

Drug–Drug Interactions Involving Dexamethasone in Clinical Practice: Myth or Reality?
 , ,
, , 
Abstract
:1. Introduction
2. Dexamethasone, an Increasingly Used Molecule
3. Dexamethasone Pharmacokinetics, Metabolism, and Drug Interactions

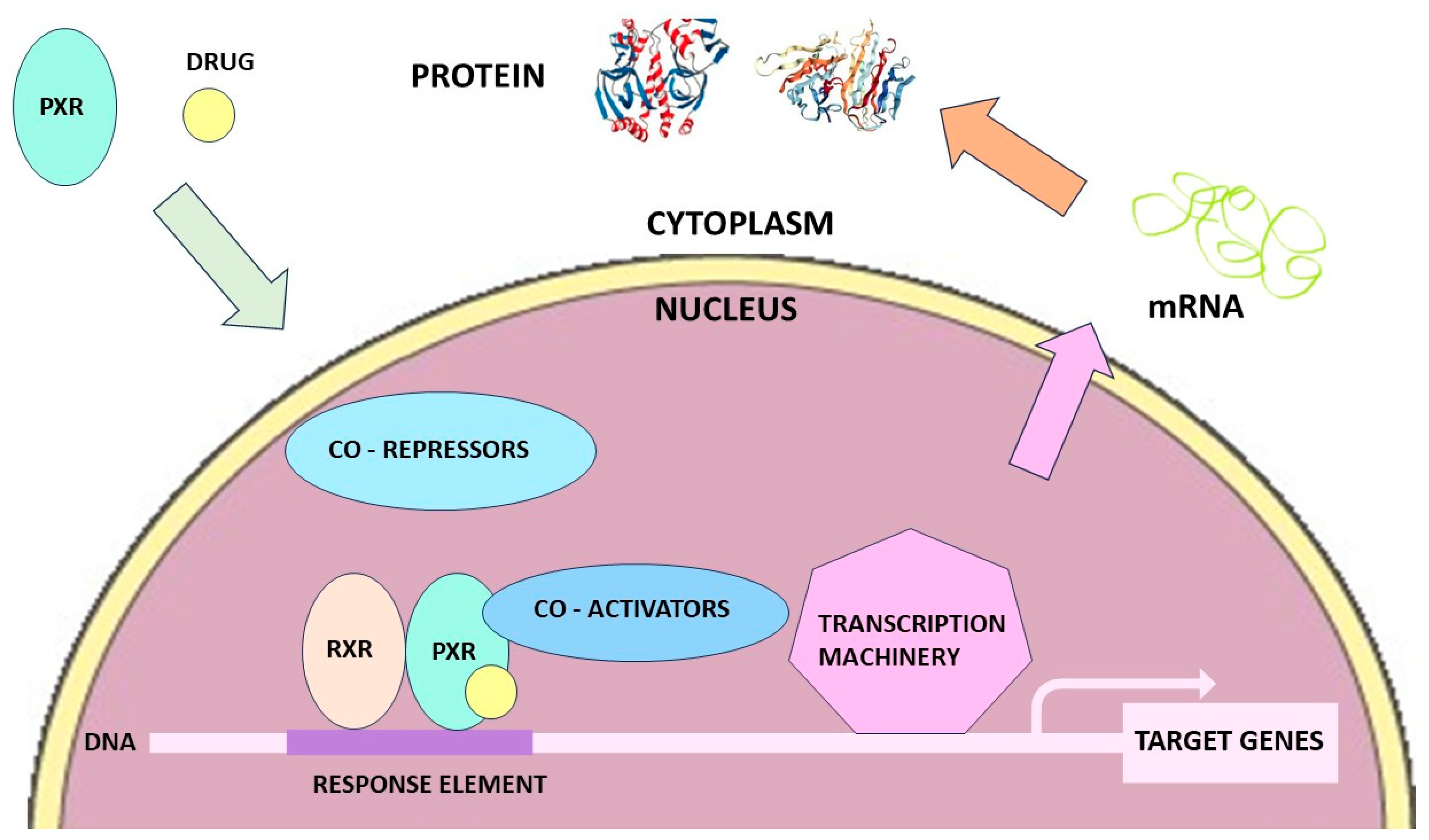{kind=link}
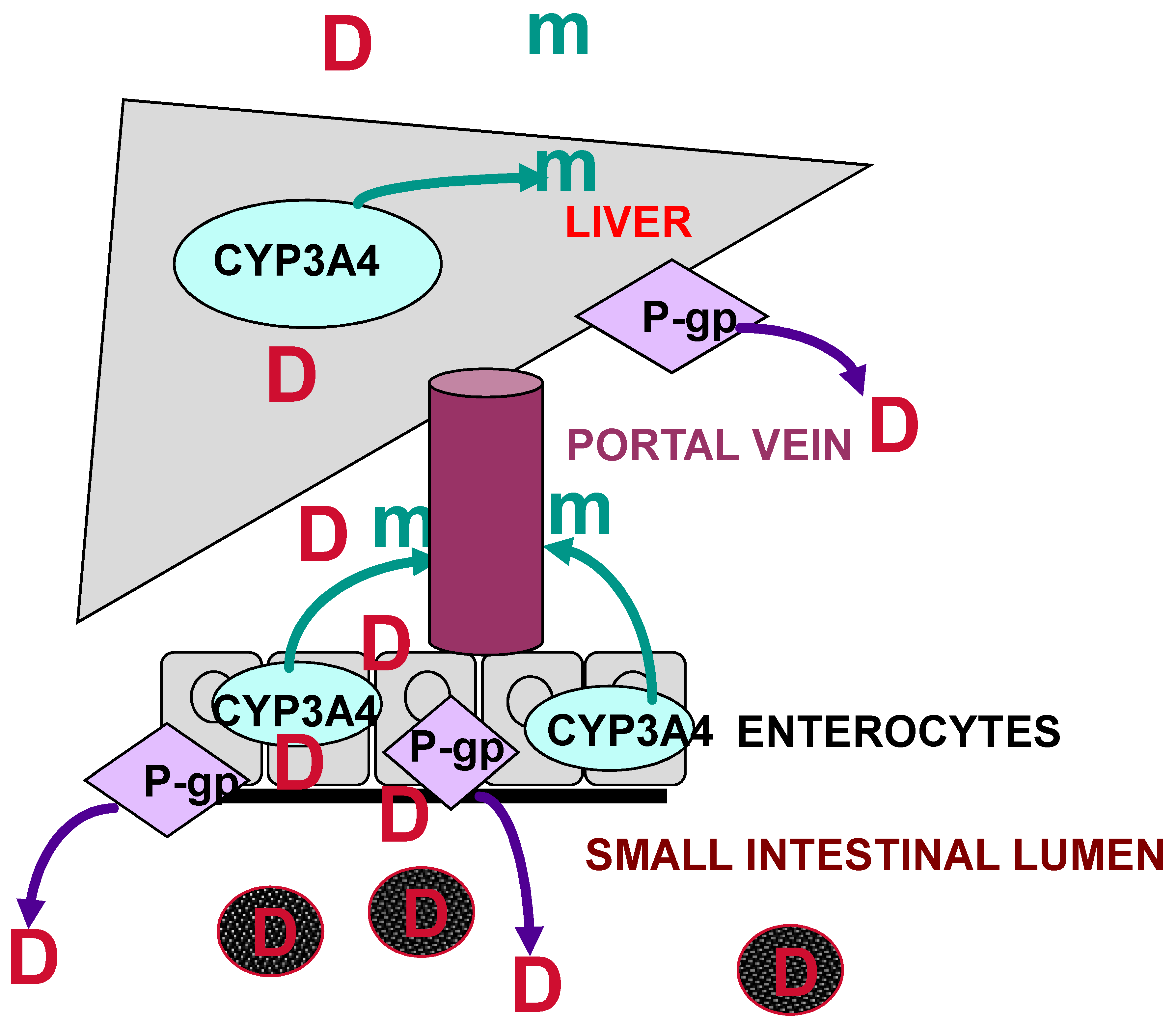{kind=link}
| Drug | Route | F (%) | Cmax (μg/L/1 mg-dose) | tmax (h) | t1/2 (h) | Vd (L) * | CL (L/h) * | ke (h−1) |
|---|---|---|---|---|---|---|---|---|
| Dexamethasone after dexamethasone sodium phosphate | IV | 90 | 10.5 ± 2.8 (10.2–10.8) | 4.6 ± 1.2 | 65.7 ± 17.3 (27.0–98) | 12 ± 4 (5–21) | 0.21 ± 0.03 | |
| Dexamethasone | Oral | 76 ± 10 (61–86) | 8.4 ± 3.6 | 1.5 (1.0–2.0) | 4.0 ± 0.9 | 76.3 | 7.7 (5.2–9.7) | 0.16 |
| Pharmacokinetic Parameter | Arithmetic Means (±Standard Deviation *) |
|---|---|
| AUC(0–36) (h·µg/L) | 1116.86 (±346.20) |
| AUC(0–∞) (h·µg/L) | 1140.30 (±366.43) |
| Cmax (µg/L) | 125.93 (±23.06) |
| tmax (h) | 3.43 (1.8–8.0) |
| Half-life (h) | 4.60 (±1.26) |
4. Clinical Rationale for the Study of Pharmacokinetic Drug–Drug Interactions Involving Dexamethasone
5. In Vitro Evidence of Drug–Drug Interactions Involving Dexamethasone
- (A)
- Induction of CYPs by Dexamethasone: Role of Nuclear Receptors
- (1)
- Induction of Liver CYP3A4
- (2)
- Induction of Other CYPs
- (B)
- Regulation of Expression and Activity of Transporters by Dexamethasone: Potential Implications for Drug–Drug Interactions
- (1)
- The Major Role Played by P-glycoprotein
- (2)
- Induction of P-glycoprotein by Dexamethasone
- (3)
- Effect of Dexamethasone on Other Transporters and Metabolism Enzymes
- (C)
- In Vitro Interaction between Dexamethasone and Other Treatments
- (D)
- In Vitro Interaction with Dexamethasone as a CYP3A4 and P-gp Substrate
- (E)
- In Vitro Studies General Conclusion
6. In Vivo Evidence of Drug–Drug Interactions Involving Dexamethasone
- (A)
- Relative Contribution of CYP3A4 and P-gp in Drug–Drug Interactions Involving Dexamethasone in Animal Studies
- (B)
- Drug-Drug Interactions with Other Transporters in Animals
- (C)
- Interaction by Unknown or Unspecified Mechanisms
7. Drug–Drug Interactions Involving Dexamethasone in Humans
- (A)
- Clinical Rationale
- (B)
- The Role of CYP3A Activity and P-gp in Drug–Drug Interactions Involving Dexamethasone in Humans: Published Evidence
- (C)
- Drug–Drug Interactions Involving Dexamethasone with Other CYPs in Humans
- (D)
- Dexamethasone Pharmacokinetics May Also Be Affected by Well-Known and Potent CYP3A/P-gp Modulators in Humans
- (E)
- Impact of Pharmacogenomics on the Potentiality and Prediction of Dexamethasone Induced DDIs
8. Overall Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Samer, C.F.; Lorenzini, K.I.; Rollason, V.; Daali, Y.; Desmeules, J.A. Applications of CYP450 testing in the clinical setting. Mol. Diagn. Ther. 2013, 17, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Liddle, G.W. Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1960, 20, 1539–1560. [Google Scholar] [CrossRef]
- Alexanian, R.; Barlogie, B.; Dixon, D. High-dose glucocorticoid treatment of resistant myeloma. Ann. Intern. Med. 1986, 105, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Delarue, R.; Haioun, C.; Ribrag, V.; Brice, P.; Delmer, A.; Tilly, H.; Salles, G.; Van Hoof, A.; Casasnovas, O.; Brousse, N.; et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: A phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood 2013, 121, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Tomazini, B.M.; Maia, I.S.; Cavalcanti, A.B.; Berwanger, O.; Rosa, R.G.; Veiga, V.C.; Avezum, A.; Lopes, R.D.; Bueno, F.R.; Silva, M.V.A.O.; et al. Effect of Dexamethasone on Days Alive and Ventilator-Free in Patients With Moderate or Severe Acute Respiratory Distress Syndrome and COVID-19: The CoDEX Randomized Clinical Trial. JAMA 2020, 324, 1307–1316. [Google Scholar] [CrossRef]
- Alunno, A.; Najm, A.; Mariette, X.; De Marco, G.; Emmel, J.; Mason, L.; McGonagle, D.G.; Machado, P.M. Immunomodulatory therapies for SARS-CoV-2 infection: A systematic literature review to inform EULAR points to consider. Ann. Rheum. Dis. 2021, 80, 803–815. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Potential therapeutic use of corticosteroids as SARS-CoV-2 main protease inhibitors: A computational study. J. Biomol. Struct. Dyn. 2022, 40, 2053–2066. [Google Scholar] [CrossRef]
- Wei, Y.; Ji, X.B.; Wang, Y.W.; Wang, J.X.; Yang, E.Q.; Wang, Z.C.; Sang, Y.Q.; Bi, Z.M.; Ren, C.A.; Zhou, F.; et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: A prospective multicenter randomized trial. Blood 2016, 127, 296–302. [Google Scholar] [CrossRef]
- Mithoowani, S.; Gregory-Miller, K.; Goy, J.; Miller, M.C.; Wang, G.; Noroozi, N.; Kelton, J.G.; Arnold, D.M. High-dose dexamethasone compared with prednisone for previously untreated primary immune thrombocytopenia: A systematic review and meta-analysis. Lancet Haematol. 2016, 3, e489–e496. [Google Scholar] [CrossRef]
- Al-Musawe, L.; Torre, C.; Guerreiro, J.P.; Rodrigues, A.T.; Raposo, J.F.; Mota-Filipe, H.; Martins, A.P. Drug-drug interactions and inappropriate medicines impact on glycemic control and kidney function in older adults with diabetes-attending specialty care institution. Eur. J. Clin. Pharmacol. 2021, 77, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Récoché, I.; Rousseau, V.; Bourrel, R.; Lapeyre-Mestre, M.; Chebane, L.; Despas, F.; Montastruc, J.L.; Bondon-Guitton, E. Drug-drug interactions with imatinib: An observational study. Medicine 2016, 95, e5076. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, H.; Wang, Y.; Wang, N.; Li, Y.; Yang, Q.; Liu, C.; Zan, Y.; Feng, S.; Xie, J. Potentially Hazardous Drug-Drug Interactions Associated With Oral Antineoplastic Agents Prescribed in Chinese Tertiary Care Teaching Hospital Settings: A Multicenter Cross-Sectional Study. Front. Pharmacol. 2022, 13, 808848. [Google Scholar] [CrossRef]
