

Coagulation Disorders in Sepsis and COVID-19—Two Sides of the Same Coin? A Review of Inflammation–Coagulation Crosstalk in Bacterial Sepsis and COVID-19
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
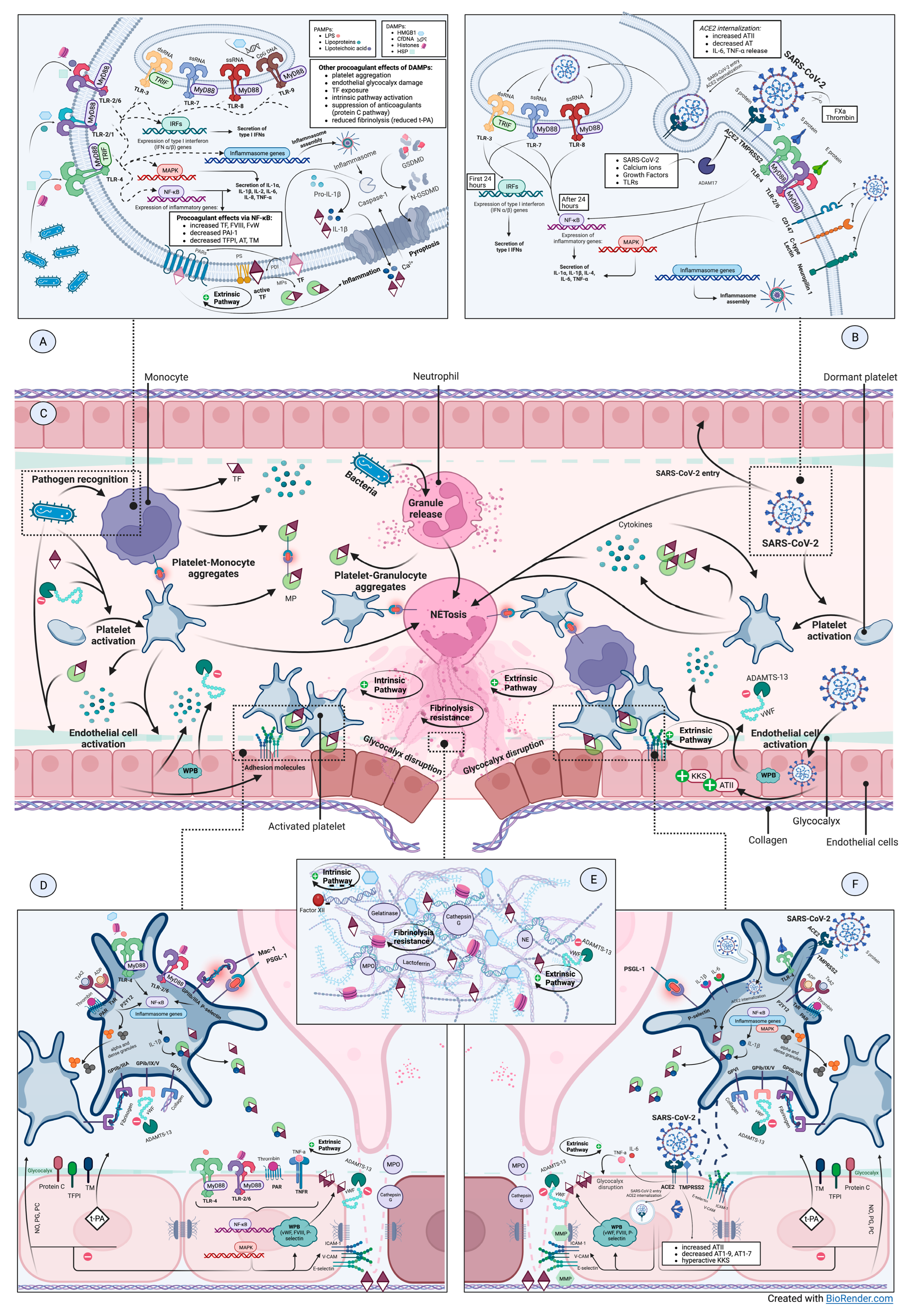
3. Molecular Mechanisms of Inflammation–Coagulation Crosstalkin Sepsis and COVID-19
3.1. The Triggers—PAMPs/DAMPs/PRRs Interplay
3.1.1. Pattern Recognition Receptors
3.1.2. Pathogen-Associated Danger Signals
3.1.3. Endogenous Danger Signals
3.1.4. How Does SARS-CoV-2 Infect Cells?
3.2. Effector Cells—Monocytes, Neutrophils, Platelets, Endothelial Cells
3.2.1. Monocytes—The Main Source of TF in Inflammation-Induced Coagulopathy
3.2.2. Activated Neutrophils—At the Intersection of Hemostatic Pathways
3.2.3. Platelets’ Function—Much More Than Primary Hemostasis
3.2.4. Endothelium Coordinates Both Inflammatory Responses and Coagulation
4. From Theory to Practice—Clinical Aspects of Coagulopathy in Sepsis and COVID-19
4.1. Progression of Coagulation Disorders—The Pathways towards DIC
4.1.1. What Is DIC?
4.1.2. Is Coagulopathy of COVID-19 a Form of DIC?
4.2. Searching for the Proof—Coagulation Studies
4.2.1. Standard Coagulation Tests
4.2.2. Advanced Coagulation Studies—Viscoelastic Tests in SIC and CAC
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, Regional, and National Sepsis Incidence and Mortality, 1990–2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Opal, S. The Current Understanding of Sepsis and Research Priorities for the Future. Virulence 2014, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- WHO Coronavirus (COVID-19) Dashboard | WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. Available online: https://covid19.who.int/ (accessed on 24 October 2022).
- Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-Specific Immune Response and the Pathogenesis of COVID-19. Int. J. Mol. Sci. 2022, 23, 1716. [Google Scholar] [CrossRef]
- Iba, T.; Warkentin, T.E.; Thachil, J.; Levi, M.; Levy, J.H. Proposal of the Definition for COVID-19-Associated Coagulopathy. J. Clin. Med. 2021, 10, 191. [Google Scholar] [CrossRef]
- Koçak Tufan, Z.; Kayaaslan, B.; Mer, M. COVID-19 and Sepsis. Turk. J. Med. Sci. 2021, 51, 3301–3311. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Wu, R.; Wang, N.; Comish, P.B.; Tang, D.; Kang, R. Inflammasome-Dependent Coagulation Activation in Sepsis. Front. Immunol. 2021, 12, 641750. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct Target 2021, 6, 291. [Google Scholar] [CrossRef]
- Raymond, S.L.; Holden, D.C.; Mira, J.C.; Stortz, J.A.; Loftus, T.J.; Mohr, A.M.; Moldawer, L.L.; Moore, F.A.; Larson, S.D.; Efron, P.A. Microbial Recognition and Danger Signals in Sepsis and Trauma. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2564–2573. [Google Scholar] [CrossRef]
- Swiatkowska, M.; Szemraj, J.; Cierniewski, C.S. Induction of PAI-1 Expression by Tumor Necrosis Factor Alpha in Endothelial Cells Is Mediated by Its Responsive Element Located in the 4G/5G Site. FEBS J. 2005, 272, 5821–5831. [Google Scholar] [CrossRef]
- Angus, D.C.; van der Poll, T. Severe Sepsis and Septic Shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef]
- Begbie, M.; Notley, C.; Tinlin, S.; Sawyer, L.; Lillicrap, D. The Factor VIII Acute Phase Response Requires the Participation of NFkappaB and C/EBP. Thromb. Haemost. 2000, 84, 216–222. [Google Scholar] [CrossRef]
- Blanch-Ruiz, M.A.; Ortega-Luna, R.; Martínez-Cuesta, M.Á.; Álvarez, Á. The Neutrophil Secretome as a Crucial Link between Inflammation and Thrombosis. Int. J. Mol. Sci. 2021, 22, 4170. [Google Scholar] [CrossRef]
- Liu, S.F.; Malik, A.B. NF-ΚB Activation as a Pathological Mechanism of Septic Shock and Inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, 622–645. [Google Scholar] [CrossRef]
- Wu, C.; Lu, W.; Zhang, Y.; Zhang, G.; Shi, X.; Hisada, Y.; Grover, S.P.; Zhang, X.; Li, L.; Xiang, B.; et al. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity 2019, 50, 1401–1411.e4. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J. Immunol. 2020, 205, 307. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 Spike Protein Interacts with and Activates TLR41. Cell Res. 2021, 31, 818. [Google Scholar] [CrossRef]
- Shirato, K.; Kizaki, T. SARS-CoV-2 Spike Protein S1 Subunit Induces pro-Inflammatory Responses via Toll-like Receptor 4 Signaling in Murine and Human Macrophages. Heliyon 2021, 7, E06187. [Google Scholar] [CrossRef]
- Khadke, S.; Ahmed, N.; Ahmed, N.; Ratts, R.; Raju, S.; Gallogly, M.; de Lima, M.; Sohail, M.R. Harnessing the Immune System to Overcome Cytokine Storm and Reduce Viral Load in COVID-19: A Review of the Phases of Illness and Therapeutic Agents. Virol. J. 2020, 17, 154. [Google Scholar] [CrossRef]
- Sohn, K.M.; Lee, S.G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 Patients Upregulate Toll-like Receptor 4-Mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J. Korean Med. Sci. 2020, 35, e343. [Google Scholar] [CrossRef]
- Bortolotti, D.; Gentili, V.; Rizzo, S.; Schiuma, G.; Beltrami, S.; Strazzabosco, G.; Fernandez, M.; Caccuri, F.; Caruso, A.; Rizzo, R. Tlr3 and Tlr7 Rna Sensor Activation during SARS-CoV-2 Infection. Microorganisms 2021, 9, 1820. [Google Scholar] [CrossRef]
- Barbu, E.C.; Chiţu-Tişu, C.E.; Lazăr, M.; Olariu, C.; Bojincă, M.; Ionescu, R.A.; Ion, D.A.; Bădărău, I.A. Hepatic Osteodystrophy: A Global (Re)View of the Problem. Acta Clin. Croat. 2017, 56, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Zhang, W.; Wu, C.; Wang, S.; Smith, F.G.; Jin, S.; Zhang, P. The Novel Role of Metabolism-Associated Molecular Patterns in Sepsis. Front. Cell Infect. Microbiol. 2022, 12, 915099. [Google Scholar] [CrossRef]
- Martin, S.J. Cell Death and Inflammation: The Case for IL-1 Family Cytokines as the Canonical DAMPs of the Immune System. FEBS J. 2016, 283, 2599–2615. [Google Scholar] [CrossRef] [Green Version]
- Rajaee, A.; Barnett, R.; Cheadle, W.G. Pathogen- A Nd Danger-Associated Molecular Patterns and the Cytokine Response in Sepsis. Surg. Infect. 2018, 19, 107–116. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel Role of PKR in Inflammasome Activation and HMGB1 Release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of Chromatin Protein HMGB1 by Necrotic Cells Triggers Inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Sha, Y.; Zmijewski, J.; Xu, Z.; Abraham, E. HMGB1 Develops Enhanced Proinflammatory Activity by Binding to Cytokines. J. Immunol. 2008, 180, 2531–2537. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ward, M.F.; Sama, A.E. Targeting HMGB1 in the Treatment of Sepsis. Expert Opin. Ther. Targets 2014, 18, 257–268. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; de Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated Platelets Present High Mobility Group Box 1 to Neutrophils, Inducing Autophagy and Promoting the Extrusion of Neutrophil Extracellular Traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Kawahara, K.; Nakamura, T.; Yamada, S.; Nakamura, T.; Abeyama, K.; Hashiguchi, T.; Maruyama, I. High-Mobility Group Box 1 Protein Promotes Development of Microvascular Thrombosis in Rats. J. Thromb. Haemost. 2007, 5, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Roviello, G.N.; Montesarchio, D. An Overview on HMGB1 Inhibitors as Potential Therapeutic Agents in HMGB1-Related Pathologies. Pharmacol. Ther. 2014, 141, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Yuseok, O.; Yang, S.; Lee, B.S.; Lee, J.H.; Park, E.K.; Baek, M.C.; Song, G.Y.; Bae, J.S. JH-4 Reduces HMGB1-Mediated Septic Responses and Improves Survival Rate in Septic Mice. J. Cell Biochem. 2019, 120, 6277–6289. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Ku, S.K.; Bae, J.S. Zingerone Reduces HMGB1-Mediated Septic Responses and Improves Survival in Septic Mice. Toxicol. Appl. Pharmacol. 2017, 329, 202–211. [Google Scholar] [CrossRef]
- Stevens, N.E.; Chapman, M.J.; Fraser, C.K.; Kuchel, T.R.; Hayball, J.D.; Diener, K.R. Therapeutic Targeting of HMGB1 during Experimental Sepsis Modulates the Inflammatory Cytokine Profile to One Associated with Improved Clinical Outcomes. Sci. Rep. 2017, 7, 5850. [Google Scholar] [CrossRef] [Green Version]
- Al-kuraishy, H.M.; Al-Gareeb, A.I.; Alkazmi, L.; Habotta, O.A.; Batiha, G.E.S. High-Mobility Group Box 1 (HMGB1) in COVID-19: Extrapolation of Dangerous Liaisons. Inflammopharmacology 2022, 30, 811. [Google Scholar] [CrossRef]
- Oehmcke, S.; Mörgelin, M.; Herwald, H. Activation of the Human Contact System on Neutrophil Extracellular Traps. J. Innate Immun. 2009, 1, 225–230. [Google Scholar] [CrossRef]
- Gansler, J.; Jaax, M.; Leiting, S.; Appel, B.; Greinacher, A.; Fischer, S.; Preissner, K.T. Structural Requirements for the Procoagulant Activity of Nucleic Acids. PLoS ONE 2012, 7, e50399. [Google Scholar] [CrossRef]
