
Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases

 ,
,  ,
,
Abstract
:1. Introduction
2. Gene Therapy Strategies
2.1. Gene Replacement Prevents and Cures Congenital Deafness
2.2. RNA Interference Therapy for Hearing Loss
2.3. Gene-Editing Therapy for Hearing Loss
3. Viral Vectors
3.1. Adenovirus and Lentivirus
3.2. Adeno-Associated Virus

{kind=link}
| Capsid | Injection Stage | Injection Route | Inner Ear Hair Cell Transduction (%) | Transduction of Other Inner Ear Cells | References | ||
|---|---|---|---|---|---|---|---|
| IHC | OHC | VHC | |||||
| AAV1 | Neonatal | RW | 0–67 | 0–14 | 0 | Inner phalangeal cells and Deiters’ cells | Askew et al., 2015 [19]; György et al., 2017 [24]; Landegger et al., 2017 [91]; Pan et al., 2017 [27]; Emptoz et al., 2017 [7] |
| CO | 36 | 17 | NR | Marginal cells | Chang et al., 2015 [26]; György et al., 2017 [24] | ||
| Mature | RW with PSCC fenestration | 10 | <5 | <10 | Stria vascularis cells | Omichi et al., 2020 [92] | |
| PSCC | 6 | 0 | NR | NR | Tao et al., 2018 [93] | ||
| AAV2 | Neonatal | RW | 0–78 | 0–50 | NR | NR | Emptoz et al., 2017 [7]; Askew et al., 2015 [19]; Landegger et al., 2017 [91]; Geng et al., 2017 [94] |
| PSCC | 44 | 54 | NR | Pillar cells | Isgrig et al., 2019 [95] | ||
| Mature | RW with PSCC fenestration | 95 | 80 | 0 | 0 | Omichi et al., 2020 [92] | |
| PSCC | 85 | 10 | 7 | NR | Tao et al., 2018 [93] | ||
| AAV5 | Neonatal | RW | 0 | 0 | 0 | Supporting and mesothelial cells | Emptoz et al., 2017 [7] |
| CO | 0 | 0 | 0 | Supporting, mesothelial, and Reissner’s membrane cells | Iizuka et al., 2015 [96] | ||
| AAV6 | Neonatal | RW | 15–20 | 5–10 | NR | NR | Askew et al., 2015 [19]; Landegger et al., 2017 [91] |
| Mature | PSCC | 5 | 0 | NR | NR | Tao et al., 2018 [93] | |
| AAV8 | Neonatal | RW | 10–90 | 5–28 | 90 | Spiral ganglion neurons | Askew et al., 2015 [19]; Chien et al., 2015 [97]; Emptoz et al., 2017 [7]; Landegger et al., 2017 [91]; Geng et al., 2017 [94]; Dulon et al., 2018 [32]; Xia et al., 2012 [98] |
| PSCC | 49–86 | 13–52 | 53 | Marginal, vestibular supporting, and pillar cells | Isgrig et al., 2019 [95]; Guo et al., 2017 [99] | ||
| Mature | RW with PSCC fenestration | 90 | <10 | 35 | Stria vascularis cells and spiral ganglion neurons | Omichi et al., 2020 [92] | |
| PSCC | 75 | 0 | 41 | NR | Tao et al., 2018 [93] | ||
| AAV9 | Neonatal | RW | 5 | 5 | NR | NR | Askew et al., 2015 [19] |
| CO | 57 | 15 | 12 | NR | Gu et al., 2019 [100] | ||
| Mature | RW + PSCC | 95 | <5 | good | NR | Yoshimura et al., 2018 [101] | |
| RW | 30 | 0 | 0 | NR | Yoshimura et al., 2018 [101] | ||