- Shini Rubina, S.K.; Anuba, P.A.; Swetha, B.; Kalala, K.P.; Pm, A.; Sabarathinam, S. Drug interaction risk between cardioprotective drugs and drugs used in treatment of COVID-19: A evidence-based review from six databases. Diabetes Metab. Syndr. 2022, 16, 102451. [Google Scholar] [CrossRef]
- Czock, D.; Keller, F.; Rasche, F.M.; Häussler, U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin. Pharmacokinet. 2005, 44, 61–98. [Google Scholar] [CrossRef]
- Hichens, M.; Hogans, A.F. Radioimmunoassay for dexamethasone in plasma. Clin. Chem. 1974, 20, 266–271. [Google Scholar] [CrossRef]
- Dollery, C.; Boobis, A.R. Therapeutic Drugs, 2nd ed.; Churchill Livingstone: Edinburgh, UK, 1999. [Google Scholar]
- Spoorenberg, S.M.; Deneer, V.H.; Grutters, J.C.; Pulles, A.E.; Voorn, G.P.; Rijkers, G.T.; Bos, W.J.; van de Garde, E.M. Pharmacokinetics of oral vs. intravenous dexamethasone in patients hospitalized with community-acquired pneumonia. Br. J. Clin. Pharmacol. 2014, 78, 78–83. [Google Scholar] [CrossRef]
- Munch, M.W.; Myatra, S.N.; Vijayaraghavan, B.K.T.; Saseedharan, S.; Benfield, T.; Wahlin, R.R.; Rasmussen, B.S.; Andreasen, A.S.; Poulsen, L.M.; Cioccari, L.; et al. Effect of 12 mg vs 6 mg of Dexamethasone on the Number of Days Alive Without Life Support in Adults With COVID-19 and Severe Hypoxemia: The COVID STEROID 2 Randomized Trial. JAMA 2021, 326, 1807–1817. [Google Scholar] [CrossRef]
- Blette, B.S.; Granholm, A.; Li, F.; Shankar-Hari, M.; Lange, T.; Munch, M.W.; Møller, M.H.; Perner, A.; Harhay, M.O. Causal Bayesian machine learning to assess treatment effect heterogeneity by dexamethasone dose for patients with COVID-19 and severe hypoxemia. Sci. Rep. 2023, 13, 6570. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P-450 3A4: Regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 1–17. [Google Scholar] [CrossRef]
- McCune, J.S.; Hawke, R.L.; LeCluyse, E.L.; Gillenwater, H.H.; Hamilton, G.; Ritchie, J.; Lindley, C. In vivo and in vitro induction of human cytochrome P4503A4 by dexamethasone. Clin. Pharmacol. Ther. 2000, 68, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Shou, M.; Hayashi, M.; Pan, Y.; Xu, Y.; Morrissey, K.; Xu, L.; Skiles, G.L. Modeling, prediction, and in vitro in vivo correlation of CYP3A4 induction. Drug Metab. Dispos. 2008, 36, 2355–2370. [Google Scholar] [CrossRef] [PubMed]
- El-Sankary, W.; Plant, N.J.; Gibson, G.G.; Moore, D.J. Regulation of the CYP3A4 gene by hydrocortisone and xenobiotics: Role of the glucocorticoid and pregnane X receptors. Drug Metab. Dispos. 2000, 28, 493–496. [Google Scholar] [PubMed]
- Shi, D.; Yang, D.; Yan, B. Dexamethasone transcriptionally increases the expression of the pregnane X receptor and synergistically enhances pyrethroid esfenvalerate in the induction of cytochrome P450 3A23. Biochem. Pharmacol. 2010, 80, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Ripp, S.L.; Mills, J.B.; Fahmi, O.A.; Trevena, K.A.; Liras, J.L.; Maurer, T.S.; de Morais, S.M. Use of immortalized human hepatocytes to predict the magnitude of clinical drug-drug interactions caused by CYP3A4 induction. Drug Metab. Dispos. 2006, 34, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Chiba, K.; Horikawa, M.; Sugiyama, Y. The quantitative prediction of in vivo enzyme-induction caused by drug exposure from in vitro information on human hepatocytes. Drug Metab. Pharmacokinet. 2005, 20, 236–243. [Google Scholar] [CrossRef]
- Pascussi, J.M.; Drocourt, L.; Gerbal-Chaloin, S.; Fabre, J.M.; Maurel, P.; Vilarem, M.J. Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Sequential role of glucocorticoid receptor and pregnane X receptor. Eur. J. Biochem. 2001, 268, 6346–6358. [Google Scholar] [CrossRef]
- Mouly, S.; Meune, C.; Bergmann, J.F. Mini-series: I. Basic science. Uncertainty and inaccuracy of predicting CYP-mediated in vivo drug interactions in the ICU from in vitro models: Focus on CYP3A4. Intensive Care Med. 2009, 35, 417–429. [Google Scholar] [CrossRef]
- Ferguson, S.S.; Chen, Y.; LeCluyse, E.L.; Negishi, M.; Goldstein, J.A. Human CYP2C8 is transcriptionally regulated by the nuclear receptors constitutive androstane receptor, pregnane X receptor, glucocorticoid receptor, and hepatic nuclear factor 4alpha. Mol. Pharmacol. 2005, 68, 747–757. [Google Scholar] [CrossRef]
- Gerbal-Chaloin, S.; Pascussi, J.M.; Pichard-Garcia, L.; Daujat, M.; Waechter, F.; Fabre, J.M.; Carrère, N.; Maurel, P. Induction of CYP2C genes in human hepatocytes in primary culture. Drug Metab. Dispos. 2001, 29, 242–251. [Google Scholar]
- Pascussi, J.M.; Gerbal-Chaloin, S.; Fabre, J.M.; Maurel, P.; Vilarem, M.J. Dexamethasone enhances constitutive androstane receptor expression in human hepatocytes: Consequences on cytochrome P450 gene regulation. Mol. Pharmacol. 2000, 58, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Onica, T.; Nichols, K.; Larin, M.; Ng, L.; Maslen, A.; Dvorak, Z.; Pascussi, J.M.; Vilarem, M.J.; Maurel, P.; Kirby, G.M. Dexamethasone-mediated up-regulation of human CYP2A6 involves the glucocorticoid receptor and increased binding of hepatic nuclear factor 4 alpha to the proximal promoter. Mol. Pharmacol. 2008, 73, 451–460. [Google Scholar] [CrossRef] [PubMed]
