The ASPEN study. Blood 2020, 136, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Johnston, N.D.; Schuster, S.J.; de Vos, S.; Salles, G.; Jurczak, W.J.; Flowers, C.R.; Viardot, A.; Flinn, I.W.; Martin, P.; Xing, G.; et al. Outcomes of patients with up to 6 years of follow-up from a phase 2 study of idelalisib for relapsed indolent lymphomas. Leuk. Lymphoma 2021, 62, 1077–1087. [Google Scholar] [CrossRef]
- Panayiotidis, P.; Follows, G.A.; Mollica, L.; Nagler, A.; Özcan, M.; Santoro, A.; Stevens, D.; Trevarthen, D.; Hiemeyer, F.; Garcia-Vargas, J.; et al. Efficacy and safety of copanlisib in patients with relapsed or refractory marginal zone lymphoma. Blood Adv. 2021, 5, 823–828. [Google Scholar] [CrossRef]
- Davids, M.S.; O’Connor, O.A.; Jurczak, W.; Samaniego, F.; Fenske, T.S.; Zinzani, P.L.; Patel, M.R.; Ghosh, N.; Cheson, B.D.; Derenzini, E.; et al. Integrated safety analysis of umbralisib, a dual PI3Kδ/CK1ε inhibitor, in relapsed/refractory lymphoid malignancies. Blood Adv. 2021, 5, 5332–5343. [Google Scholar] [CrossRef]
- Trotman, J.; Buske, C.; Tedeschi, A.; Matous, J.V.; MacDonald, D.; Tam, C.S.; Tournilhac, O.; Ma, S.; Treon, S.P.; Oriol, A.; et al. Single-Agent Ibrutinib for Rituximab-Refractory Waldenström Macroglobulinemia: Final Analysis of the Substudy of the Phase III Innovate(TM) Trial. Clin. Cancer Res. 2021, 27, 5793–5800. [Google Scholar] [CrossRef]
- Bertoni, F.; Rossi, D.; Raderer, M.; Zucca, E. Marginal Zone Lymphomas. Cancer J. 2020, 26, 336–347. [Google Scholar] [CrossRef]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK signalling in cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]
- Li, Z.M.; Rinaldi, A.; Cavalli, A.; Mensah, A.A.; Ponzoni, M.; Gascoyne, R.D.; Bhagat, G.; Zucca, E.; Bertoni, F. MYD88 somatic mutations in MALT lymphomas. Br. J. Haematol. 2012, 158, 662–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascione, L.; Rinaldi, A.; Bruscaggin, A.; Tarantelli, C.; Arribas, A.J.; Kwee, I.; Pecciarini, L.; Mensah, A.A.; Spina, V.; Chung, E.Y.L.; et al. Novel insights into the genetics and epigenetics of MALT lymphoma unveiled by next generation sequencing analyses. Haematologica 2019, 104, e558–e561. [Google Scholar] [CrossRef]
- Yan, Q.; Huang, Y.; Watkins, A.J.; Kocialkowski, S.; Zeng, N.; Hamoudi, R.A.; Isaacson, P.G.; de Leval, L.; Wotherspoon, A.; Du, M.Q. BCR and TLR signaling pathways are recurrently targeted by genetic changes in splenic marginal zone lymphomas. Haematologica 2012, 97, 595–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Watanabe, N.; Tzioni, M.M.; Akarca, A.; Zhang, C.; Li, Y.; Chen, Z.; Cucco, F.; Carmell, N.; Noh, J.Y.; et al. Thyroid MALT lymphoma: Self-harm to gain potential T-cell help. Leukemia 2021, 35, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef] [PubMed]
- de Leval, L.; Alizadeh, A.A.; Bergsagel, P.L.; Campo, E.; Davies, A.; Dogan, A.; Fitzgibbon, J.; Horwitz, S.M.; Melnick, A.M.; Morice, W.G.; et al. Genomic profiling for clinical decision making in lymphoid neoplasms. Blood 2022, 140, 2193–2227. [Google Scholar] [CrossRef]