| RW with PSCC fenestration | 100 | 0 | 20 | Stria vascularis cells, and spiral ganglion neurons | Omichi et al., 2020 [92] | ||
| PSCC | 60 | 0 | 20 | NR | Tao et al., 2018 [93] | ||
| Anc80L65 | Neonatal | RW | 90–100 | 80–95 | 95 | Pillar and Deiters’ cells | Pan et al., 2017 [27]; Landegger et al., 2017 [91]; Lee et al., 2020 [86] |
| CO | 100 | 90 | NR | Supporting cells | Gu et al., 2019 [100] | ||
| PSCC | 94 | 67 | NR | Pillar cells | Isgrig et al., 2019 [95] | ||
| Utricle | 100 | 30–90 | robust | Pillar and Deiters’ cells | Lee et al., 2020 [86] | ||
| Mature | PSCC post | 95–100 | 40–50 | 40 | NR | Suzuki et al., 2017 [102]; Tao et al., 2018 [93] | |
| RW + PSCC | 90 | - | good | NR | Yoshimura et al., 2018 [101] | ||
| RW with PSCC fenestration | 100 | 50 | 35 | Stria vascularis cells, and spiral ganglion neurons | Omichi et al., 2020 [92] | ||
| Utricle | 100 | 0–20 | moderate | NR | Lee et al., 2020 [86] | ||
| AAV2 quadY-F | Mature | RW | 85 | NR | NR | NR | Akil et al., 2019 [13] |
| AAV2.7m8 | Neonatal | PSCC | 84 | 83 | NR | Pillar and internal phalangeal cells | Isgrig et al., 2019 [95] |
| Utricle | 40–100 | 40 | NR | NR | Lee et al., 2020 [86] | ||
| Mature | RW | 84 | 75 | NR | NR | Isgrig et al., 2019 [95] | |
| AAV8BP2 | Neonatal | PSCC | 56 | 44 | NR | NR | Isgrig et al., 2019 [95] |
| AAV9-PHP.B | Neonatal | RW | 70–100 | 35–70 | NR | NR | György et al., 2019 [33]; Lee et al., 2020 [86] |
| Utricle | 100 | 100 | robust | NR | Lee et al., 2020 [86] | ||
| Mature | PSCC | 100 | 0 | robust | NR | György et al., 2019 [33] | |
| Utricle | 100 | 20–80 | robust | NR | Lee et al., 2020 [86] | ||
| AAVrh.39 | Mature | PSCC | 55 | 0 | NR | NR | Tao et al., 2018 [93] |
| AAVrh.43 | Mature | PSCC | 95 | 0 | NR | NR | Tao et al., 2018 [93] |
| AAV-S | Neonatal | RW | 100 | 50–75 | robust | Interdental, inner and outer sulcus, Claudius cells, and spiral ganglion neurons | Ivanchenko et al., 2021 [88] |
| AAV-ie | Neonatal | RW | 100 | 60–100 | 100% | All cell types of supporting cells | Tan et al., 2019 [103] |
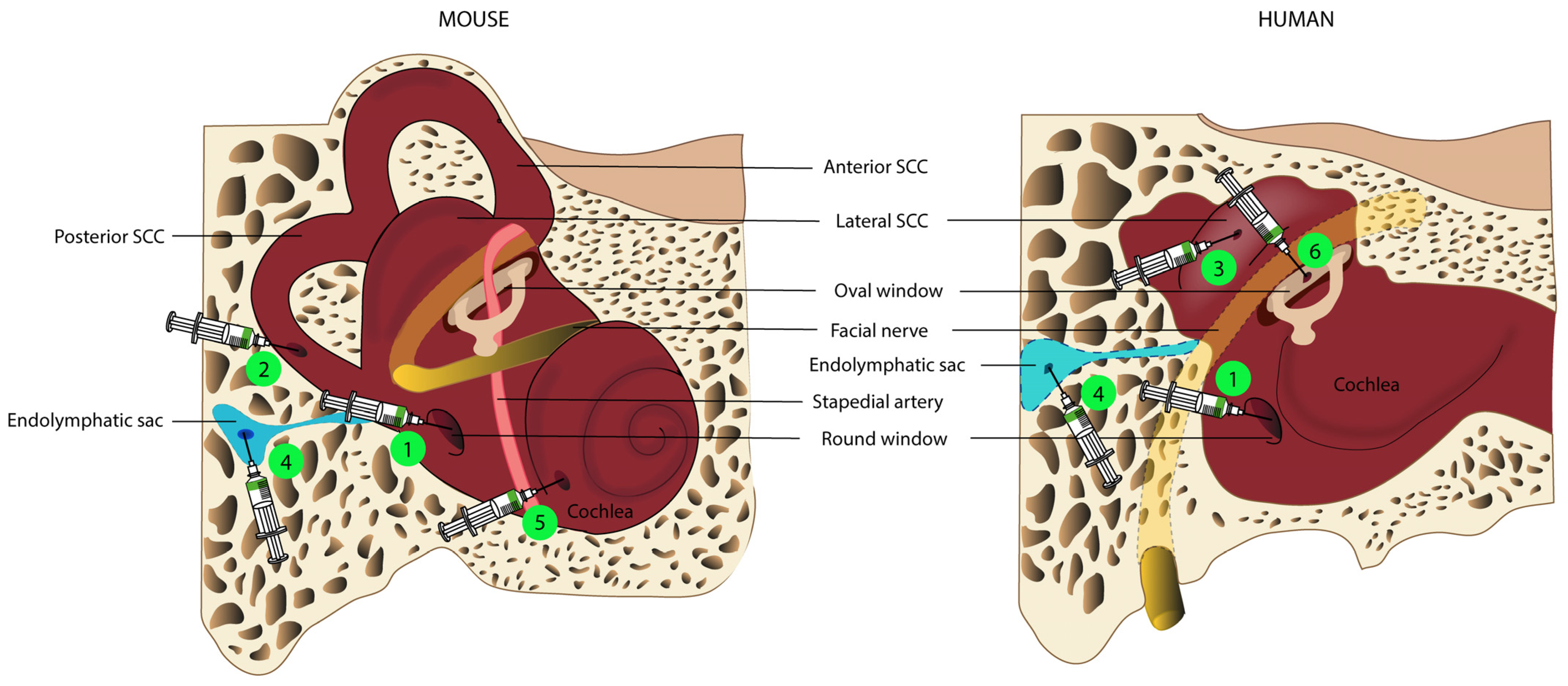
4. Routes for Inner Ear Gene Delivery
4.1. Delivery to the Endolymphatic Space: Cochleostomy, Endolymphatic Sac, and Utricle Administration
4.2. Delivery to the Perilymphatic Space: Round Window, Posterior Semi-Circular Canal, and Oval Window Administration
4.3. Volume Injected and Its Flow Rate
5. Unresolved Issues
5.1. The Temporal Window for Therapeutic Intervention