- van de Kerkhof, E.G.; de Graaf, I.A.; Ungell, A.L.; Groothuis, G.M. Induction of metabolism and transport in human intestine: Validation of precision-cut slices as a tool to study induction of drug metabolism in human intestine in vitro. Drug Metab. Dispos. 2008, 36, 604–613. [Google Scholar] [CrossRef]
- Mouly, S.; Lloret-Linares, C.; Sellier, P.O.; Sene, D.; Bergmann, J.F. Is the clinical relevance of drug-food and drug-herb interactions limited to grapefruit juice and Saint-John’s Wort? Pharmacol. Res. 2017, 118, 82–92. [Google Scholar] [CrossRef]
- Hoffmeyer, S.; Burk, O.; von Richter, O.; Arnold, H.P.; Brockmöller, J.; Johne, A.; Cascorbi, I.; Gerloff, T.; Roots, I.; Eichelbaum, M.; et al. Functional polymorphisms of the human multidrug-resistance gene: Multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Wacher, V.J.; Wu, C.Y.; Benet, L.Z. Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: Implications for drug delivery and activity in cancer chemotherapy. Mol. Carcinog. 1995, 13, 129–134. [Google Scholar] [CrossRef]
- Annaert, P.P.; Brouwer, K.L. Assessment of drug interactions in hepatobiliary transport using rhodamine 123 in sandwich-cultured rat hepatocytes. Drug Metab. Dispos. 2005, 33, 388–394. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, M.; Sun, X.; Li, C.; Kuang, X.; Ruan, X. Expression and activity of p-glycoprotein elevated by dexamethasone in cultured retinal pigment epithelium involve glucocorticoid receptor and pregnane X receptor. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3508–3515. [Google Scholar] [CrossRef]
- Demeule, M.; Jodoin, J.; Beaulieu, E.; Brossard, M.; Béliveau, R. Dexamethasone modulation of multidrug transporters in normal tissues. FEBS Lett. 1999, 442, 208–214. [Google Scholar] [CrossRef]
- Manceau, S.; Giraud, C.; Declèves, X.; Batteux, F.; Chéreau, C.; Chouzenoux, S.; Scherrmann, J.M.; Weill, B.; Perrot, J.Y.; Tréluyer, J.M. Expression and induction by dexamethasone of ABC transporters and nuclear receptors in a human T-lymphocyte cell line. J. Chemother. 2012, 24, 48–55. [Google Scholar] [CrossRef]
- Manceau, S.; Giraud, C.; Declèves, X.; Scherrmann, J.M.; Artiguebieille, F.; Goffinet, F.; Chappuy, H.; Vinot, C.; Tréluyer, J.M. ABC drug transporter and nuclear receptor expression in human cytotrophoblasts: Influence of spontaneous syncytialization and induction by glucocorticoids. Placenta 2012, 33, 927–932. [Google Scholar] [CrossRef]
- Pułaski, L.; Kania, K.; Ratajewski, M.; Uchiumi, T.; Kuwano, M.; Bartosz, G. Differential regulation of the human MRP2 and MRP3 gene expression by glucocorticoids. J. Steroid Biochem. Mol. Biol. 2005, 96, 229–234. [Google Scholar] [CrossRef] [PubMed]
- El-Sheikh, A.A.; Greupink, R.; Wortelboer, H.M.; van den Heuvel, J.J.; Schreurs, M.; Koenderink, J.B.; Masereeuw, R.; Russel, F.G. Interaction of immunosuppressive drugs with human organic anion transporter (OAT) 1 and OAT3, and multidrug resistance-associated protein (MRP) 2 and MRP4. Transl. Res. 2013, 162, 398–409. [Google Scholar] [CrossRef]
- Bruyère, A.; Declèves, X.; Bouzom, F.; Ball, K.; Marques, C.; Treton, X.; Pocard, M.; Valleur, P.; Bouhnik, Y.; Panis, Y.; et al. Effect of variations in the amounts of P-glycoprotein (ABCB1), BCRP (ABCG2) and CYP3A4 along the human small intestine on PBPK models for predicting intestinal first pass. Mol. Pharm. 2010, 7, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Elahian, F.; Kalalinia, F.; Behravan, J. Evaluation of indomethacin and dexamethasone effects on BCRP-mediated drug resistance in MCF-7 parental and resistant cell lines. Drug Chem. Toxicol. 2010, 33, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhou, J.; Guo, J.; Hu, W.; Chen, G.; Li, B.; Wen, Y.; Jiang, Y.; Fu, K.; Bi, H.; et al. Dexamethasone induces an imbalanced fetal-placental-maternal bile acid circulation: Involvement of placental transporters. BMC Med. 2021, 19, 87. [Google Scholar] [CrossRef] [PubMed]
- Lye, P.; Bloise, E.; Nadeem, L.; Gibb, W.; Lye, S.J.; Matthews, S.G. Glucocorticoids modulate multidrug resistance transporters in the first trimester human placenta. J. Cell. Mol. Med. 2018, 22, 3652–3660. [Google Scholar] [CrossRef]
- Gui, C.; Miao, Y.; Thompson, L.; Wahlgren, B.; Mock, M.; Stieger, B.; Hagenbuch, B. Effect of pregnane X receptor ligands on transport mediated by human OATP1B1 and OATP1B3. Eur. J. Pharmacol. 2008, 584, 57–65. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Fisch, T.; Oswald, M.; Hagenbuch, B.; Meier, P.J.; Beuers, U.; Paumgartner, G. Dehydroepiandrosterone sulfate (DHEAS): Identification of a carrier protein in human liver and brain. FEBS Lett. 1998, 424, 173–176. [Google Scholar] [CrossRef]
- Duanmu, Z.; Locke, D.; Smigelski, J.; Wu, W.; Dahn, M.S.; Falany, C.N.; Kocarek, T.A.; Runge-Morris, M. Effects of dexamethasone on aryl (SULT1A1)- and hydroxysteroid (SULT2A1)-sulfotransferase gene expression in primary cultured human hepatocytes. Drug Metab. Dispos. 2002, 30, 997–1004. [Google Scholar] [CrossRef]
- Bian, H.S.; Ngo, S.Y.; Tan, W.; Wong, C.H.; Boelsterli, U.A.; Tan, T.M. Induction of human sulfotransferase 1A3 (SULT1A3) by glucocorticoids. Life Sci. 2007, 81, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Kanou, M.; Usui, T.; Ueyama, H.; Sato, H.; Ohkubo, I.; Mizutani, T. Stimulation of transcriptional expression of human UDP-glucuronosyltransferase 1A1 by dexamethasone. Mol. Biol. Rep. 2004, 31, 151–158. [Google Scholar] [CrossRef]
- Martínez-Guzmán, C.; Cortés-Reynosa, P.; Pérez-Salazar, E.; Elizondo, G. Dexamethasone induces human glutathione S transferase alpha 1 (hGSTA1) expression through the activation of glucocorticoid receptor (hGR). Toxicology 2017, 385, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kamataki, T.; Yokoi, T.; Fujita, K.; Ando, Y. Preclinical approach for identifying drug interactions. Cancer Chemother. Pharmacol. 1998, 42, S50–S53. [Google Scholar] [CrossRef]