- Komissarov, A.A.; Florova, G.; Idell, S. Effects of Extracellular DNA on Plasminogen Activation and Fibrinolysis. J. Biol. Chem. 2011, 286, 41949–41962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA Traps Promote Thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Fourrier, F. Severe Sepsis, Coagulation, and Fibrinolysis: Dead End or One Way? Crit. Care Med. 2012, 40, 2704–2708. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Iba, T. Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. Anesthesiology 2020, 132, 1238–1283. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Inoue, H.; Ono, C.; Hashimoto, S.; Kioi, Y.; Matsumoto, H.; Matsuura, H.; Matsubara, T.; Shimizu, K.; et al. IL-6 Trans-Signaling Induces Plasminogen Activator Inhibitor-1 from Vascular Endothelial Cells in Cytokine Release Syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 22351–22356. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.A.; Hisada, Y.; Denorme, F.; Grover, S.P.; Bouck, E.G.; Middleton, E.A.; Wolberg, A.S.; Rondina, M.T.; Mackman, N. Comparison of the Coagulopathies Associated with COVID-19 and Sepsis. Res. Pract. Thromb. Haemost. 2021, 5, e12525. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Li, Y.; Wan, D.; Luo, X.; Song, T.; Wang, Y.; Yu, Q.; Jiang, L.; Liao, R.; Zhao, W.; Su, B. Circulating Histones in Sepsis: Potential Outcome Predictors and Therapeutic Targets. Front. Immunol. 2021, 12, 650184. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones Induce Rapid and Profound Thrombocytopenia in Mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, M.; Ito, T.; Kawahara, K.-I.; Yamamoto, M.; Nagasato, T.; Shrestha, B.; Yamada, S.; Miyauchi, T.; Higuchi, K.; Takenaka, T.; et al. Recombinant Thrombomodulin Protects Mice against Histone-Induced Lethal Thromboembolism. PLoS ONE 2013, 8, e75961. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal Coupling of Coagulation and Innate Immunity via Neutrophil Serine Proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular Histones Increase Plasma Thrombin Generation by Impairing Thrombomodulin-Dependent Protein C Activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef]
- Biswas, I.; Panicker, S.R.; Cai, X.S.; Giri, H.; Rezaie, A.R. Extracellular Histones Bind Vascular Glycosaminoglycans and Inhibit the Anti-Inflammatory Function of Antithrombin. Cell Physiol. Biochem. 2021, 55, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-Spike Protein Is a Novel Route for SARS-CoV-2 Infection to Host Cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856. [Google Scholar] [CrossRef] [PubMed]
- Thépaut, M.; Luczkowiak, J.; Vivès, C.; Labiod, N.; Bally, I.; Lasala, F.; Grimoire, Y.; Fenel, D.; Sattin, S.; Thielens, N.; et al. DC/L-SIGN Recognition of Spike Glycoprotein Promotes SARS-CoV-2 Trans-Infection and Can Be Inhibited by a Glycomimetic Antagonist. PLoS Pathog. 2021, 17, e1009576. [Google Scholar] [CrossRef]
- Shirato, K.; Takanari, J.; Kizaki, T. Standardized Extract of Asparagus Officinalis Stem Attenuates SARS-CoV-2 Spike Protein-Induced IL-6 and IL-1β Production by Suppressing P44/42 MAPK and Akt Phosphorylation in Murine Primary Macrophages. Molecules 2021, 26, 6189. [Google Scholar] [CrossRef]
- Geng, J.; Chen, L.; Yuan, Y.; Wang, K.; Wang, Y.; Qin, C.; Wu, G.; Chen, R.; Zhang, Z.; Wei, D.; et al. CD147 Antibody Specifically and Effectively Inhibits Infection and Cytokine Storm of SARS-CoV-2 and Its Variants Delta, Alpha, Beta, and Gamma. Signal Transduct. Target. Ther. 2021, 6, 347. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Mercadante, M.; Nilsson-Payant, B.; Johnson, J.L.; Jaimes, J.A.; Muecksch, F.; Weisblum, Y.; Bram, Y.; Chandar, V.; Whittaker, G.R.; et al. Coagulation Factors Directly Cleave SARS-CoV-2 Spike and Enhance Viral Entry. Elife 2022, 11, 77444. [Google Scholar] [CrossRef]
- Khawaja, U.A.; Shamsoddin, E.; Desideri, L.F.; Tovani-Palone, M.R. Infection of Red Blood Cells by SARS-CoV-2: New Evidence. Einstein 2021, 19, eCE6285. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Tellone, E.; Barreca, D.; Ficarra, S.; Laganà, G. Implication of COVID-19 on Erythrocytes Functionality: Red Blood Cell Biochemical Implications and Morpho-Functional Aspects. Int. J. Mol. Sci. 2022, 23, 2171. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, H. COVID-19: Attacks the 1-Beta Chain of Hemoglobin and Captures the Porphyrin to Inhibit Human Heme Metabolism. Biol. Med. Chem. 2020. preprint. [Google Scholar] [CrossRef]
- Ninivaggi, M.; de Laat, M.; Lancé, M.M.D.; Kicken, C.H.; Pelkmans, L.; Bloemen, S.; Dirks, M.L.; van Loon, L.J.C.; Govers-Riemslag, J.W.P.; Lindhout, T.; et al. Hypoxia Induces a Prothrombotic State Independently of the Physical Activity. PLoS ONE 2015, 10, e0141797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeper, M.M.; Granton, J. Intensive Care Unit Management of Patients with Severe Pulmonary Hypertension and Right Heart Failure. Am. J. Respir. Crit. Care Med. 2011, 184, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.R.; Geng, R.; Li, Q.; Chen, Y.; Li, S.F.; Wang, Q.; Min, J.; Yang, Y.; Li, B.; Jiang, R.-D.; et al. ACE2-Independent Infection of T Lymphocytes by SARS-CoV-2. Signal Transduct. Target. Ther. 2022, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Zhang, J.; Fang, Y.; Lu, S.; Wu, J.; Zheng, X.; Deng, F. SARS-CoV-2 Interacts with Platelets and Megakaryocytes via ACE2-Independent Mechanism. J. Hematol. Oncol. 2021, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Savla, S.R.; Prabhavalkar, K.S.; Bhatt, L.K. Cytokine Storm Associated Coagulation Complications in COVID-19 Patients: Pathogenesis and Management. Expert Rev. Anti Infect. Ther. 2021, 19, 1397–1413. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 Binds Platelet ACE2 to Enhance Thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Li, Y.; Wang, Z.; Ma, F.; Luo, R.; Xu, X.; Zhou, G.; Wang, J.; Niu, J.; et al. Platelets Mediate Inflammatory Monocyte Activation by SARS-CoV-2 Spike Protein. J. Clin. Invest. 2022, 132, e150101. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-ΚB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, E.E.; Jørgensen, M.J.; Nore, K.G.; Dahl, T.B.; Yang, K.; Ranheim, T.; Huse, C.; Lind, A.; Nur, S.; Stiksrud, B.; et al. Critical COVID-19 Is Associated with Distinct Leukocyte Phenotypes and Transcriptome Patterns. J. Intern. Med. 2021, 290, 677. [Google Scholar] [CrossRef] [PubMed]
- Paffen, E.; Vos, H.L.; Bertina, R.M. C-Reactive Protein Does Not Directly Induce Tissue Factor in Human Monocytes. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 975–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åberg, M.; Siegbahn, A. Tissue Factor Non-Coagulant Signaling—Molecular Mechanisms and Biological Consequences with a Focus on Cell Migration and Apoptosis. J. Thromb. Haemost. 2013, 11, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, E.; Hartung, T.; Morath, S.; Egesten, A. Highly Purified Lipoteichoic Acid from Staphylococcus Aureus Induces Procoagulant Activity and Tissue Factor Expression in Human Monocytes but Is a Weak Inducer in Whole Blood: Comparison with Peptidoglycan. Infect. Immun. 2004, 72, 4322–4326. [Google Scholar] [CrossRef] [Green Version]
- Hottz, E.D.; Martins-Gonçalves, R.; Palhinha, L.; Azevedo-Quintanilha, I.G.; de Campos, M.M.; Sacramento, C.Q.; Temerozo, J.R.; Soares, V.C.; Gomes Dias, S.S.; Teixeira, L.; et al. Platelet-Monocyte Interaction Amplifies Thromboinflammation through Tissue Factor Signaling in COVID-19. Blood Adv. 2022, 6, 5085. [Google Scholar] [CrossRef]
- Egorina, E.M.; Sovershaev, M.A.; Hansen, J.B. The Role of Tissue Factor in Systemic Inflammatory Response Syndrome. Blood Coagul. Fibrinolysis 2011, 22, 451–456. [Google Scholar] [CrossRef]
- Stiel, L.; Meziani, F.; Helms, J. Neutrophil Activation during Septic Shock. Shock 2018, 49, 371–384. [Google Scholar] [CrossRef]
- Brown, K.; Brain, S.; Pearson, J.; Edgeworth, J.; Lewis, S.; Treacher, D. Neutrophils in Development of Multiple Organ Failure in Sepsis. Lancet 2006, 368, 157–169. [Google Scholar] [CrossRef]
- Bian, X.W.; Yao, X.H.; Ping, Y.F.; Yu, S.; Shi, Y.; Luo, T.; He, Z.C.; Tang, R.; Chen, C.; Fu, W.J.; et al. Autopsy of COVID-19 Patients in China. Natl. Sci. Rev. 2020, 7, 1414–1418. [Google Scholar] [CrossRef]
- Lazar, M.; Barbu, E.C.; Chitu, C.E.; Anghel, A.M.J.; Niculae, C.M.; Manea, E.D.; Damalan, A.C.; Bel, A.A.; Patrascu, R.E.; Hristea, A.; et al. Pericardial Involvement in Severe COVID-19 Patients. Medicina 2022, 58, 1093. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe 2020, 27, 883–890.e2. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Mo, P.; Liu, J.; Wang, H.; Wang, F.; Zhao, Q. Neutrophil to CD4+ Lymphocyte Ratio as a Potential Biomarker in Predicting Virus Negative Conversion Time in COVID-19. Int. Immunopharmacol. 2020, 85, 106683. [Google Scholar] [CrossRef]
- Cai, J.; Li, H.; Zhang, C.; Chen, Z.; Liu, H.; Lei, F.; Qin, J.J.; Liu, Y.M.; Zhou, F.; Song, X.; et al. The Neutrophil-to-Lymphocyte Ratio Determines Clinical Efficacy of Corticosteroid Therapy in Patients with COVID-19. Cell Metab. 2021, 33, 258–269.e3. [Google Scholar] [CrossRef] [PubMed]