5.2. Does the Inner Ear Have Immune Privilege?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, R.J.H.; Bale, J.F.; White, K.R. Sensorineural Hearing Loss in Children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Haile, L.M.; Kamenov, K.; Briant, P.S.; Orji, A.U.; Steinmetz, J.D.; Abdoli, A.; Abdollahi, M.; Abu-Gharbieh, E.; Afshin, A.; Ahmed, H.; et al. Hearing Loss Prevalence and Years Lived with Disability, 1990–2019: Findings from the Global Burden of Disease Study 2019. Lancet 2021, 397, 996–1009. [Google Scholar] [CrossRef]
- François, M.; Boukhris, M.; Noel-Petroff, N. Schooling of Hearing-Impaired Children and Benefit of Early Diagnosis. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2015, 132, 251–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.R.; Yaffe, K.; Xia, J.; Xue, Q.L.; Harris, T.B.; Purchase-Helzner, E.; Satterfield, S.; Ayonayon, H.N.; Ferrucci, L.; Simonsick, E.M. Hearing Loss and Cognitive Decline in Older Adults. JAMA Intern. Med. 2013, 173, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, L.L.; Tucci, D.L. Hearing Loss in Adults. N. Engl. J. Med. 2017, 377, 2465–2473. [Google Scholar] [CrossRef]
- Guinand, N.; Boselie, F.; Guyot, J.; Kingma, H. Quality of Life of Patients with Bilateral Vestibulopathy. Ann. Otol. Rhinol. Laryngol. 2012, 121, 471–477. [Google Scholar] [CrossRef]
- Emptoz, A.; Michel, V.; Lelli, A.; Akil, O.; de Monvel, J.B.; Lahlou, G.; Meyer, A.; Dupont, T.; Nouaille, S.; Ey, E.; et al. Local Gene Therapy Durably Restores Vestibular Function in a Mouse Model of Usher Syndrome Type 1G. Proc. Natl. Acad. Sci. USA 2017, 114, 9695–9700. [Google Scholar] [CrossRef] [Green Version]
- Akil, O.; Seal, R.P.; Burke, K.; Wang, C.; Alemi, A.; During, M.; Edwards, R.H.; Lustig, L.R. Restoration of Hearing in the VGLUT3 Knockout Mouse Using Virally-Mediated Gene Therapy. Neuron 2012, 75, 283–293. [Google Scholar] [CrossRef] [Green Version]
- György, B.; Nist-Lund, C.; Pan, B.; Asai, Y.; Karavitaki, K.D.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Solanes, P.; Spataro, S.; et al. Allele-Specific Gene Editing Prevents Deafness in a Model of Dominant Progressive Hearing Loss. Nat. Med. 2019, 25, 1123–1130. [Google Scholar] [CrossRef]
- Delmaghani, S.; Defourny, J.; Aghaie, A.; Beurg, M.; Dulon, D.; Thelen, N.; Perfettini, I.; Zelles, T.; Aller, M.; Meyer, A.; et al. Hypervulnerability to Sound Exposure through Impaired Adaptive Proliferation of Peroxisomes. Cell 2015, 163, 894–906. [Google Scholar] [CrossRef]
- Lentz, J.J.; Pan, B.; Ponnath, A.; Tran, C.M.; Nist-Lund, C.; Galvin, A.; Goldberg, H.; Robillard, K.N.; Jodelka, F.M.; Farris, H.E.; et al. Direct Delivery of Antisense Oligonucleotides to the Middle and Inner Ear Improves Hearing and Balance in Usher Mice. Mol. Ther. 2020, 28, 2662–2676. [Google Scholar] [CrossRef]