- Marre, F.; Sanderink, G.J.; de Sousa, G.; Gaillard, C.; Martinet, M.; Rahmani, R. Hepatic biotransformation of docetaxel (Taxotere) in vitro: Involvement of the CYP3A subfamily in humans. Cancer Res. 1996, 56, 1296–1302. [Google Scholar]
- Dukhanina, E.A.; Portseva, T.N.; Dukhanin, A.S.; Georgieva, S.G. Studying of the Mechanisms of Combined Effect of Dexamethasone, Doxorubicin, and Docetaxel on Breast Cancer Cells. Bull. Exp. Biol. Med. 2018, 166, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Dukhanina, E.A.; Portseva, T.N.; Pankratova, E.V.; Soshnikova, N.V.; Stepchenko, A.G.; Dukhanin, A.S.; Georgieva, S.G. Oct-1 modifies S100A4 exchange between intra- and extracellular compartments in Namalwa cells and increases their sensitivity to glucocorticoids. Cell Cycle 2016, 15, 1471–1478. [Google Scholar] [CrossRef]
- Wagenblast, J.; Arnoldner, C.; Gstöttner, W.; Bisdas, S.; Mörtel, S.; May, A.; Hambek, M. Does dexamethasone inhibit the antineoplastic effect of cisplatin and docetaxel in head and neck cancer cells? Anticancer Res. 2010, 30, 123–127. [Google Scholar]
- Zayed, A.L.; Hamadneh, G.N.; Hroot, J.A.; Mayyas, A.; Jaber, S.A.; Qinna, N.A. HPLC methods for studying pharmacokinetics of tivozanib and in vitro metabolic interaction with dexamethasone in rat. J. Pharm. Biomed. Anal. 2023, 232, 115423. [Google Scholar] [CrossRef]
- Yu, R.; Li, X.; DuBois, D.C.; Almon, R.R.; Cao, Y.; Jusko, W.J. Interactions of Tofacitinib and Dexamethasone on Lymphocyte Proliferation. Pharm. Res. 2020, 37, 105. [Google Scholar] [CrossRef]
- Yates, C.R.; Chang, C.; Kearbey, J.D.; Yasuda, K.; Schuetz, E.G.; Miller, D.D.; Dalton, J.T.; Swaan, P.W. Structural determinants of P-glycoprotein-mediated transport of glucocorticoids. Pharm. Res. 2003, 20, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Maillefert, J.F.; Duchamp, O.; Solary, E.; Genne, P.; Tavernier, C. Effects of cyclosporin at various concentrations on dexamethasone intracellular uptake in multidrug resistant cells. Ann. Rheum. Dis. 2000, 59, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Korhonova, M.; Doricakova, A.; Dvorak, Z. Optical Isomers of Atorvastatin, Rosuvastatin and Fluvastatin Enantiospecifically Activate Pregnane X Receptor PXR and Induce CYP2A6, CYP2B6 and CYP3A4 in Human Hepatocytes. PLoS ONE 2015, 10, e0137720. [Google Scholar] [CrossRef] [PubMed]
- Novotna, A.; Dvorak, Z. Omeprazole and lansoprazole enantiomers induce CYP3A4 in human hepatocytes and cell lines via glucocorticoid receptor and pregnane X receptor axis. PLoS ONE 2014, 9, e105580. [Google Scholar] [CrossRef]
- Nishimura, M.; Koeda, A.; Suzuki, E.; Kawano, Y.; Nakayama, M.; Satoh, T.; Narimatsu, S.; Naito, S. Regulation of mRNA expression of MDR1, MRP1, MRP2 and MRP3 by prototypical microsomal enzyme inducers in primary cultures of human and rat hepatocytes. Drug Metab. Pharmacokinet. 2006, 21, 297–307. [Google Scholar] [CrossRef]
- Perloff, M.D.; von Moltke, L.L.; Greenblatt, D.J. Ritonavir and dexamethasone induce expression of CYP3A and P-glycoprotein in rats. Xenobiotica 2004, 34, 133–150. [Google Scholar] [CrossRef]
- Yokogawa, K.; Shimada, T.; Higashi, Y.; Itoh, Y.; Masue, T.; Ishizaki, J.; Asahi, M.; Miyamoto, K. Modulation of mdr1a and CYP3A gene expression in the intestine and liver as possible cause of changes in the cyclosporin A disposition kinetics by dexamethasone. Biochem. Pharmacol. 2002, 63, 777–783. [Google Scholar] [CrossRef]
- Jin, M.; Shimada, T.; Yokogawa, K.; Nomura, M.; Ishizaki, J.; Piao, Y.; Kato, Y.; Tsuji, A.; Miyamoto, K. Site-dependent contributions of P-glycoprotein and CYP3A to cyclosporin A absorption, and effect of dexamethasone in small intestine of mice. Biochem. Pharmacol. 2006, 72, 1042–1050. [Google Scholar] [CrossRef]
- Jin, M.; Shimada, T.; Yokogawa, K.; Nomura, M.; Kato, Y.; Tsuji, A.; Miyamoto, K. Contributions of intestinal P-glycoprotein and CYP3A to oral bioavailability of cyclosporin A in mice treated with or without dexamethasone. Int. J. Pharm. 2006, 309, 81–86. [Google Scholar] [CrossRef]
- Lin, J.H.; Chiba, M.; Chen, I.W.; Nishime, J.A.; deLuna, F.A.; Yamazaki, M.; Lin, Y.J. Effect of dexamethasone on the intestinal first-pass metabolism of indinavir in rats: Evidence of cytochrome P-450 3A [correction of P-450 A] and p-glycoprotein induction. Drug Metab. Dispos. 1999, 27, 1187–1193. [Google Scholar]
- Deeken, J.F.; Beumer, J.H.; Anders, N.M.; Wanjiku, T.; Rusnak, M.; Rudek, M.A. Preclinical assessment of the interactions between the antiretroviral drugs, ritonavir and efavirenz, and the tyrosine kinase inhibitor erlotinib. Cancer Chemother. Pharmacol. 2015, 76, 813–819. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.F.; Chen, X.Y.; Zhong, D.F. CYP3A4 inducer and inhibitor strongly affect the pharmacokinetics of triptolide and its derivative in rats. Acta Pharmacol. Sin. 2018, 39, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Paudel, S.; Shrestha, A.; Cho, P.; Shrestha, R.; Kim, Y.; Lee, T.; Kim, J.H.; Jeong, T.C.; Lee, E.S.; Lee, S. Assessing Drug Interaction and Pharmacokinetics of Loxoprofen in Mice Treated with CYP3A Modulators. Pharmaceutics 2019, 11, 479. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Ben-Eltriki, M.; Adomat, H.; Chin, M.Y.; Tomlinson Guns, E.S. Effect of Dexamethasone on Abiraterone Pharmacokinetics in Mice: Determined by LC/MS Analysis. Medicines 2023, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, J.; Yang, T.; Fan, K.; Gu, J.; Yin, G. Pharmacokinetics of dexamethasone and nefopam administered alone or in combination using a newly developed prefilled multi-drug injector in rats. Pharmacology 2014, 93, 220–224. [Google Scholar] [CrossRef]