- Moisa, E.; Corneci, D.; Negoita, S.; Filimon, C.R.; Serbu, A.; Negutu, M.I.; Grintescu, I.M. Dynamic Changes of the Neutrophil-to-Lymphocyte Ratio, Systemic Inflammation Index, and Derived Neutrophil-to-Lymphocyte Ratio Independently Predict Invasive Mechanical Ventilation Need and Death in Critically Ill COVID-19 Patients. Biomedicines 2021, 9, 1656. [Google Scholar] [CrossRef] [PubMed]
- Anghel, A.M.J.; Niculae, C.M.; Manea, E.D.; Lazar, M.; Popescu, M.; Damalan, A.C.; Bel, A.A.; Nedelcu, I.M.; Patrascu, R.E.; Hristea, A. The Impact of Tocilizumab on Radiological Changes Assessed by Quantitative Chest CT in Severe COVID-19 Patients. J. Clin. Med. 2022, 11, 1247. [Google Scholar] [CrossRef]
- Abrams, S.T.; Morton, B.; Alhamdi, Y.; Alsabani, M.; Lane, S.; Welters, I.D.; Wang, G.; Toh, C.H. A Novel Assay for Neutrophil Extracellular Trap Formation Independently Predicts Disseminated Intravascular Coagulation and Mortality in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 2019, 200, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Gupta, E.; Kaushik, S.; Srivastava, V.K.; Saxena, J.; Mehta, S.; Jyoti, A. Quantification of NETs Formation in Neutrophil and Its Correlation with the Severity of Sepsis and Organ Dysfunction. Clin. Chim. Acta 2019, 495, 606–610. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, S.; Wang, Y.; Rahman, M.; Syk, I.; Zhang, E.; Thorlacius, H. Proinflammatory Role of Neutrophil Extracellular Traps in Abdominal Sepsis. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L586–L596. [Google Scholar] [CrossRef]
- Czaikoski, P.G.; Mota, J.M.S.C.; Nascimento, D.C.; Sônego, F.; Castanheira, F.V.E.S.; Melo, P.H.; Scortegagna, G.T.; Silva, R.L.; Barroso-Sousa, R.; Souto, F.O.; et al. Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PLoS ONE 2016, 11, e0148142. [Google Scholar] [CrossRef]
- Becker, K.; Beythien, G.; de Buhr, N.; Stanelle-Bertram, S.; Tuku, B.; Kouassi, N.M.; Beck, S.; Zickler, M.; Allnoch, L.; Gabriel, G.; et al. Vasculitis and Neutrophil Extracellular Traps in Lungs of Golden Syrian Hamsters With SARS-CoV-2. Front. Immunol. 2021, 12, 640842. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Anders, H.J.; Bilyy, R.; Bowlin, G.L.; Daniel, C.; de Lorenzo, R.; Egeblad, M.; Henneck, T.; Hidalgo, A.; Hoffmann, M.; et al. Patients with COVID-19: In the Dark-NETs of Neutrophils. Cell Death Differ. 2021, 28, 3125–3139. [Google Scholar] [CrossRef]
- Obermayer, A.; Jakob, L.M.; Haslbauer, J.D.; Matter, M.S.; Tzankov, A.; Stoiber, W. Neutrophil Extracellular Traps in Fatal COVID-19-Associated Lung Injury. Dis. Markers 2021, 2021, 5566826. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, L.E.; Pekkarinen, P.T.; Alander, M.; Nowlan, K.H.A.; Nguyen, N.A.; Jokiranta, S.; Kuivanen, S.; Patjas, A.; Mero, S.; Pakkanen, S.H.; et al. Characterization of Low-Density Granulocytes in COVID-19. PLoS Pathog. 2021, 17, e1009721. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, S.M.; Geller, A.E.; Hu, X.; Tieri, D.; Ding, C.; Klaes, C.K.; Cooke, E.A.; Woeste, M.R.; Martin, Z.C.; Chen, O.; et al. A Specific Low-Density Neutrophil Population Correlates with Hypercoagulation and Disease Severity in Hospitalized COVID-19 Patients. JCI Insight 2021, 6, e148435. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated Endothelial Cells Induce Neutrophil Extracellular Traps and Are Susceptible to NETosis-Mediated Cell Death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [Green Version]
- Yipp, B.G.; Kubes, P. NETosis: How Vital Is It? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus Aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [Green Version]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-Triggered Neutrophil Extracellular Traps Mediate COVID-19 Pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Sung, P.S.; Hsieh, S.L. C-Type Lectins and Extracellular Vesicles in Virus-Induced NETosis. J. Biomed. Sci. 2021, 28, 46. [Google Scholar] [CrossRef] [PubMed]
- Arcanjo, A.; Logullo, J.; Menezes, C.C.B.; de Souza Carvalho Giangiarulo, T.C.; dos Reis, M.C.; de Castro, G.M.M.; da Silva Fontes, Y.; Todeschini, A.R.; Freire-de-Lima, L.; Decoté-Ricardo, D.; et al. The Emerging Role of Neutrophil Extracellular Traps in Severe Acute Respiratory Syndrome Coronavirus 2 (COVID-19). Sci. Rep. 2020, 10, 19630. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.-S.; Yang, S.-P.; Peng, Y.-C.; Sun, C.-P.; Tao, M.-H.; Hsieh, S.-L. CLEC5A and TLR2 Are Critical in SARS-CoV-2-Induced NET Formation and Lung Inflammation. J. Biomed. Sci. 2022, 29, 52. [Google Scholar] [CrossRef]
- Yaqinuddin, A.; Kashir, J. Novel Therapeutic Targets for SARS-CoV-2-Induced Acute Lung Injury: Targeting a Potential IL-1β/Neutrophil Extracellular Traps Feedback Loop. Med. Hypotheses 2020, 143, 109906. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.; Leunig, A.; Pekayvaz, K.; Popp, O.; Joppich, M.; Polewka, V.; Escaig, R.; Anjum, A.; Hoffknecht, M.L.; Gold, C.; et al. Self-Sustaining IL-8 Loops Drive a Prothrombotic Neutrophil Phenotype in Severe COVID-19. JCI Insight 2021, 6, e150862. [Google Scholar] [CrossRef]
- McDonald, B.; Davis, R.P.; Kim, S.J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and Neutrophil Extracellular Traps Collaborate to Promote Intravascular Coagulation during Sepsis in Mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and Tissue Factor-Enriched Neutrophil Extracellular Traps Are Key Drivers in COVID-19 Immunothrombosis. J. Clin. Invest. 2020, 130, 6151–6157. [Google Scholar] [CrossRef] [PubMed]
- von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, Neutrophils, and Platelets Cooperate to Initiate and Propagate Venous Thrombosis in Mice in Vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.C.; Weitz, J.I.; Liaw, P.C. Neutrophil Extracellular Traps Promote Thrombin Generation through Platelet-Dependent and Platelet-Independent Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Lee, J.K. Role of HMGB1 in the Interplay between NETosis and Thrombosis in Ischemic Stroke: A Review. Cells 2020, 9, 1794. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-Selectin Promotes Neutrophil Extracellular Trap Formation in Mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Gould, T.J.; Vu, T.T.; Stafford, A.R.; Dwivedi, D.J.; Kim, P.Y.; Fox-Robichaud, A.E.; Weitz, J.I.; Liaw, P.C. Cell-Free DNA Modulates Clot Structure and Impairs Fibrinolysis in Sepsis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2544–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iba, T.; Levy, J.H. Inflammation and Thrombosis: Roles of Neutrophils, Platelets and Endothelial Cells and Their Interactions in Thrombus Formation during Sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Qi, H.; Kan, K.; Chen, J.; Xie, H.; Guo, X.; Zhang, L. Neutrophil Extracellular Traps Promote Hypercoagulability in Patients with Sepsis. Shock 2017, 47, 132–139. [Google Scholar] [CrossRef]
- Osada, K.; Minami, T.; Arioka, T.; Sakai, T.; Tawara, S.; Kawasaki, K.; Fareed, J.; Matsuzaki, O. Thrombomodulin Alfa Attenuates the Procoagulant Effect and Cytotoxicity of Extracellular Histones through the Promotion of Protein C Activation. Thromb. Res. 2017, 160, 51–57. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.-B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In Vitro Activation of Coagulation by Human Neutrophil DNA and Histone Proteins but Not Neutrophil Extracellular Traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Maroney, S.A.; Mast, A.E. Tissue Factor Pathway Inhibitor and Bacterial Infection. J. Thromb. Haemost. 2011, 9, 119. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.; Pittet, J.F. The Coagulopathy of Acute Sepsis. Curr. Opin. Anaesthesiol. 2015, 28, 227. [Google Scholar] [CrossRef] [Green Version]
- Scapini, P.; Nesi, L.; Morini, M.; Tanghetti, E.; Belleri, M.; Noonan, D.; Presta, M.; Albini, A.; Cassatella, M.A. Generation of Biologically Active Angiostatin Kringle 1-3 by Activated Human Neutrophils. J. Immunol. 2002, 168, 5798–5804. [Google Scholar] [CrossRef]
- Da Cruz, D.B.; Helms, J.; Aquino, L.R.; Stiel, L.; Cougourdan, L.; Broussard, C.; Chafey, P.; Riès-Kautt, M.; Meziani, F.; Toti, F.; et al. DNA-Bound Elastase of Neutrophil Extracellular Traps Degrades Plasminogen, Reduces Plasmin Formation, and Decreases Fibrinolysis: Proof of Concept in Septic Shock Plasma. FASEB J. 2019, 33, 14270–14280. [Google Scholar] [CrossRef] [Green Version]
- Varjú, I.; Longstaff, C.; Szabó, L.; Farkas, Á.Z.; Varga-Szabó, V.J.; Tanka-Salamon, A.; Machovich, R.; Kolev, K. DNA, Histones and Neutrophil Extracellular Traps Exert Anti-Fibrinolytic Effects in a Plasma Environment. Thromb. Haemost. 2015, 113, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Longstaff, C.; Varjú, I.; Sótonyi, P.; Szabó, L.; Krumrey, M.; Hoell, A.; Bóta, A.; Varga, Z.; Komorowicz, E.; Kolev, K. Mechanical Stability and Fibrinolytic Resistance of Clots Containing Fibrin, DNA, and Histones. J. Biol. Chem. 2013, 288, 6946–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil Extracellular Traps Infiltrate the Lung Airway, Interstitial, and Vascular Compartments in Severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef] [PubMed]