- Bonfiglio, F.; Bruscaggin, A.; Guidetti, F.; Terzi di Bergamo, L.; Faderl, M.; Spina, V.; Condoluci, A.; Bonomini, L.; Forestieri, G.; Koch, R.; et al. Genetic and phenotypic attributes of splenic marginal zone lymphoma. Blood 2022, 139, 732–747. [Google Scholar] [CrossRef]
- Saikh, K.U. MyD88 and beyond: A perspective on MyD88-targeted therapeutic approach for modulation of host immunity. Immunol. Res. 2021, 69, 117–128. [Google Scholar] [CrossRef]
- Erika Shiratori, M.I.; Ohtaka, M.; Nogami, S.; Tohda, S. Mechanisms of Suppressive Effects of MYD88 Inhibitors on the Growth of Lymphoma and Leukemia Cells. Blood 2016, 128, 2773. [Google Scholar] [CrossRef]
- Zhang, J.; Fu, L.; Shen, B.; Liu, Y.; Wang, W.; Cai, X.; Kong, L.; Yan, Y.; Meng, R.; Zhang, Z.; et al. Assessing IRAK4 Functions in ABC DLBCL by IRAK4 Kinase Inhibition and Protein Degradation. Cell Chem. Biol. 2020, 27, 1500–1509.e1513. [Google Scholar] [CrossRef]
- Chen, Y.; Bai, G.; Ning, Y.; Cai, S.; Zhang, T.; Song, P.; Zhou, J.; Duan, W.; Ding, J.; Xie, H.; et al. Design and synthesis of Imidazo[1,2-b]pyridazine IRAK4 inhibitors for the treatment of mutant MYD88 L265P diffuse large B-cell lymphoma. Eur. J. Med. Chem. 2020, 190, 112092. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; Romero, D.L.; Yang, Y.; Shaffer, A.L., 3rd; Chaudhary, D.; Robinson, S.; Miao, W.; Rui, L.; Westlin, W.F.; Kapeller, R.; et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 2015, 212, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
- Gummadi, V.R.; Boruah, A.; Ainan, B.R.; Vare, B.R.; Manda, S.; Gondle, H.P.; Kumar, S.N.; Mukherjee, S.; Gore, S.T.; Krishnamurthy, N.R.; et al. Discovery of CA-4948, an Orally Bioavailable IRAK4 Inhibitor for Treatment of Hematologic Malignancies. ACS Med. Chem. Lett. 2020, 11, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- Booher, R.N.; Samson, M.E.S.; Borek, M.; Modafferi, H.; Martell, R.E. PS991. CA-4948, an IRAK4/FLT3 inhibitor, shows antileukemic activity in mouse models of FLT3 wild-type and FLT3 mutated Acute Myeloid Leukemia (AML). Hemasphere 2019, 3, 445–446. [Google Scholar] [CrossRef]
- von Roemeling, C.A.; Doonan, B.P.; Hoang-Minh, L.; Tun, H.W.; Martinez, E.; Soikes, R.; von Roemeling, R.; Mitchell, D.A. Abstract P243: The IRAK4 inhibitor CA-4948 demonstrates antitumor activity in a preclinical model of CNS lymphoma. Mol. Cancer Ther. 2021, 20, P243. [Google Scholar] [CrossRef]
- Booher, R.N.; Samson, M.E.; Xu, G.-X.; Cheng, H.; Tuck, D.P. Abstract 1168: Efficacy of the IRAK4 inhibitor CA-4948 in patient-derived xenograft models of diffuse large B cell lymphoma. Cancer Res. 2017, 77, 1168. [Google Scholar] [CrossRef]
- Booher, R.N.; Nowakowski, G.S.; Patel, K.; Lunning, M.A.; Samson, M.E.S.; Atoyan, R.; Ma, A.W.; Xu, G.X.; Dellarocca, S.; Modafferi, H.; et al. Preclinical Activity of IRAK4 Kinase Inhibitor CA-4948 Alone or in Combination with Targeted Therapies and Preliminary Phase 1 Clinical Results in Non-Hodgkin Lymphoma. Blood 2018, 132, 4168. [Google Scholar] [CrossRef]