- Miyazaki, N.; Misaka, S.; Ogata, H.; Fukushima, T.; Kimura, J. Effects of itraconazole, dexamethasone and naringin on the pharmacokinetics of nadolol in rats. Drug Metab. Pharmacokinet. 2013, 28, 356–361. [Google Scholar] [CrossRef]
- Yumoto, R.; Murakami, T.; Sanemasa, M.; Nasu, R.; Nagai, J.; Takano, M. Pharmacokinetic interaction of cytochrome P450 3A-related compounds with rhodamine 123, a P-glycoprotein substrate, in rats pretreated with dexamethasone. Drug Metab. Dispos. 2001, 29, 145–151. [Google Scholar]
- Li, L.; Li, Z.Q.; Deng, C.H.; Ning, M.R.; Li, H.Q.; Bi, S.S.; Zhou, T.Y.; Lu, W. A mechanism-based pharmacokinetic/pharmacodynamic model for CYP3A1/2 induction by dexamethasone in rats. Acta Pharmacol. Sin. 2012, 33, 127–136. [Google Scholar] [CrossRef]
- Chiou, W.L.; Jeong, H.Y.; Wu, T.C.; Ma, C. Use of the erythromycin breath test for in vivo assessments of cytochrome P4503A activity and dosage individualization. Clin. Pharmacol. Ther. 2001, 70, 305–310. [Google Scholar] [CrossRef]
- Sugiyama, E.; Kikuchi, A.; Inada, M.; Sato, H. The use of 13C-erythromycin as an in vivo probe to evaluate CYP3A-mediated drug interactions in rats. J. Pharm. Sci. 2011, 100, 3995–4005. [Google Scholar] [CrossRef]
- Areskog, M.; von Samson-Himmelstjerna, G.; Alvinerie, M.; Sutra, J.F.; Höglund, J. Dexamethasone treatment interferes with the pharmacokinetics of ivermectin in young cattle. Vet. Parasitol. 2012, 190, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Nicolaï, J.; Williamson, B. The role of the efflux transporter, P-glycoprotein, at the blood-brain barrier in drug discovery. Biopharm. Drug Dispos. 2023, 44, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Misaka, S.; Kurosawa, S.; Uchida, S.; Yoshida, A.; Kato, Y.; Kagawa, Y.; Yamada, S. Evaluation of the pharmacokinetic interaction of midazolam with ursodeoxycholic acid, ketoconazole and dexamethasone by brain benzodiazepine receptor occupancy. J. Pharm. Pharmacol. 2011, 63, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, P.W.; Clarke, C.; Dawson, K.B. The effect of phenytoin, phenobarbitone, dexamethasone and flurbiprofen on misonidazole neurotoxicity in mice. Br. J. Cancer 1984, 49, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Narang, V.S.; Fraga, C.; Kumar, N.; Shen, J.; Throm, S.; Stewart, C.F.; Waters, C.M. Dexamethasone increases expression and activity of multidrug resistance transporters at the rat blood-brain barrier. Am. J. Physiol. Cell Physiol. 2008, 295, C440–C450. [Google Scholar] [CrossRef]
- Fuksa, L.; Brcakova, E.; Kolouchova, G.; Hirsova, P.; Hroch, M.; Cermanova, J.; Staud, F.; Micuda, S. Dexamethasone reduces methotrexate biliary elimination and potentiates its hepatotoxicity in rats. Toxicology 2010, 267, 165–171. [Google Scholar] [CrossRef]
- Petropoulos, S.; Gibb, W.; Matthews, S.G. Glucocorticoid regulation of placental breast cancer resistance protein (Bcrp1) in the mouse. Reprod. Sci. 2011, 18, 631–639. [Google Scholar] [CrossRef]
- Palomares-Alonso, F.; Toledo, A.; Palencia Hernández, G.; Jung-Cook, H.; Fleury, A. Effect of dexamethasone on albendazole cysticidal activity in experimental cysticercosis by Taenia crassiceps in BALB/c mice: In vitro and in vivo evaluation. Exp. Parasitol. 2020, 208, 107801. [Google Scholar] [CrossRef]
- Pawluk, S.A.; Roels, C.A.; Wilby, K.J.; Ensom, M.H. A review of pharmacokinetic drug-drug interactions with the anthelmintic medications albendazole and mebendazole. Clin. Pharmacokinet. 2015, 54, 371–383. [Google Scholar] [CrossRef]
- Hou, W.J.; Guan, J.H.; Dong, Q.; Han, Y.H.; Zhang, R. Dexamethasone inhibits the effect of paclitaxel on human ovarian carcinoma xenografts in nude mice. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2902–2908. [Google Scholar]
- Yu, R.; Song, D.; DuBois, D.C.; Almon, R.R.; Jusko, W.J. Modeling Combined Anti-Inflammatory Effects of Dexamethasone and Tofacitinib in Arthritic Rats. AAPS J. 2019, 21, 93. [Google Scholar] [CrossRef]
- Matoulková, P.; Pávek, P.; Malý, J.; Vlček, J. Cytochrome P450 enzyme regulation by glucocorticoids and consequences in terms of drug interaction. Expert Opin. Drug Metab. Toxicol. 2014, 10, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Antona, C.; Savieo, J.L.; Lauschke, V.M.; Sangkuhl, K.; Drögemöller, B.I.; Wang, D.; van Schaik, R.H.N.; Gilep, A.A.; Peter, A.P.; Boone, E.C.; et al. PharmVar GeneFocus: CYP3A5. Clin. Pharmacol. Ther. 2022, 112, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Rollins, K.D.; Kashuba, A.D.; Paine, M.F.; Nelsen, A.C.; Williams, E.E.; Moran, C.; Lamba, J.K.; Schuetz, E.G.; Hawke, R.L. The influence of CYP3A5 genotype on dexamethasone induction of CYP3A activity in African Americans. Drug Metab. Dispos. 2008, 36, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Teo, Y.L.; Saetaew, M.; Chanthawong, S.; Yap, Y.S.; Chan, E.C.; Ho, H.K.; Chan, A. Effect of CYP3A4 inducer dexamethasone on hepatotoxicity of lapatinib: Clinical and in vitro evidence. Breast Cancer Res. Treat. 2012, 133, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Kuroda, Y.; Shibayama, H.; Hino, M.; Tajima, T.; Corrado, C.; Lin, R.; Waldron, E.; Binlich, F.; Suzuki, K. Panobinostat PK/PD profile in combination with bortezomib and dexamethasone in patients with relapsed and relapsed/refractory multiple myeloma. Eur. J. Clin. Pharmacol. 2016, 72, 153–161. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Richardson, P.G.; Günther, A.; Sezer, O.; Siegel, D.; Bladé, J.; LeBlanc, R.; Sutherland, H.; Sopala, M.; Mishra, K.K.; et al. Phase Ib study of panobinostat and bortezomib in relapsed or relapsed and refractory multiple myeloma. J. Clin. Oncol. 2013, 31, 3696–3703. [Google Scholar] [CrossRef]