- Caillon, A.; Trimaille, A.; Favre, J.; Jesel, L.; Morel, O.; Kauffenstein, G. Role of Neutrophils, Platelets, and Extracellular Vesicles and Their Interactions in COVID-19-Associated Thrombopathy. J. Thromb. Haemost. 2022, 20, 17–31. [Google Scholar] [CrossRef]
- Rabinovitch, M. NETs Activate Pulmonary Arterial Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2035–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil Extracellular Traps Induce Endothelial Dysfunction in Systemic Lupus Erythematosus through the Activation of Matrix Metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [Green Version]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin, G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.Y.; Zhang, J.H.; Cheng, W.; Chen, J.W.; Cui, N. Effects of Neutrophil Extracellular Traps in Patients with Septic Coagulopathy and Their Interaction with Autophagy. Front. Immunol. 2021, 12, 757041. [Google Scholar] [CrossRef]
- Osterud, B.; Rao, L.; Olsen, J. Induction of Tissue Factor Expression in Whole Blood: Lack of Evidence for the Presence of Tissue Factor Expression in Granulocytes—PubMed. Thromb. Haemost. 2000, 83, 861–867. [Google Scholar] [CrossRef]
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J.D. A Novel C5a Receptor-Tissue Factor Cross-Talk in Neutrophils Links Innate Immunity to Coagulation Pathways. J. Immunol. 2006, 177, 4794–4802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, N.; Brambilla, M.; Camera, M.; Carbone, A.; Tremoli, E.; Donati, M.B.; de Gaetano, G.; Cerletti, C. Human Polymorphonuclear Leukocytes Produce and Express Functional Tissue Factor upon Stimulation. J. Thromb. Haemost. 2006, 4, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, L.; Braun, O.; Westman, J.; Madhi, R.; Herwald, H.; Mörgelin, M.; Thorlacius, H. Neutrophil Extracellular Trap-Microparticle Complexes Enhance Thrombin Generation via the Intrinsic Pathway of Coagulation in Mice. Sci. Rep. 2018, 8, 4020. [Google Scholar] [CrossRef] [Green Version]
- Pitanga, T.N.; de Aragão França, L.; Rocha, V.C.J.; Meirelles, T.; Matos Borges, V.; Gonçalves, M.S.; Pontes-de-Carvalho, L.C.; Noronha-Dutra, A.A.; Dos-Santos, W.L.C. Neutrophil-Derived Microparticles Induce Myeloperoxidase-Mediated Damage of Vascular Endothelial Cells. BMC Cell Biol. 2014, 15, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belani, P.; Schefflein, J.; Kihira, S.; Rigney, B.; Delman, B.N.; Mahmoudi, K.; Mocco, J.; Majidi, S.; Yeckley, J.; Aggarwal, A.; et al. COVID-19 Is an Independent Risk Factor for Acute Ischemic Stroke. AJNR Am. J. Neuroradiol. 2020, 41, 1361–1364. [Google Scholar] [CrossRef]
- Zhang, C.; Shen, L.; Le, K.-J.; Pan, M.-M.; Kong, L.-C.; Gu, Z.-C.; Xu, H.; Zhang, Z.; Ge, W.-H.; Lin, H.-W. Incidence of Venous Thromboembolism in Hospitalized Coronavirus Disease 2019 Patients: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2020, 7, 151. [Google Scholar] [CrossRef]
- Du, Y.; Tu, L.; Zhu, P.; Mu, M.; Wang, R.; Yang, P.; Wang, X.; Hu, C.; Ping, R.; Hu, P.; et al. Clinical Features of 85 Fatal Cases of COVID-19 from Wuhan. A Retrospective Observational Study. Am. J. Respir. Crit. Care Med. 2020, 201, 1372–1379. [Google Scholar] [CrossRef] [Green Version]
- Linden, M.D. Platelet Physiology. Methods Mol. Biol. 2013, 992, 13–30. [Google Scholar] [CrossRef]
- Coughlin, S.R. Thrombin Signalling and Protease-Activated Receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Kral, J.B.; Schrottmaier, W.C.; Salzmann, M.; Assinger, A. Platelet Interaction with Innate Immune Cells. Transfus. Med. Hemother. 2016, 43, 78–88. [Google Scholar] [CrossRef]
- Bockmeyer, C.L.; Claus, R.A.; Budde, U.; Kentouche, K.; Schneppenheim, R.; Lösche, W.; Reinhart, K.; Brunkhorst, F.M. Inflammation-Associated ADAMTS13 Deficiency Promotes Formation of Ultra-Large von Willebrand Factor. Haematologica 2008, 93, 137–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaid, Y.; Guessous, F.; Puhm, F.; Elhamdani, W.; Chentoufi, L.; Morris, A.C.; Cheikh, A.; Jalali, F.; Boilard, E.; Flamand, L. Platelet Reactivity to Thrombin Differs between Patients with COVID-19 and Those with ARDS Unrelated to COVID-19. Blood Adv. 2021, 5, 635. [Google Scholar] [CrossRef] [PubMed]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; ben El Haj, R.; et al. Platelets Can Associate with SARS-CoV-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Kanth Manne, B.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet Gene Expression and Function in Patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Grobbelaar, L.M.; Venter, C.; Vlok, M.; Ngoepe, M.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. SARS-CoV-2 Spike Protein S1 Induces Fibrin(Ogen) Resistant to Fibrinolysis: Implications for Microclot Formation in COVID-19. Biosci. Rep. 2021, 41, 20210611. [Google Scholar] [CrossRef] [PubMed]
- Bouck, E.G.; Denorme, F.; Holle, L.A.; Middelton, E.A.; Blair, A.M.; de Laat, B.; Schiffman, J.D.; Yost, C.C.; Rondina, M.T.; Wolberg, A.S.; et al. COVID-19 and Sepsis Are Associated with Different Abnormalities in Plasma Procoagulant and Fibrinolytic Activity. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 401. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-Derived HMGB1 Is a Critical Mediator of Thrombosis. J. Clin. Invest. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [Green Version]
- Berthet, J.; Damien, P.; Hamzeh-Cognasse, H.; Arthaud, C.A.; Eyraud, M.A.; Zéni, F.; Pozzetto, B.; McNicol, A.; Garraud, O.; Cognasse, F. Human Platelets Can Discriminate between Various Bacterial LPS Isoforms via TLR4 Signaling and Differential Cytokine Secretion. Clin. Immunol. 2012, 145, 189–200. [Google Scholar] [CrossRef]
- Zhang, G.; Han, J.; Welch, E.J.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B.; Du, X.; Li, Z. Lipopolysaccharide Stimulates Platelet Secretion and Potentiates Platelet Aggregation via TLR4/MyD88 and the CGMP-Dependent Protein Kinase Pathway. J. Immunol. 2009, 182, 7997–8004. [Google Scholar] [CrossRef] [Green Version]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular Histones Promote Thrombin Generation through Platelet-Dependent Mechanisms: Involvement of Platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Arman, M.; Krauel, K.; Tilley, D.O.; Weber, C.; Cox, D.; Greinacher, A.; Kerrigan, S.W.; Watson, S.P. Amplification of Bacteria-Induced Platelet Activation Is Triggered by FcγRIIA, Integrin AIIbβ3, and Platelet Factor 4. Blood 2014, 123, 3166–3174. [Google Scholar] [CrossRef] [PubMed]
- Miajlovic, H.; Zapotoczna, M.; Geoghegan, J.A.; Kerrigan, S.W.; Speziale, P.; Foster, T.J. Direct Interaction of Iron-Regulated Surface Determinant IsdB of Staphylococcus Aureus with the GPIIb/IIIa Receptor on Platelets. Microbiology 2010, 156, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, M.P.; Loughman, A.; Devocelle, M.; Arasu, S.; Chubb, A.J.; Foster, T.J.; Cox, D. Elucidating the Role of Staphylococcus Epidermidis Serine-Aspartate Repeat Protein G in Platelet Activation. J. Thromb. Haemost. 2009, 7, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.; Bridge, A.; le Mercier, P. A Potential Role for Integrins in Host Cell Entry by SARS-CoV-2. Antivir. Res. 2020, 177, 104759. [Google Scholar] [CrossRef]
- Garciarena, C.D.; McHale, T.M.; Watkin, R.L.; Kerrigan, S.W. Coordinated Molecular Cross-Talk between Staphylococcus Aureus, Endothelial Cells and Platelets in Bloodstream Infection. Pathogens 2015, 4, 869. [Google Scholar] [CrossRef] [Green Version]
- Bury, L.; Camilloni, B.; Castronari, R.; Piselli, E.; Malvestiti, M.; Borghi, M.; KuchiBotla, H.; Falcinelli, E.; Petito, E.; Amato, F.; et al. Search for SARS-CoV-2 RNA in Platelets from COVID-19 Patients. Platelets 2021, 32, 284–287. [Google Scholar] [CrossRef]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Tanriverdi, K.; Somasundaran, M.; Liu, P.; Soofi, S.; Bhandari, R.; Godwin, M.; Parsi, K.M.; et al. SARS-CoV-2 Initiates Programmed Cell Death in Platelets. Circ. Res. 2021, 129, 631. [Google Scholar] [CrossRef]
- Jevtic, S.D.; Nazy, I. The COVID Complex: A Review of Platelet Activation and Immune Complexes in COVID-19. Front. Immunol. 2022, 13, 594. [Google Scholar] [CrossRef]
- Brambilla, M.; Canzano, P.; Becchetti, A.; Tremoli, E.; Camera, M. Alterations in Platelets during SARS-CoV-2 Infection. Platelets 2021, 33, 1. [Google Scholar] [CrossRef]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yu, A.; Zhou, X. Elevation of Plasma Angiotensin II Level Is a Potential Pathogenesis for the Critically Ill COVID-19 Patients. Crit. Care 2020, 24, 290. [Google Scholar] [CrossRef]
- Maugeri, N.; de Lorenzo, R.; Clementi, N.; Antonia Diotti, R.; Criscuolo, E.; Godino, C.; Tresoldi, C.; Bio Angels For Covid-BioB Study Group; Bonini, C.; Clementi, M.; et al. Unconventional CD147-dependent Platelet Activation Elicited by SARS-CoV-2 in COVID-19. J. Thromb. Haemost. 2022, 20, 434. [Google Scholar] [CrossRef] [PubMed]