- Kim, M.A.; Cho, H.J.; Bae, S.H.; Lee, B.; Oh, S.K.; Kwon, T.J.; Ryoo, Z.Y.; Kim, H.Y.; Cho, J.H.; Kim, U.K.; et al. Methionine Sulfoxide Reductase B3-Targeted in Utero Gene Therapy Rescues Hearing Function in a Mouse Model of Congenital Sensorineural Hearing Loss. Antioxid. Redox Signal. 2015, 24, 590–602. [Google Scholar] [CrossRef] [Green Version]
- Akil, O.; Dyka, F.; Calvet, C.; Emptoz, A.; Lahlou, G.; Nouaille, S.; de Monvel, J.B.; Hardelin, J.-P.; Hauswirth, W.W.; Avan, P.; et al. Dual AAV-Mediated Gene Therapy Restores Hearing in a DFNB9 Mouse Model. Proc. Natl. Acad. Sci. USA 2019, 116, 4496–4501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Yang, Z.; Tang, X. Chemical Modifications of Nucleic Acid Drugs and Their Delivery Systems for Gene-Based Therapy. Med. Res. Rev. 2018, 38, 829–869. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Bauer, D.E.; Chiarle, R. Assessing and Advancing the Safety of CRISPR-Cas Tools: From DNA to RNA Editing. Nat. Commun. 2023, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Miwa, T.; Minoda, R.; Ise, M.; Yamada, T.; Yumoto, E. Mouse Otocyst Transuterine Gene Transfer Restores Hearing in Mice with Connexin 30 Deletion-Associated Hearing Loss. Mol. Ther. 2013, 21, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Crispino, G.; Galindo Ramirez, F.; Campioni, M.; Zorzi, V.; Praetorius, M.; di Pasquale, G.; Chiorini, J.A.; Mammano, F. In Vivo Genetic Manipulation of Inner Ear Connexin Expression by Bovine Adeno-Associated Viral Vectors. Sci. Rep. 2017, 7, 6567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Wang, Y.; Chang, Q.; Wang, J.; Gong, S.; Li, H.; Lin, X. Virally Expressed Connexin26 Restores Gap Junction Function in the Cochlea of Conditional Gjb2 Knockout Mice. Gene Ther. 2014, 21, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askew, C.; Rochat, C.; Pan, B.; Asai, Y.; Ahmed, H.; Child, E.; Schneider, B.L.; Aebischer, P.; Holt, J.R. Tmc Gene Therapy Restores Auditory Function in Deaf Mice. Sci. Transl. Med. 2015, 7, 295ra108. [Google Scholar] [CrossRef] [PubMed]
- Nist-Lund, C.A.; Pan, B.; Patterson, A.; Asai, Y.; Chen, T.; Zhou, W.; Zhu, H.; Romero, S.; Resnik, J.; Polley, D.B.; et al. Improved TMC1 Gene Therapy Restores Hearing and Balance in Mice with Genetic Inner Ear Disorders. Nat. Commun. 2019, 10, 236. [Google Scholar] [CrossRef]