- Villikka, K.; Kivistö, K.T.; Neuvonen, P.J. The effect of dexamethasone on the pharmacokinetics of triazolam. Pharmacol. Toxicol. 1998, 83, 135–138. [Google Scholar] [CrossRef]
- Moore, M.J.; Hardy, R.W.; Thiessen, J.J.; Soldin, S.J.; Erlichman, C. Rapid development of enhanced clearance after high-dose cyclophosphamide. Clin. Pharmacol. Ther. 1988, 44, 622–628. [Google Scholar] [CrossRef]
- Kovarik, J.M.; Purba, H.S.; Pongowski, M.; Gerbeau, C.; Humbert, H.; Mueller, E.A. Pharmacokinetics of dexamethasone and valspodar, a P-glycoprotein (mdr1) modulator: Implications for coadministration. Pharmacotherapy 1998, 18, 1230–1236. [Google Scholar] [CrossRef]
- Potere, N.; Candeloro, M.; Porreca, E.; Marinari, S.; Federici, C.; Auciello, R.; Di Nisio, M. Direct oral anticoagulant plasma levels in hospitalized COVID-19 patients treated with dexamethasone. J. Thromb. Thrombolysis 2022, 53, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, O.V.; Boyce, R.D.; Gomez-Lumbreras, A.; Kocis, P.T.; Villa Zapata, L.; Tan, M.; Leonard, C.E.; Andersen, K.M.; Mehta, H.; Alexander, G.C.; et al. Drug-drug interaction between dexamethasone and direct-acting oral anticoagulants: A nested case-control study in the National COVID Cohort Collaborative (N3C). BMJ Open 2022, 12, e066846. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.L.; Filipek, R.L.; La Hoz, R.M.; Williamson, J.C. Subtherapeutic voriconazole concentrations associated with concomitant dexamethasone: Case report and review of the literature. J. Clin. Pharm. Ther. 2016, 41, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Taghvaye Masoumi, H.; Hadjibabaie, M.; Gholami, K.; Zarif-Yeganeh, M.; Ghavamzadeh, A. Significant drug interaction between voriconazole and dexamethasone: A case report. J. Oncol. Pharm. Pract. 2019, 25, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.J.; Gao, K.Q.; Huang, P.H.; Guo, R.; Zuo, X.C.; Xia, Q.; Hu, S.Y.; Yu, Z.; Xie, Y.L. Interactive Effects of Glucocorticoids and Cytochrome P450 Polymorphisms on the Plasma Trough Concentrations of Voriconazole. Front. Pharmacol. 2021, 12, 666296. [Google Scholar] [CrossRef] [PubMed]
- Li-Wan-Po, A.; Girard, T.; Farndon, P.; Cooley, C.; Lithgow, J. Pharmacogenetics of CYP2C19: Functional and clinical implications of a new variant CYP2C19*17. Br. J. Clin. Pharmacol. 2010, 69, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.Y.; Lichtor, T.; Brown, F.D. Severe thrombocytopenia associated with phenytoin and cimetidine therapy. Surg. Neurol. 1985, 23, 169–172. [Google Scholar] [CrossRef]
- Yue, C.P.; Mann, K.S.; Chan, K.H. Severe thrombocytopenia due to combined cimetidine and phenytoin therapy. Neurosurgery 1987, 20, 963–965. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Goldstein, A.M.; Gordon, D. Thrombocytopenia following administration of phenytoin, dexamethasone and cimetidine: A case report and a potential mechanism. J. Intern. Med. 1993, 234, 91–94. [Google Scholar] [CrossRef]
- Thorning, G.; Raghavan, K. Fatal phenytoin-induced thrombocytopaenia in a neurosurgical patient. Eur. J. Anaesthesiol. 2007, 24, 897–898. [Google Scholar] [CrossRef]
- Pandey, M.; Yarlagadda, L. Drug-induced thrombocytopenia: A less known interaction. Blood Coagul. Fibrinolysis 2012, 23, 778–780. [Google Scholar] [CrossRef] [PubMed]
- Gattis, W.A.; May, D.B. Possible interaction involving phenytoin, dexamethasone, and antineoplastic agents: A case report and review. Ann. Pharmacother. 1996, 30, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Varis, T.; Kivistö, K.T.; Backman, J.T.; Neuvonen, P.J. The cytochrome P450 3A4 inhibitor itraconazole markedly increases the plasma concentrations of dexamethasone and enhances its adrenal-suppressant effect. Clin. Pharmacol. Ther. 2000, 68, 487–494. [Google Scholar] [CrossRef]
- McCrea, J.B.; Majumdar, A.K.; Goldberg, M.R.; Iwamoto, M.; Gargano, C.; Panebianco, D.L.; Hesney, M.; Lines, C.R.; Petty, K.J.; Deutsch, P.J.; et al. Effects of the neurokinin1 receptor antagonist aprepitant on the pharmacokinetics of dexamethasone and methylprednisolone. Clin. Pharmacol. Ther. 2003, 74, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Marbury, T.C.; Ngo, P.L.; Shadle, C.R.; Jin, B.; Panebianco, D.; Caro, L.; Valentine, J.; Murphy, G. Pharmacokinetics of oral dexamethasone and midazolam when administered with single-dose intravenous 150 mg fosaprepitant in healthy adult subjects. J. Clin. Pharmacol. 2011, 51, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Hatano, K.; Fujiwara, S.I.; Umino, K.; Ikeda, T.; Nakano, H.; Mashima, K.; Kawaguchi, S.I.; Ito, S.; Toda, Y.; Nagayama, T.; et al. Clinical interaction between dexamethasone and aprepitant in chemotherapy for lymphoma. Ann. Hematol. 2022, 101, 1211–1216. [Google Scholar] [CrossRef]
- Nijstad, A.L.; de Vos-Kerkhof, E.; Enters-Weijnen, C.F.; van de Wetering, M.D.; Tissing, W.J.E.; Tibben, M.M.; Rosing, H.; Lalmohamed, A.; Huitema, A.D.R.; Zwaan, C.M. Overestimation of the effect of (fos)aprepitant on intravenous dexamethasone pharmacokinetics requires adaptation of the guidelines for children with chemotherapy-induced nausea and vomiting. Support. Care Cancer 2022, 30, 9991–9999. [Google Scholar] [CrossRef]