- Schwertz, H.; Tolley, N.D.; Foulks, J.M.; Denis, M.M.; Risenmay, B.W.; Buerke, M.; Tilley, R.E.; Rondina, M.T.; Harris, E.M.; Kraiss, L.W.; et al. Signal-Dependent Splicing of Tissue Factor Pre-MRNA Modulates the Thrombogenicity of Human Platelets. J. Exp. Med. 2006, 203, 2433–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.T.; Narayanan, P.; Li, W.; Silverstein, R.L.; McIntyre, T.M. Lipopolysaccharide Stimulates Platelets through an IL-1β Autocrine Loop. J. Immunol. 2013, 191, 5196–5203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.T.; McIntyre, T.M. Lipopolysaccharide Signaling without a Nucleus: Kinase Cascades Stimulate Platelet Shedding of Proinflammatory IL-1β-Rich Microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated Platelets Mediate Inflammatory Signaling by Regulated Interleukin 1beta Synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Denis, M.M.; Tolley, N.D.; Bunting, M.; Schwertz, H.; Jiang, H.; Lindemann, S.; Yost, C.C.; Rubner, F.J.; Albertine, K.H.; Swoboda, K.J.; et al. Escaping the Nuclear Confines: Signal-Dependent Pre-MRNA Splicing in Anucleate Platelets. Cell 2005, 122, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, L.M.; Lin, E.; Mick, E.; Koupenova, M.; Weinberg, E.O.; Kramer, C.D.; Genco, C.A.; Tanriverdi, K.; Larson, M.G.; Benjamin, E.J.; et al. Interleukin 1 Receptor 1 and Interleukin 1β Regulate Megakaryocyte Maturation, Platelet Activation, and Transcript Profile during Inflammation in Mice and Humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Wu, X.; Luo, Q.; Wei, G.; Xu, M.; Wu, Y.; Liu, Y.; Li, X.; Zi, J.; Ju, W.; et al. NLRP3 Regulates Platelet Integrin AIIbβ3 Outside-in Signaling, Hemostasis and Arterial Thrombosis. Haematologica 2018, 103, 1568–1576. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Apta, B.H.R.; Bonna, A.M.; Harper, M.T. Platelet P-Selectin Triggers Rapid Surface Exposure of Tissue Factor in Monocytes. Sci. Rep. 2019, 9, 13397. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the Extrinsic Pathway of Blood Coagulation in Hemostasis and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 Infection: The Role of Cytokines in COVID-19 Disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Patko, Z.; Csaszar, A.; Acsady, G.; Peter, K.; Schwarz, M. Roles of Mac-1 and Glycoprotein IIb/IIIa Integrins in Leukocyte-Platelet Aggregate Formation: Stabilization by Mac-1 and Inhibition by GpIIb/IIIa Blockers. Platelets 2012, 23, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, R.; Ustinov, V.; Chen, Z.; Zhang, X.; Rao, R.; Luscinskas, F.W.; Lopez, J.; Plow, E.; Simon, D.I. Targeting Platelet-Leukocyte Interactions: Identification of the Integrin Mac-1 Binding Site for the Platelet Counter Receptor Glycoprotein Ibalpha. J. Exp. Med. 2003, 198, 1077–1088. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.F.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet Glycoprotein Ibalpha Is a Counterreceptor for the Leukocyte Integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Lipińska-Gediga, M. Platelets in Sepsis—Are There Any New Aspects? Anaesthesiol. Intensive Ther. 2017, 49, 167–172. [Google Scholar] [CrossRef]
- Taus, F.; Salvagno, G.; Canè, S.; Fava, C.; Mazzaferri, F.; Carrara, E.; Petrova, V.; Barouni, R.M.; Dima, F.; Dalbeni, A.; et al. Platelets Promote Thromboinflammation in SARS-CoV-2 Pneumonia. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2975. [Google Scholar] [CrossRef]
- Zhang, J.; Tecson, K.M.; McCullough, P.A. Endothelial Dysfunction Contributes to COVID-19-Associated Vascular Inflammation and Coagulopathy. Rev. Cardiovasc. Med. 2020, 21, 315–319. [Google Scholar] [CrossRef]
- le Joncour, A.; Biard, L.; Vautier, M.; Bugaut, H.; Mekinian, A.; Maalouf, G.; Vieira, M.; Marcelin, A.G.; Rosenzwajg, M.; Klatzmann, D.; et al. Neutrophil–Platelet and Monocyte–Platelet Aggregates in COVID-19 Patients. Thromb. Haemost. 2020, 120, 1733. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet Activation and Platelet-Monocyte Aggregate Formation Trigger Tissue Factor Expression in Patients with Severe COVID-19. Blood 2020, 136, 1330. [Google Scholar] [CrossRef]
- Semple, J.W.; Italiano, J.E.; Freedman, J. Platelets and the Immune Continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Reid, V.L.; Webster, N.R. Role of Microparticles in Sepsis. Br. J. Anaesth. 2012, 109, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Stoppelaar, S.F.; van ’t Veer, C.; van der Poll, T. The Role of Platelets in Sepsis. Thromb. Haemost. 2014, 112, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; et al. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic Transl. Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Rosell, A.; Havervall, S.; von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thålin, C. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated With Severity and Mortality-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Zahran, A.M.; El-Badawy, O.; Ali, W.A.; Mahran, Z.G.; Mahran, E.E.M.O.; Rayan, A. Circulating Microparticles and Activated Platelets as Novel Prognostic Biomarkers in COVID-19; Relation to Cancer. PLoS ONE 2021, 16, e0246806. [Google Scholar] [CrossRef] [PubMed]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as New Vesicular Lipid Transporters Involved in Cell-Cell Communication and Various Pathophysiologies. Biochim. Biophys. Acta 2014, 1841, 108–120. [Google Scholar] [CrossRef]
- Köffel, R.; Wolfmeier, H.; Larpin, Y.; Besançon, H.; Schoenauer, R.; Babiychuk, V.S.; Drücker, P.; Pabst, T.; Mitchell, T.J.; Babiychuk, E.B.; et al. Host-Derived Microvesicles Carrying Bacterial Pore-Forming Toxins Deliver Signals to Macrophages: A Novel Mechanism of Shaping Immune Responses. Front. Immunol. 2018, 9, 1688. [Google Scholar] [CrossRef] [Green Version]
- Varon, D.; Shai, E. Platelets and Their Microparticles as Key Players in Pathophysiological Responses. J. Thromb. Haemost. 2015, 13 (Suppl. 1), S40–S46. [Google Scholar] [CrossRef]
- Kuckleburg, C.J.; Tiwari, R.; Czuprynski, C.J. Endothelial Cell Apoptosis Induced by Bacteria-Activated Platelets Requires Caspase-8 and -9 and Generation of Reactive Oxygen Species. Thromb. Haemost. 2008, 99, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, L.C.P.; Janiszewski, M.; Pontieri, V.; de Almeida Pedro, M.; Bassi, E.; Tucci, P.J.F.; Laurindo, F.R.M. Platelet-Derived Exosomes from Septic Shock Patients Induce Myocardial Dysfunction. Crit. Care 2007, 11, R120. [Google Scholar] [CrossRef]
- Janiszewski, M.; do Carmo, A.O.; Pedro, M.A.; Silva, E.; Knobel, E.; Laurindo, F.R.M. Platelet-Derived Exosomes of Septic Individuals Possess Proapoptotic NAD(P)H Oxidase Activity: A Novel Vascular Redox Pathway. Crit. Care Med. 2004, 32, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, D.; Negru, A.; Mihailescu, R.; Tiliscan, C.; Tudor, A.-M.; Lazăr, M.; Aramă, Ș.S.; Ion, D.; Popescu, C.; Aramă, V. Evaluation of Bone Mineral Density and Correlations with Inflammation Markers in Romanian HIV-Positive Patients Undergoing Combined Antiretroviral Therapy. Farmacia 2017, 65, 114–119. [Google Scholar]
- Barbu, E.C.; Chiţu-Tișu, C.E.; Lazăr, M.; Olariu, C.M.; Olteanu, D.; Bojincă, M.; Abagiu, A.O.; Aramă, V.; Ion, D.A.; Bădărău, I.A. Body Composition Changes in Patients with Chronic Hepatitis C. J. Gastrointest. Liver Dis. 2016, 25, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Garcia, C.; Compagnon, B.; Poëtte, M.; Gratacap, M.P.; Lapébie, F.X.; Voisin, S.; Minville, V.; Payrastre, B.; Vardon-Bounes, F.; Ribes, A. Platelet Versus Megakaryocyte: Who Is the Real Bandleader of Thromboinflammation in Sepsis? Cells 2022, 11, 1507. [Google Scholar] [CrossRef] [PubMed]
- Pariser, D.N.; Hilt, Z.T.; Ture, S.K.; Blick-Nitko, S.K.; Looney, M.R.; Cleary, S.J.; Roman-Pagan, E.; Saunders, J.; Georas, S.N.; Veazey, J.; et al. Lung Megakaryocytes Are Immune Modulatory Cells. J. Clin. Invest. 2021, 131, e137377. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 Stimulates Thrombopoiesis through Thrombopoietin: Role in Inflammatory Thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef] [Green Version]
- Valet, C.; Magnen, M.; Qiu, L.; Cleary, S.J.; Wang, K.M.; Ranucci, S.; Grockowiak, E.; Boudra, R.; Conrad, C.; Seo, Y.; et al. Sepsis Promotes Splenic Production of a Protective Platelet Pool with High CD40 Ligand Expression. J. Clin. Invest. 2022, 132, e153920. [Google Scholar] [CrossRef]
- Zufferey, A.; Speck, E.R.; Machlus, K.R.; Aslam, R.; Guo, L.; McVey, M.J.; Kim, M.; Kapur, R.; Boilard, E.; Italiano, J.E.; et al. Mature Murine Megakaryocytes Present Antigen-MHC Class I Molecules to T Cells and Transfer Them to Platelets. Blood Adv. 2017, 1, 1773–1785. [Google Scholar] [CrossRef]
- Wang, J.; Xie, J.; Wang, D.; Han, X.; Chen, M.; Shi, G.; Jiang, L.; Zhao, M. CXCR4high Megakaryocytes Regulate Host-Defense Immunity against Bacterial Pathogens. Elife 2022, 11, e78662. [Google Scholar] [CrossRef]