- Ugolkov, A.; Hok, R.; von Roemeling, R.; Martell, R.E. EP390. IRAK4 inhibitor CA-4948 potentiates antitumor effects of azacitidine and venetoclax in human acute myeloid leukemia. HemaSphere 2021, 5, 153–154. [Google Scholar]
- Joffe, E.; Nowakowski, G.S.; Tun, H.W.; Rosenthal, A.C.; Lunning, M.A.; Ramchandren, R.; Li, C.-C.; Zhou, L.; Martinez, E.; Roemeling, R.W.V.; et al. Open-label, dose-escalation, and expansion trial of CA-4948 in combination with ibrutinib in patients with relapsed or refractory hematologic malignancies. J. Clin. Oncol. 2022, 40, 7575. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Winer, E.S.; DeAngelo, D.J.; Tarantolo, S.R.; Sallman, D.A.; Dugan, J.; Groepper, S.; Giagounidis, A.; Gotze, K.S.; Metzeler, K.; et al. Phase 1/2a study of the IRAK4 inhibitor CA-4948 as monotherapy or in combination with azacitidine or venetoclax in patients with relapsed/refractory (R/R) acute myeloid leukemia or lyelodysplastic syndrome. J. Clin. Oncol. 2022, 40, 7016. [Google Scholar] [CrossRef]
- Spriano, F.; Chung, E.Y.L.; Gaudio, E.; Tarantelli, C.; Cascione, L.; Napoli, S.; Jessen, K.; Carrassa, L.; Priebe, V.; Sartori, G.; et al. The ETS Inhibitors YK-4-279 and TK-216 Are Novel Antilymphoma Agents. Clin. Cancer Res. 2019, 25, 5167–5176. [Google Scholar] [CrossRef] [PubMed]
- Arribas, A.J.; Napoli, S.; Cascione, L.; Sartori, G.; Barnabei, L.; Gaudio, E.; Tarantelli, C.; Mensah, A.A.; Spriano, F.; Zucchetto, A.; et al. Resistance to PI3Kdelta inhibitors in marginal zone lymphoma can be reverted by targeting the IL-6/PDGFRA axis. Haematologica 2022, 107, 2685–2697. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, E.; Tarantelli, C.; Spriano, F.; Guidetti, F.; Sartori, G.; Bordone, R.; Arribas, A.J.; Cascione, L.; Bigioni, M.; Merlino, G.; et al. Targeting CD205 with the antibody drug conjugate MEN1309/OBT076 is an active new therapeutic strategy in lymphoma models. Haematologica 2020, 105, 2584–2591. [Google Scholar] [CrossRef] [Green Version]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/mTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin. Cancer Res. 2018, 24, 120–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.T.; Wooten, D.J.; Paudel, B.B.; Bauer, J.; Hardeman, K.N.; Westover, D.; Lovly, C.M.; Harris, L.A.; Tyson, D.R.; Quaranta, V. Quantifying Drug Combination Synergy along Potency and Efficacy Axes. Cell Syst 2019, 8, 97–108.e116. [Google Scholar] [CrossRef] [Green Version]
- Bliss, C.I. The toxicity of poisons applied jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Mensah, A.A.; Kwee, I.; Gaudio, E.; Rinaldi, A.; Ponzoni, M.; Cascione, L.; Fossati, G.; Stathis, A.; Zucca, E.; Caprini, G.; et al. Novel HDAC inhibitors exhibit pre-clinical efficacy in lymphoma models and point to the importance of CDKN1A expression levels in mediating their anti-tumor response. Oncotarget 2015, 6, 5059–5071. [Google Scholar] [CrossRef] [Green Version]