- Hancock, K.W.; Levell, M.J. Primidone/dexamethasone interaction. Lancet 1978, 2, 97–98. [Google Scholar] [CrossRef]
- Young, M.C.; Hughes, I.A. Loss of therapeutic control in congenital adrenal hyperplasia due to interaction between dexamethasone and primidone. Acta Paediatr. Scand. 1991, 80, 120–124. [Google Scholar] [CrossRef]
- Kyriazopoulou, V.; Vagenakis, A.G. Abnormal overnight dexamethasone suppression test in subjects receiving rifampicin therapy. J. Clin. Endocrinol. Metab. 1992, 75, 315–317. [Google Scholar] [CrossRef]
- Privitera, M.R.; Greden, J.F.; Gardner, R.W.; Ritchie, J.C.; Carroll, B.J. Interference by carbamazepine with the dexamethasone suppression test. Biol. Psychiatry 1982, 17, 611–620. [Google Scholar]
- Dimaraki, E.V.; Jaffe, C.A. Troglitazone induces CYP3A4 activity leading to falsely abnormal dexamethasone suppression test. J. Clin. Endocrinol. Metab. 2003, 88, 3113–3116. [Google Scholar] [CrossRef] [PubMed]
- Malki, M.A.; Pearson, E.R. Drug-drug-gene interactions and adverse drug reactions. Pharmacogenomics J. 2020, 20, 355–366. [Google Scholar] [CrossRef]
- Claassens, D.M.F.; Vos, G.J.A.; Bergmeijer, T.O.; Hermanides, R.S.; van’t Hof, A.W.J.; van der Harst, P.; Barbato, E.; Morisco, C.; Tjon Joe Gin, R.M.; Asselbergs, F.W.; et al. A Genotype-Guided Strategy for Oral P2Y. N. Engl. J. Med. 2019, 381, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Oslin, D.W.; Lynch, K.G.; Shih, M.C.; Ingram, E.P.; Wray, L.O.; Chapman, S.R.; Kranzler, H.R.; Gelernter, J.; Pyne, J.M.; Stone, A.; et al. Effect of Pharmacogenomic Testing for Drug-Gene Interactions on Medication Selection and Remission of Symptoms in Major Depressive Disorder: The PRIME Care Randomized Clinical Trial. JAMA 2022, 328, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; van der Wouden, C.H.; Manson, L.E.; Abdullah-Koolmees, H.; Blagec, K.; Blagus, T.; Böhringer, S.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; et al. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: An open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet 2023, 401, 347–356. [Google Scholar] [CrossRef]
- Borro, M.; Gentile, G.; Preissner, S.H.; Pomes, L.M.; Gohlke, B.O.; Del Casale, A.; Eckert, A.; Marchetti, P.; Preissner, S.; Preissner, R.; et al. Individualized Drugs’ Selection by Evaluation of Drug Properties, Pharmacogenomics and Clinical Parameters: Performance of a Bioinformatic Tool Compared to a Clinically Established Counselling Process. Pharmgenomics Pers. Med. 2021, 14, 955–962. [Google Scholar] [CrossRef]
- Preissner, S.H.; Marchetti, P.; Simmaco, M.; Gohlke, B.O.; Eckert, A.; Preissner, S.; Preissner, R. Man vs. machine: Comparison of pharmacogenetic expert counselling with a clinical medication support system in a study with 200 genotyped patients. Eur. J. Clin. Pharmacol. 2022, 78, 579–587. [Google Scholar] [CrossRef]
- Thervet, E.; Anglicheau, D.; King, B.; Schlageter, M.H.; Cassinat, B.; Beaune, P.; Legendre, C.; Daly, A.K. Impact of cytochrome p450 3A5 genetic polymorphism on tacrolimus doses and concentration-to-dose ratio in renal transplant recipients. Transplantation 2003, 76, 1233–1235. [Google Scholar] [CrossRef]
- Bissada, J.E.; Truong, V.; Abouda, A.A.; Wines, K.J.; Crouch, R.D.; Jackson, K.D. Interindividual Variation in CYP3A Activity Influences Lapatinib Bioactivation. Drug Metab. Dispos. 2019, 47, 1257–1269. [Google Scholar] [CrossRef]
- Werk, A.N.; Cascorbi, I. Functional gene variants of CYP3A4. Clin. Pharmacol. Ther. 2014, 96, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Mulder, T.A.M.; van Eerden, R.A.G.; de With, M.; Elens, L.; Hesselink, D.A.; Matic, M.; Bins, S.; Mathijssen, R.H.J.; van Schaik, R.H.N. Genotyping in Clinical Practice: Ready for Implementation? Front. Genet. 2021, 12, 711943. [Google Scholar] [CrossRef]
- Chinn, L.W.; Kroetz, D.L. ABCB1 pharmacogenetics: Progress, pitfalls, and promise. Clin. Pharmacol. Ther. 2007, 81, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xie, W.; Krasowski, M.D. PXR: A xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics 2008, 9, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Botelho Barra, C.; Villela, T.R.; Soares, N.F.; Colosimo, E.A.; Belisário, A.R.; Silva, A.C.S.E.; Silva, I.N. Pharmacogenomic markers of glucocorticoid response in congenital adrenal hyperplasia. PLoS ONE 2022, 17, e0279298. [Google Scholar] [CrossRef] [PubMed]
- Nebesio, T.D.; Renbarger, J.L.; Nabhan, Z.M.; Ross, S.E.; Slaven, J.E.; Li, L.; Walvoord, E.C.; Eugster, E.A. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: A pilot study in children with congenital adrenal hyperplasia. Int. J. Pediatr. Endocrinol. 2016, 2016, 17. [Google Scholar] [CrossRef]
- Jackson, R.K.; Irving, J.A.; Veal, G.J. Personalization of dexamethasone therapy in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2016, 173, 13–24. [Google Scholar] [CrossRef]
- Shinohara, T.; Urayama, K.Y.; Watanabe, A.; Akahane, K.; Goi, K.; Huang, M.; Kagami, K.; Abe, M.; Sugita, K.; Okada, Y.; et al. Inherited genetic variants associated with glucocorticoid sensitivity in leukaemia cells. J. Cell. Mol. Med. 2020, 24, 12920–12932. [Google Scholar] [CrossRef]
- Vohra, M.; Sharma, A.R.; Satyamoorthy, K.; Rai, P.S. Pharmacogenomic considerations for repurposing of dexamethasone as a potential drug against SARS-CoV-2 infection. Per. Med. 2021, 18, 389–398. [Google Scholar] [CrossRef]
- Jakobsen Falk, I.; Lund, J.; Gréen, H.; Gruber, A.; Alici, E.; Lauri, B.; Blimark, C.; Mellqvist, U.H.; Swedin, A.; Forsberg, K.; et al. Pharmacogenetic study of the impact of ABCB1 single-nucleotide polymorphisms on lenalidomide treatment outcomes in patients with multiple myeloma: Results from a phase IV observational study and subsequent phase II clinical trial. Cancer Chemother. Pharmacol. 2018, 81, 183–193. [Google Scholar] [CrossRef]