- Chiţu-Tişu, C.-E.; Barbu, E.-C.; Lazăr, M.; Ionescu, R.; Bojinca, M.; Ion, D.; Bădărău, I.A. An Overview of Bone Disease in HIV-Infected Patients. Acta Med. Mediterr. 2015, 31, 1139–1151. [Google Scholar]
- Comer, S.P.; Cullivan, S.; Szklanna, P.B.; Weiss, L.; Cullen, S.; Kelliher, S.; Smolenski, A.; Murphy, C.; Altaie, H.; Curran, J.; et al. COVID-19 Induces a Hyperactive Phenotype in Circulating Platelets. PLoS Biol. 2021, 19, e3001109. [Google Scholar] [CrossRef]
- Ushiyama, A.; Kataoka, H.; Iijima, T. Glycocalyx and Its Involvement in Clinical Pathophysiologies. J. Intensive Care 2016, 4, 59. [Google Scholar] [CrossRef]
- Endo, S.; Shimazaki, R.; Gando, S.; Oda, S.; Ootomo, Y.; Miura, M.; Ogura, S.; Eguchi, Y.; Kotani, J.; Yamashita, N.; et al. An Open-Label, Randomized, Phase 3 Study of the Efficacy and Safety of Antithrombin Gamma in Patients with Sepsis-Induced Disseminated Intravascular Coagulation Syndrome. J. Intensive Care 2018, 6, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Maruyama, I.; Shimazaki, S.; Yamamoto, Y.; Aikawa, N.; Hirayama, A.; Honda, G.; Saito, H. Effects of Thrombomodulin Alfa on Hemostatic Parameters in Disseminated Intravascular Coagulation: Post Hoc Analysis of a Phase 3 Randomized Controlled Trial. Res. Pract. Thromb. Haemost. 2020, 4, 1141–1149. [Google Scholar] [CrossRef]
- Higgins, S.J.; de Ceunynck, K.; Kellum, J.A.; Chen, X.; Gu, X.; Chaudhry, S.A.; Schulman, S.; Libermann, T.A.; Lu, S.; Shapiro, N.I.; et al. Tie2 Protects the Vasculature against Thrombus Formation in Systemic Inflammation. J. Clin. Invest. 2018, 128, 1471–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120. [Google Scholar] [CrossRef] [PubMed]
- Khakpour, S.; Wilhelmsen, K.; Hellman, J. Vascular Endothelial Cell Toll-like Receptor Pathways in Sepsis. Innate Immun. 2015, 21, 827–846. [Google Scholar] [CrossRef] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Smadja, D.M.; Guerin, C.L.; Chocron, R.; Yatim, N.; Boussier, J.; Gendron, N.; Khider, L.; Hadjadj, J.; Goudot, G.; Debuc, B.; et al. Angiopoietin-2 as a Marker of Endothelial Activation Is a Good Predictor Factor for Intensive Care Unit Admission of COVID-19 Patients. Angiogenesis 2020, 23, 611–620. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.S.; Chien, S. Shear Stress-Initiated Signaling and Its Regulation of Endothelial Function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Orr, A.W.; Sanders, J.M.; Bevard, M.; Coleman, E.; Sarembock, I.J.; Schwartz, M.A. The Subendothelial Extracellular Matrix Modulates NF-KappaB Activation by Flow: A Potential Role in Atherosclerosis. J. Cell Biol. 2005, 169, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutikhin, A.G.; Sinitsky, M.Y.; Yuzhalin, A.E.; Velikanova, E.A. Shear Stress: An Essential Driver of Endothelial Progenitor Cells. J. Mol. Cell Cardiol. 2018, 118, 46–69. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.M.; Gonagle, D.M.; Ward, S.E.; Preston, R.J.S.; O’Donnell, J.S. Endothelial Cells Orchestrate COVID-19 Coagulopathy. Lancet Haematol. 2020, 7, e553–e555. [Google Scholar] [CrossRef] [PubMed]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 Spike Protein Alters Barrier Function in 2D Static and 3D Microfluidic in-Vitro Models of the Human Blood-Brain Barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Pickering, R.J.; Tsorotes, D.; Koitka, A.; Sheehy, K.; Bernardi, S.; Toffoli, B.; Nguyen-Huu, T.P.; Head, G.A.; Fu, Y.; et al. Genetic Ace2 Deficiency Accentuates Vascular Inflammation and Atherosclerosis in the ApoE Knockout Mouse. Circ. Res. 2010, 107, 888–897. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Thippegowda, P.B.; Hu, G.; Bachmaier, K.; Christman, J.W.; Malik, A.B.; Tiruppathi, C. NF-KappaB Regulates Thrombin-Induced ICAM-1 Gene Expression in Cooperation with NFAT by Binding to the Intronic NF-KappaB Site in the ICAM-1 Gene. Physiol.Genom. 2009, 38, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The Vasculature Unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Kouijzer, I.J.E.; de Nooijer, A.H.; van der Hoeven, H.G.; Maas, C.; Netea, M.G.; Brüggemann, R.J.M.; van de Veerdonk, F.L. Outcomes Associated with Use of a Kinin B2 Receptor Antagonist Among Patients With COVID-19. JAMA Netw. Open 2020, 3, e2017708. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Michels, A.; Albánez, S.; Mewburn, J.; Nesbitt, K.; Gould, T.J.; Liaw, P.C.; James, P.D.; Swystun, L.L.; Lillicrap, D. Histones Link Inflammation and Thrombosis through the Induction of Weibel-Palade Body Exocytosis. J. Thromb. Haemost. 2016, 14, 2274–2286. [Google Scholar] [CrossRef]
- Nightingale, T.; Cutler, D. The Secretion of von Willebrand Factor from Endothelial Cells; an Increasingly Complicated Story. J. Thromb. Haemost. 2013, 11 (Suppl. 1), 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escher, R.; Breakey, N.; Lämmle, B. ADAMTS13 Activity, von Willebrand Factor, Factor VIII and D-Dimers in COVID-19 Inpatients. Thromb. Res. 2020, 192, 174–175. [Google Scholar] [CrossRef] [PubMed]
- Booth, K.K.; Terrell, D.R.; Vesely, S.K.; George, J.N. Systemic Infections Mimicking Thrombotic Thrombocytopenic Purpura. Am. J. Hematol. 2011, 86, 743–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, M.; van der Poll, T. Inflammation and Coagulation. Crit. Care Med. 2010, 38, 26–34. [Google Scholar] [CrossRef] [PubMed]
- van der Poll, T.; Parker, R.I. Platelet Activation and Endothelial Cell Dysfunction. Crit. Care Clin. 2020, 36, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Keith, P.; Day, M.; Perkins, L.; Moyer, L.; Hewitt, K.; Wells, A. A Novel Treatment Approach to the Novel Coronavirus: An Argument for the Use of Therapeutic Plasma Exchange for Fulminant COVID-19. Crit. Care 2020, 24, 128. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Levy, J.H. Derangement of the Endothelial Glycocalyx in Sepsis. J. Thromb. Haemost. 2019, 17, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Bruegger, D.; Schwartz, L.; Chappell, D.; Jacob, M.; Rehm, M.; Vogeser, M.; Christ, F.; Reichart, B.; Becker, B.F. Release of Atrial Natriuretic Peptide Precedes Shedding of the Endothelial Glycocalyx Equally in Patients Undergoing On- and off-Pump Coronary Artery Bypass Surgery. Basic Res. Cardiol. 2011, 106, 1111–1121. [Google Scholar] [CrossRef]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef]
- Ito, T.; Kakuuchi, M.; Maruyama, I. Endotheliopathy in Septic Conditions: Mechanistic Insight into Intravascular Coagulation. Crit. Care 2021, 25, 95. [Google Scholar] [CrossRef]
- Vollenberg, R.; Tepasse, P.R.; Ochs, K.; Floer, M.; Strauss, M.; Rennebaum, F.; Kabar, I.; Rovas, A.; Nowacki, T. Indications of Persistent Glycocalyx Damage in Convalescent COVID-19 Patients: A Prospective Multicenter Study and Hypothesis. Viruses 2021, 13, 2324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, L.; Chen, Y.; Ma, J.; Yang, Y.; Aodeng, S.; Cui, Q.; Wen, K.; Xiao, M.; Xie, J.; et al. Syndecan-1, an Indicator of Endothelial Glycocalyx Degradation, Predicts Outcome of Patients Admitted to an ICU with COVID-19. Mol. Med. 2021, 27, 151. [Google Scholar] [CrossRef] [PubMed]
- Eslamifar, Z.; Behzadifard, M.; Soleimani, M.; Behzadifard, S. Coagulation Abnormalities in SARS-CoV-2 Infection: Overexpression Tissue Factor. Thromb. J. 2020, 18, 38. [Google Scholar] [CrossRef]
- Rodríguez, Y.; Novelli, L.; Rojas, M.; de Santis, M.; Acosta-Ampudia, Y.; Monsalve, D.M.; Ramírez-Santana, C.; Costanzo, A.; Ridgway, W.M.; Ansari, A.A.; et al. Autoinflammatory and Autoimmune Conditions at the Crossroad of COVID-19. J. Autoimmun. 2020, 114, 102506. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune Mechanisms of Pulmonary Intravascular Coagulopathy in COVID-19 Pneumonia. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar] [CrossRef]
- Venkata, C.; Kashyap, R.; Christopher Farmer, J.; Afessa, B. Thrombocytopenia in Adult Patients with Sepsis: Incidence, Risk Factors, and Its Association with Clinical Outcome. J. Intensive Care 2013, 1, 9. [Google Scholar] [CrossRef]
- Levi, M.; Schultz, M.; van der Poll, T. Sepsis and Thrombosis. Semin. Thromb. Hemost. 2013, 39, 559–566. [Google Scholar] [CrossRef]
- Taylor, F.B., Jr.; Toh, C.-H.; Hoots, W.K.; Wada, H.; Levi, M. Towards Definition, Clinical and Laboratory Criteria, and a Scoring System for Disseminated Intravascular Coagulation. Thromb. Haemost. 2001, 86, 1327–1330. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, F.C.F.; Manolov, V.; Morgenstern, J.; Fleming, T.; Heitmeier, S.; Uhle, F.; Al-Saeedi, M.; Hackert, T.; Bruckner, T.; Schöchl, H.; et al. Acute Fibrinolysis Shutdown Occurs Early in Septic Shock and Is Associated with Increased Morbidity and Mortality: Results of an Observational Pilot Study. Ann. Intensive Care 2019, 9, 19. [Google Scholar] [CrossRef]
- Iba, T.; Umemura, Y.; Wada, H.; Levy, J.H. Roles of Coagulation Abnormalities and Microthrombosis in Sepsis: Pathophysiology, Diagnosis, and Treatment. Arch. Med. Res. 2021, 52, 788–797. [Google Scholar] [CrossRef]