- Barnabei, L.; Lamrini, H.; Castela, M.; Jeremiah, N.; Stolzenberg, M.-C.; Chentout, L.; Jacques, S.; Bouafia, A.; Magérus-Chatinet, A.; Moncan, M.; et al. Heterozygous RELA mutations cause early-onset systemic lupus erythematosus by hijacking the NF-κB pathway towards transcriptional activation of type-I Interferon genes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Inokuchi, K.; Abo, J.; Takahashi, H.; Miyake, K.; Inokuchi, S.; Dan, K.; Nomura, T. Establishment and characterization of a villous lymphoma cell line from splenic B-cell lymphoma. Leuk. Res. 1995, 19, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Climent, J.A.; Sanchez-Izquierdo, D.; Sarsotti, E.; Blesa, D.; Benet, I.; Climent, J.; Vizcarra, E.; Marugan, I.; Terol, M.J.; Sole, F.; et al. Genomic abnormalities acquired in the blastic transformation of splenic marginal zone B-cell lymphoma. Leuk. Lymphoma 2003, 44, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Arribas, A.J.; Napoli, S.; Gaudio, E.; Cascione, L.; Di Veroli, A.; Tarantelli, C.; Spriano, F.; Zucchetto, A.; Rossi, F.M.; Rinaldi, A.; et al. Secreted Factors Determine Resistance to Idelalisib in Marginal Zone Lymphoma Models of Resistance. Blood 2019, 134, 2569. [Google Scholar] [CrossRef]
- Arribas, A.J.; Napoli, S.; Gaudio, E.; Cascione, L.; Veroli, A.D.; Tarantelli, C.; Spriano, F.; Zucchetto, A.; Rossi, F.; Sartori, G.; et al. Abstract A127: Secretion of IL16 is associated with resistance to ibrutinib in pre-clinical models of lymphoma. Mol. Cancer Ther. 2019, 18, A127. [Google Scholar] [CrossRef]
- Arribas, A.; Napoli, S.; Cascione, L.; Gaudio, E.; Bordone-Pittau, R.; Barreca, M.; Sartori, G.; Tarantelli, C.; Spriano, F.; Rinaldi, A.; et al. Secondary resistance to the PI3K inhibitor copanlisib in marginal zone lymphoma. Eur. J. Cancer 2020, 138, S40. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guidetti, F.; Arribas, A.J.; Sartori, G.; Spriano, F.; Barnabei, L.; Tarantelli, C.; Von Roemeling, R.; Martinez, E.; Zucca, E.; Bertoni, F. Targeting IRAK4 with Emavusertib in Lymphoma Models with Secondary Resistance to PI3K and BTK Inhibitors. J. Clin. Med. 2023, 12, 399. https://doi.org/10.3390/jcm12020399
Guidetti F, Arribas AJ, Sartori G, Spriano F, Barnabei L, Tarantelli C, Von Roemeling R, Martinez E, Zucca E, Bertoni F. Targeting IRAK4 with Emavusertib in Lymphoma Models with Secondary Resistance to PI3K and BTK Inhibitors. Journal of Clinical Medicine. 2023; 12(2):399. https://doi.org/10.3390/jcm12020399
Chicago/Turabian StyleGuidetti, Francesca, Alberto J. Arribas, Giulio Sartori, Filippo Spriano, Laura Barnabei, Chiara Tarantelli, Reinhard Von Roemeling, Elizabeth Martinez, Emanuele Zucca, and Francesco Bertoni. 2023. "Targeting IRAK4 with Emavusertib in Lymphoma Models with Secondary Resistance to PI3K and BTK Inhibitors" Journal of Clinical Medicine 12, no. 2: 399. https://doi.org/10.3390/jcm12020399
APA StyleGuidetti, F., Arribas, A. J., Sartori, G., Spriano, F., Barnabei, L., Tarantelli, C., Von Roemeling, R., Martinez, E., Zucca, E., & Bertoni, F. (2023). Targeting IRAK4 with Emavusertib in Lymphoma Models with Secondary Resistance to PI3K and BTK Inhibitors. Journal of Clinical Medicine, 12(2), 399. https://doi.org/10.3390/jcm12020399