- Chen, N.; Weiss, D.; Reyes, J.; Liu, L.; Kasserra, C.; Wang, X.; Zhou, S.; Kumar, G.; Weiss, L.; Palmisano, M. No clinically significant drug interactions between lenalidomide and P-glycoprotein substrates and inhibitors: Results from controlled phase I studies in healthy volunteers. Cancer Chemother. Pharmacol. 2014, 73, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Maggini, V.; Buda, G.; Martino, A.; Presciuttini, S.; Galimberti, S.; Orciuolo, E.; Barale, R.; Petrini, M.; Rossi, A.M. MDR1 diplotypes as prognostic markers in multiple myeloma. Pharmacogenet. Genom. 2008, 18, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Jamroziak, K.; Młynarski, W.; Balcerczak, E.; Mistygacz, M.; Trelinska, J.; Mirowski, M.; Bodalski, J.; Robak, T. Functional C3435T polymorphism of MDR1 gene: An impact on genetic susceptibility and clinical outcome of childhood acute lymphoblastic leukemia. Eur. J. Haematol. 2004, 72, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.S.; Van Eyck, L.; Pawlak, S.; Beckerman, B.; Linn, C.; Ginman, K.; Thay Cha, Y.; LaBadie, R.R.; Shi, H.; Damle, B. Effects of itraconazole and carbamazepine on the pharmacokinetics of nirmatrelvir/ritonavir in healthy adults. Br. J. Clin. Pharmacol. 2023, 89, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Ghasemiyeh, P.; Mortazavi, N.; Karimzadeh, I.; Vazin, A.; Mahmoudi, L.; Moghimi-Sarani, E.; MohammadSadeghi, A.; Shahisavandi, M.; Kheradmand, A.; Mohammadi-Samani, S. Psychiatric Adverse Drug Reactions and Potential Anti-COVID-19 Drug Interactions with Psychotropic Medications. Iran J. Pharm. Res. 2021, 20, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Potschka, H.; Chandra, P.P.; Tripathi, M.; Vohora, D. Management of COVID-19 in patients with seizures: Mechanisms of action of potential COVID-19 drug treatments and consideration for potential drug-drug interactions with anti-seizure medications. Epilepsy Res. 2021, 174, 106675. [Google Scholar] [CrossRef]


| Drugs Implicated | Time to Occurrence | Dexamethasone Dosage * | Nature of Interaction | Clinical Implication | Quality of Evidence |
|---|---|---|---|---|---|
| Lapatinib | Days to weeks | Low to high | Induction of lapatinib CYP3A-mediated metabolism: exposition to reactive, potentially toxic metabolites of lapatinib | Hepatotoxicity leading to lapatinib withdrawal in some cases | Moderate |
| Panobinostat | Few days | High | Decrease in panobinostat plasma concentrations by up to 20% | Possibly better disease control under a combination of dexamethasone and Panobinostat | Moderate |
| Cyclophosphamide | Few days | High | Decrease in Cyclophosphamide plasma concentrations | Theoretical decreased cyclophosphamide efficacy although no available signal so far | Low |
| Voriconazole | Few days | High to moderate | Decreased voriconazole plasma concentrations due to CYPC19, CYP2C9 and CYP3A induction | High to moderate increase in the risk of treatment failure, thus requiring voriconazole therapeutic drug monitoring | Moderate |
| Itraconazole | Few days | Moderate | Increase in dexamethasone plasma concentration by itraconazole mediated CYP3A4 inhibition | Potential increase in adrenal suppression at day 4, scant data currently available regarding potentially prolonged adrenal suppression | Moderate |
| Aprepitant/fosaprepitant | Immediate | High | Increase in dexamethasone plasma concentration by itraconazole mediated CYP3A4 inhibition | Low clinical implication due to sequential treatment and anticipated lowering of dexamethasone posology in the guidelines | Moderate |
| Phenytoine | few days | High | Induction of phenytoine metabolism by CYP2C9/CYP2C19 | High, thrombopenia with fatal cases reported | Very low |
| Primidone | <1 month | Low | Decrease in dexamethasone activity by metabolism induction | Moderate, lack of control of congenital adrenal hyperplasia | Very low |
| rifampicin | Immediate | Low | Decrease in dexamethasone activity by metabolism induction | Moderate, misdiagnosis of Cushing syndrome | Very low |
| carbamazepine | Very low | ||||
| troglitazone | Very low |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourdin, V.; Bigot, W.; Vanjak, A.; Burlacu, R.; Lopes, A.; Champion, K.; Depond, A.; Amador-Borrero, B.; Sene, D.; Comarmond, C.; et al. Drug–Drug Interactions Involving Dexamethasone in Clinical Practice: Myth or Reality? J. Clin. Med. 2023, 12, 7120. https://doi.org/10.3390/jcm12227120
Bourdin V, Bigot W, Vanjak A, Burlacu R, Lopes A, Champion K, Depond A, Amador-Borrero B, Sene D, Comarmond C, et al. Drug–Drug Interactions Involving Dexamethasone in Clinical Practice: Myth or Reality? Journal of Clinical Medicine. 2023; 12(22):7120. https://doi.org/10.3390/jcm12227120
Chicago/Turabian StyleBourdin, Venceslas, William Bigot, Anthony Vanjak, Ruxandra Burlacu, Amanda Lopes, Karine Champion, Audrey Depond, Blanca Amador-Borrero, Damien Sene, Chloe Comarmond, and et al. 2023. "Drug–Drug Interactions Involving Dexamethasone in Clinical Practice: Myth or Reality?" Journal of Clinical Medicine 12, no. 22: 7120. https://doi.org/10.3390/jcm12227120
APA StyleBourdin, V., Bigot, W., Vanjak, A., Burlacu, R., Lopes, A., Champion, K., Depond, A., Amador-Borrero, B., Sene, D., Comarmond, C., & Mouly, S. (2023). Drug–Drug Interactions Involving Dexamethasone in Clinical Practice: Myth or Reality? Journal of Clinical Medicine, 12(22), 7120. https://doi.org/10.3390/jcm12227120