- Toh, C.H.; Dennis, M. Disseminated Intravascular Coagulation: Old Disease, New Hope. BMJ 2003, 327, 974–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, M. The Coagulant Response in Sepsis. Clin. Chest Med. 2008, 29, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Asakura, H.; Takahashi, H.; Uchiyama, T.; Eguchi, Y.; Okamoto, K.; Kawasugi, K.; Madoiwa, S.; Wada, H. Proposal for New Diagnostic Criteria for DIC from the Japanese Society on Thrombosis and Hemostasis. Thromb. J. 2016, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; di Nisio, M.; Levy, J.H.; Kitamura, N.; Thachil, J. New Criteria for Sepsis-Induced Coagulopathy (SIC) Following the Revised Sepsis Definition: A Retrospective Analysis of a Nationwide Survey. BMJ Open 2017, 7, e017046. [Google Scholar] [CrossRef] [Green Version]
- Moisa, E.; Corneci, D.; Negutu, M.I.; Filimon, C.R.; Serbu, A.; Popescu, M.; Negoita, S.; Grintescu, I.M. Development and Internal Validation of a New Prognostic Model Powered to Predict 28-Day All-Cause Mortality in ICU COVID-19 Patients—The COVID-SOFA Score. J. Clin. Med. 2022, 11, 4160. [Google Scholar] [CrossRef]
- Raschke, R.A.; Agarwal, S.; Rangan, P.; Heise, C.W.; Curry, S.C. Discriminant Accuracy of the SOFA Score for Determining the Probable Mortality of Patients With COVID-19 Pneumonia Requiring Mechanical Ventilation. JAMA 2021, 325, 1469. [Google Scholar] [CrossRef] [PubMed]
- Lazar, M.; Barbu, E.C.; Chitu, C.E.; Anghel, A.M.J.; Niculae, C.M.; Manea, E.D.; Damalan, A.C.; Bel, A.A.; Patrascu, R.E.; Hristea, A.; et al. Mortality Predictors in Severe SARS-CoV-2 Infection. Medicina 2022, 58, 945. [Google Scholar] [CrossRef]
- Belen-Apak, F.B.; Sarıalioğlu, F. Pulmonary Intravascular Coagulation in COVID-19: Possible Pathogenesis and Recommendations on Anticoagulant/Thrombolytic Therapy. J. Thromb. Thrombolysis 2020, 50, 278. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines Including Interleukin-6 in COVID-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Asakura, H.; Ogawa, H. COVID-19-Associated Coagulopathy and Disseminated Intravascular Coagulation. Int. J. Hematol. 2021, 113, 45. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 Patients in Intensive Care Unit: A Report of Thromboelastography Findings and Other Parameters of Hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary Manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Kohansal Vajari, M.; Shirin, M.; Pourbagheri-Sigaroodi, A.; Akbari, M.E.; Abolghasemi, H.; Bashash, D. COVID-19-Related Coagulopathy: A Review of Pathophysiology and Pharmaceutical Management. Cell Biol. Int. 2021, 45, 1832–1850. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Connors, J.M.; Nagaoka, I.; Levy, J.H. Recent Advances in the Research and Management of Sepsis-Associated DIC. Int. J. Hematol. 2021, 113, 24. [Google Scholar] [CrossRef] [PubMed]
- Ibañez, C.; Perdomo, J.; Calvo, A.; Ferrando, C.; Reverter, J.C.; Tassies, D.; Blasi, A. High D Dimers and Low Global Fibrinolysis Coexist in COVID19 Patients: What Is Going on in There? J. Thromb. Thrombolysis 2021, 51, 308–312. [Google Scholar] [CrossRef]
- Levi, M.; Iba, T. COVID-19 Coagulopathy: Is It Disseminated Intravascular Coagulation? Intern. Emerg. Med. 2021, 16, 309. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Hardy, M.; Bareille, M.; Lecompte, T.; Mullier, F. Don’t Let D-Dimer Fool You: Elevated D-Dimer Plasma Levels Should Not Imply ‘Hyperfibrinolysis’. Thromb. Res. 2022, 214, 63. [Google Scholar] [CrossRef]
- Hosseini, S.; Behnam-Roudsari, S.; Alavinia, G.; Emami, A.; Toghyani, A.; Moradi, S.; Zadeh, M.; Mohseni, S.; Shafiee, M. Diagnostic and Prognostic Value of Sepsis-Induced Coagulopathy and International Society on Thrombosis and Hemostasis Scoring Systems in COVID-19-Associated Disseminated Intravascular Coagulopathy. J. Res. Med. Sci. 2021, 26, 102. [Google Scholar] [CrossRef]
- Lippi, G.; Plebani, M.; Henry, B.M. Thrombocytopenia Is Associated with Severe Coronavirus Disease 2019 (COVID-19) Infections: A Meta-Analysis. Clin. Chim. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef]
- Bazzan, M.; Montaruli, B.; Sciascia, S.; Cosseddu, D.; Norbiato, C.; Roccatello, D. Low ADAMTS 13 Plasma Levels Are Predictors of Mortality in COVID-19 Patients. Intern. Emerg. Med. 2020, 15, 861–863. [Google Scholar] [CrossRef]
- Levi, M.; Meijers, J.C. DIC: Which Laboratory Tests Are Most Useful. Blood Rev. 2011, 25, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Colucci, M.; Caironi, P.; Masson, S.; Ammollo, C.T.; Teli, R.; Semeraro, N.; Magnoli, M.; Salati, G.; Isetta, M.; et al. Platelet Drop and Fibrinolytic Shutdown in Patients with Sepsis. Crit. Care Med. 2018, 46, E221–E228. [Google Scholar] [CrossRef] [PubMed]
- Rodelo, J.R.; de La Rosa, G.; Valencia, M.L.; Ospina, S.; Arango, C.M.; Gómez, C.I.; García, A.; Nuñez, E.; Jaimes, F.A. D-Dimer Is a Significant Prognostic Factor in Patients with Suspected Infection and Sepsis. Am. J. Emerg. Med. 2012, 30, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Schwameis, M.; Steiner, M.M.; Schoergenhofer, C.; Lagler, H.; Buchtele, N.; Jilma-Stohlawetz, P.; Boehm, T.; Jilma, B. D-Dimer and Histamine in Early Stage Bacteremia: A Prospective Controlled Cohort Study. Eur. J. Intern. Med. 2015, 26, 782–786. [Google Scholar] [CrossRef] [Green Version]
- Levi, M.; Vincent, J.L.; Tanaka, K.; Radford, A.H.; Kayanoki, T.; Fineberg, D.A.; Hoppensteadt, D.; Fareed, J. Effect of a Recombinant Human Soluble Thrombomodulin on Baseline Coagulation Biomarker Levels and Mortality Outcome in Patients with Sepsis-Associated Coagulopathy. Crit. Care Med. 2020, 48, 1140–1147. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Caironi, P.; Masson, S.; Latini, R.; Panigada, M.; Semeraro, N.; Gattinoni, L.; Colucci, M. Low D-Dimer Levels in Sepsis: Good or Bad? Thromb. Res. 2019, 174, 13–15. [Google Scholar] [CrossRef]
- Han, Y.Q.; Yan, L.; Zhang, L.; Ouyang, P.H.; Li, P.; Lippi, G.; Hu, Z. de Performance of D-Dimer for Predicting Sepsis Mortality in the Intensive Care Unit. Biochem. Med. 2021, 31, 020709. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiari, K.; Meijers, J.C.M.; de Jonge, E.; Levi, M. Prospective Validation of the International Society of Thrombosis and Haemostasis Scoring System for Disseminated Intravascular Coagulation. Crit. Care Med. 2004, 32, 2416–2421. [Google Scholar] [CrossRef]
- Levi, M.; de Jonge, E.; Meijers, J. The Diagnosis of Disseminated Intravascular Coagulation. Blood Rev. 2002, 16, 217–223. [Google Scholar] [CrossRef]
- Hunt, B.J.; Levi, M. Re The Source of Elevated Plasma D-dimer Levels in COVID-19 Infection. Br. J. Haematol. 2020, 190, e133. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Levi, M.; Hunt, B.J. Thrombosis and Coagulopathy in COVID-19: An Illustrated Review. Res. Pract. Thromb. Haemost. 2020, 4, 744–751. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation Abnormalities and Thrombosis in Patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal Coagulation Parameters Are Associated with Poor Prognosis in Patients with Novel Coronavirus Pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent Changes in Blood Coagulation of Patients with SARS-CoV-2 Infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Amgalan, A.; Othman, M. Hemostatic Laboratory Derangements in COVID-19 with a Focus on Platelet Count. Platelets 2020, 31, 740–745. [Google Scholar] [CrossRef]
- Yang, X.; Yang, Q.; Wang, Y.; Wu, Y.; Xu, J.; Yu, Y.; Shang, Y. Thrombocytopenia and Its Association with Mortality in Patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1469–1472. [Google Scholar] [CrossRef]
- Wool, G.D.; Miller, J.L. The Impact of COVID-19 Disease on Platelets and Coagulation. Pathobiology 2021, 88, 15–27. [Google Scholar] [CrossRef]
- Asakura, H. Classifying Types of Disseminated Intravascular Coagulation: Clinical and Animal Models. J. Intensive Care 2014, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary Post-Mortem Findings in a Series of COVID-19 Cases from Northern Italy: A Two-Centre Descriptive Study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.C.; Meijers, J.C.M.; Vroom, M.B.; Juffermans, N.P. Utility of Thromboelastography and/or Thromboelastometry in Adults with Sepsis: A Systematic Review. Crit. Care 2014, 18, R30. [Google Scholar] [CrossRef]
- Haase, N.; Ostrowski, S.R.; Wetterslev, J.; Lange, T.; Møller, M.H.; Tousi, H.; Steensen, M.; Pott, F.; Søe-Jensen, P.; Nielsen, J.; et al. Thromboelastography in Patients with Severe Sepsis: A Prospective Cohort Study. Intensive Care Med. 2015, 41, 77–85. [Google Scholar] [CrossRef]
- Ostrowski, S.R.; Windeløv, N.A.; Ibsen, M.; Haase, N.; Perner, A.; Johansson, P.I. Consecutive Thrombelastography Clot Strength Profiles in Patients with Severe Sepsis and Their Association with 28-Day Mortality: A Prospective Study. J. Crit. Care 2013, 28, 317.e1–317.e11. [Google Scholar] [CrossRef]
- Johansson, P.I.; Stensballe, J.; Vindeløv, N.; Perner, A.; Espersen, K. Hypocoagulability, as Evaluated by Thrombelastography, at Admission to the ICU Is Associated with Increased 30-Day Mortality. Blood Coagul. Fibrinolysis 2010, 21, 168–174. [Google Scholar] [CrossRef]
- Boscolo, A.; Spiezia, L.; Campello, E.; Bertini, D.; Lucchetta, V.; Piasentini, E.; de Cassai, A.; Simioni, P. Whole-Blood Hypocoagulable Profile Correlates with a Greater Risk of Death within 28 Days in Patients with Severe Sepsis. Korean J. Anesthesiol. 2020, 73, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Adamzik, M.; Langemeier, T.; Frey, U.H.; Görlinger, K.; Saner, F.; Eggebrecht, H.; Peters, J.; Hartmann, M. Comparison of Thrombelastometry with Simplified Acute Physiology Score Ii and Sequential Organ Failure Assessment Scores for the Prediction of 30-Day Survival: A Cohort Study. Shock 2011, 35, 339–342. [Google Scholar] [CrossRef]
- Muzaffar, S.N.; Azim, A.; Siddiqui, S.S. Thromboelastography for Predicting Disseminated Intravascular Coagulation (DIC) in Sepsis. Shock 2022, 57, 759. [Google Scholar] [CrossRef]
- Brenner, T.; Schmidt, K.; Delang, M.; Mehrabi, A.; Bruckner, T.; Lichtenstern, C.; Martin, E.; Weigand, M.A.; Hofer, S. Viscoelastic and Aggregometric Point-of-Care Testing in Patients with Septic Shock—Cross-Links between Inflammation and Haemostasis. Acta Anaesthesiol. Scand. 2012, 56, 1277–1290. [Google Scholar] [CrossRef]
- Sivula, M.; Pettilä, V.; Niemi, T.T.; Varpula, M.; Kuitunen, A.H. Thromboelastometry in Patients with Severe Sepsis and Disseminated Intravascular Coagulation. Blood Coagul. Fibrinolysis 2009, 20, 419–426. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, S.-I.; Yu, G.; Kim, J.S.; Hong, S.I.; Chae, B.; Shin, Y.S.; Kim, Y.J.; Jang, S.; Kim, W.Y. Role of Thromboelastography in the Evaluation of Septic Shock Patients with Normal Prothrombin Time and Activated Partial Thromboplastin Time. Sci. Rep. 2021, 11, 11833. [Google Scholar] [CrossRef] [PubMed]
- Pavoni, V.; Gianesello, L.; Pazzi, M.; Stera, C.; Meconi, T.; Frigieri, F.C. Evaluation of Coagulation Function by Rotation Thromboelastometry in Critically Ill Patients with Severe COVID-19 Pneumonia. J. Thromb. Thrombolysis 2020, 50, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Gergi, M.; Goodwin, A.; Freeman, K.; Colovos, C.; Volod, O. Viscoelastic Hemostasis Assays (VHA) in Septic, Critically Ill COVID-19 Patients: A Practical Guide for Clinicians. Blood Coagul. Fibrinolysis 2021, 32, 225. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.L.; Vogler, T.O.; Moore, E.E.; Moore, H.B.; Wohlauer, M.V.; Urban, S.; Nydam, T.L.; Moore, P.K.; McIntyre, R.C. Fibrinolysis Shutdown Correlation with Thromboembolic Events in Severe COVID-19 Infection. J. Am. Coll. Surg. 2020, 231, 193–203.e1. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Commonalities between Sepsis and COVID-19-Associated Coagulopathies | Characteristics of Sepsis-Induced Coagulopathy | Characteristics of COVID-19-Associated Coagulopathy |
|---|---|---|
| TLRs and CLRs found on cell membranes and endosomes interact with PAMPs and DAMPs [10,11]. The synthesis of proinflammatory cytokines is activated via MAPK and NF-κB pathways [10,11,209]. Increased PAI-1 levels can be found in both COVID-19 and sepsis. [45,46] TF, FVIII, u-PA, PAI-1, TFPI, antithrombin and thrombomodulin are endpoints of the NF-κB pathway [12,13,14,15,16]. Histones promote platelet aggregation, bind prothrombotic molecules, and damage the antithrombotic properties of the endothelial glycocalyx [50,51]. Activated neutrophils generate prothrombotic and hyperinflammatory states via NETs formation and inflammasome activation, leading to immunothrombosis [88,89,90,91,92,93,94,95,96]. Intercellular interactions between platelets, endothelial cells, neutrophils and monocytes lead to MPs release and TF exposure [145,179]. Monocytes, as a source of soluble TF, promote the activation of coagulation via the extrinsic pathway [72,73]. Active platelets and MPs induce adhesion molecules’ expression and the dysfunction of endothelial cells [127,128,129,130]. The antithrombin pathway and protein C anticoagulant system become dysfunctional due to impaired protein C synthesis and activation via thrombomodulin and endothelial protein C receptor deficiency [52,53,54,226,227]. Tie2, NF-κB and MAPK signaling lead to ECs acquisitionof a proinflammatory and prothrombotic phenotype [207,208,209,210,211,212,213]. IL-6 contributes to vascular permeability and TNF-α worsens the glycocalyx disruption of endothelial cells [219]. | HMGB1 can modulate fibrinolysis by interacting with plasminogen and t-PA, can promote coagulation via TF exposure on macrophages and inhibits the protein C pathway [25,26,27,28,29,30,31,32,33,34,35,36,37,38,39]. cfDNA activates coagulation via FXI and FXII [40,41]. High cfDNA concentrations inactivate t-PA by PAI-1 [42,43,44]. Increased TF release from monocytes occurs upon stimulation by LPS [74,75,76]. Fibrinolysis resistance is enhanced via plasminogen altering within NETs, the formation of fibrin–DNA tight complexes insensitive to plasmin and the activation of PAI-1 by cfDNA [42,114,123,124]. Sepsis is associated withADAMTS13 deficiency, which generates large circulating vWF multimers, excessively activating platelets [234,235,236,237]. Platelet activation can arise via direct and indirect bacterial–platelet interactions [153]. A septic proinflammatory status induces (via IL-6 and IL-3 stimulation) the release of thrombopoietin-independent thrombocytes with more numerous TLRs and interleukin receptors, more IL-6 and TNF-α [72,196]. Endothelial cells’ activation can be triggered by TNF-α and thrombin, bacterial LPS and other PAMPs, HMGB1 and other DAMPs, cytokines, such as IFN-γ and IL-1β and shear stress [72,207,208,209,210,211,212,213,214]. Reduced TFPI and t-PA synthesis and increased PAI-1 expression by ECs potentiate the prothrombotic status [179,209,219,224,228]. Glycocalyx injury is initiated by specific enzymes (glucuronidases, hyaluronidases, plasmin, ROS) and hypervolemia (via excessive fluid administration within sepsis management) [227,228,229,230,231]. | Activated coagulation FX and thrombin may cleave S protein and promote viral entry in a potential inflammation–coagulation positive feedback loop [61]. Platelet–monocyte interactions are themain stimulus for monocyte activation with robust cytokine and chemokine secretion and TF expression [77]. Low-density granulocytes are found in greater proportions in COVID-19 patients; they are more active in generating NETs, further enhancing hypercoagulability-mediated organ damage [94,95,96]. Platelets from COVID-19 patients exhibit a particular hyperreactivity to low-dose common agonists (such as collagen, α-thrombin or ADP) [143,144,145]. IL-6 and TNF-α can directly activate platelets; IL-6 and IL-1β can prime platelets before stimulation by classical agonists [161]. Pulmonary-residing megakaryocytes are theoretically susceptible to SARS-CoV-2 infection and may transfer viral particles and cytokines when new circulating platelets are generated [144]. SARS-CoV-2 can infect endothelial cells via ACE2 and TMPRSS2, with endotheliitis as a result of direct viral entry [208]. S-protein-related damage increases adhesion molecules’ exposure, ROS synthesis and matrix metalloproteinases’ release with the disruption of the endothelial barrier [179,209,215,216,217,218,219]. COVID-19 induces a hyperactive state of the KKS pathway, promoting vascular permeability [217,220,221]. Excessive complement activation via the lectin pathway accentuates endothelial-derived immunothrombosis [217,220,221]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuculeanu, G.; Barbu, E.C.; Lazar, M.; Chitu-Tisu, C.E.; Moisa, E.; Negoita, S.I.; Ion, D.A. Coagulation Disorders in Sepsis and COVID-19—Two Sides of the Same Coin? A Review of Inflammation–Coagulation Crosstalk in Bacterial Sepsis and COVID-19. J. Clin. Med. 2023, 12, 601. https://doi.org/10.3390/jcm12020601
Tuculeanu G, Barbu EC, Lazar M, Chitu-Tisu CE, Moisa E, Negoita SI, Ion DA. Coagulation Disorders in Sepsis and COVID-19—Two Sides of the Same Coin? A Review of Inflammation–Coagulation Crosstalk in Bacterial Sepsis and COVID-19. Journal of Clinical Medicine. 2023; 12(2):601. https://doi.org/10.3390/jcm12020601
Chicago/Turabian StyleTuculeanu, Georgeana, Ecaterina Constanta Barbu, Mihai Lazar, Cristina Emilia Chitu-Tisu, Emanuel Moisa, Silvius Ioan Negoita, and Daniela Adriana Ion. 2023. "Coagulation Disorders in Sepsis and COVID-19—Two Sides of the Same Coin? A Review of Inflammation–Coagulation Crosstalk in Bacterial Sepsis and COVID-19" Journal of Clinical Medicine 12, no. 2: 601. https://doi.org/10.3390/jcm12020601
APA StyleTuculeanu, G., Barbu, E. C., Lazar, M., Chitu-Tisu, C. E., Moisa, E., Negoita, S. I., & Ion, D. A. (2023). Coagulation Disorders in Sepsis and COVID-19—Two Sides of the Same Coin? A Review of Inflammation–Coagulation Crosstalk in Bacterial Sepsis and COVID-19. Journal of Clinical Medicine, 12(2), 601. https://doi.org/10.3390/jcm12020601