- Yeh, W.H.; Shubina-Oleinik, O.; Levy, J.M.; Pan, B.; Newby, G.A.; Wornow, M.; Burt, R.; Chen, J.C.; Holt, J.R.; Liu, D.R. In Vivo Base Editing Restores Sensory Transduction and Transiently Improves Auditory Function in a Mouse Model of Recessive Deafness. Sci. Transl. Med. 2020, 12, eaay9101. [Google Scholar] [CrossRef]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Moteki, H.; Smith, R.J.H. Targeted Allele Suppression Prevents Progressive Hearing Loss in the Mature Murine Model of Human TMC1 Deafness. Mol. Ther. 2019, 27, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.; Pan, B.; Hu, Y.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of Autosomal Dominant Hearing Loss by in Vivo Delivery of Genome Editing Agents. Nature 2018, 553, 217–221. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Sage, C.; Indzhykulian, A.A.; Scheffer, D.I.; Brisson, A.R.; Tan, S.; Wu, X.; Volak, A.; Mu, D.; Tamvakologos, P.I.; et al. Rescue of Hearing by Gene Delivery to Inner-Ear Hair Cells Using Exosome-Associated AAV. Mol. Ther. 2017, 25, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Moyed, H.; Cepeda, A.P.; Jung, S.; Moser, T.; Kügler, S.; Reisinger, E. A Dual-AAV Approach Restores Fast Exocytosis and Partially Rescues Auditory Function in Deaf Otoferlin Knock-out Mice. EMBO Mol. Med. 2019, 11, e9396. [Google Scholar] [CrossRef]
- Chang, Q.; Wang, J.; Li, Q.; Kim, Y.; Zhou, B.; Wang, Y.; Li, H.; Lin, X. Virally Mediated Kcnq 1 Gene Replacement Therapy in the Immature Scala Media Restores Hearing in a Mouse Model of Human Jervell and Lange-Nielsen Deafness Syndrome. EMBO J. 2015, 7, 1077–1086. [Google Scholar]
- Pan, B.; Askew, C.; Galvin, A.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene Therapy Restores Auditory and Vestibular Function in a Mouse Model of Usher Syndrome Type 1c. Nat. Biotechnol. 2017, 35, 264–272. [Google Scholar] [CrossRef]
- Lentz, J.J.; Jodelka, F.M.; Hinrich, A.J.; Mccaffrey, K.E.; Farris, H.E.; Spalitta, M.J.; Bazan, N.G.; Duelli, D.M.; Rigo, F.; Hastings, M.L. Rescue of Hearing and Vestibular Function by Antisense Oligonucleotides in a Mouse Model of Human Deafness. Nat. Med. 2013, 19, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumar, S.; Depreux, F.F.; Jodelka, F.M.; Lentz, J.J.; Rigo, F.; Jones, T.A.; Hastings, M.L. Rescue of Peripheral Vestibular Function in Usher Syndrome Mice Using a Splice-Switching Antisense Oligonucleotide. Hum. Mol. Genet. 2017, 26, 3482–3494. [Google Scholar] [CrossRef] [Green Version]
- Chien, W.W.; Isgrig, K.; Roy, S.; Belyantseva, I.A.; Drummond, M.C.; May, L.A.; Fitzgerald, T.S.; Friedman, T.B.; Cunningham, L.L. Gene Therapy Restores Hair Cell Stereocilia Morphology in Inner Ears of Deaf Whirler Mice. Mol. Ther. 2015, 24, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Isgrig, K.; Shteamer, J.W.; Belyantseva, I.A.; Drummond, M.C.; Fitzgerald, T.S.; Vijayakumar, S.; Jones, S.M.; Griffith, A.J.; Friedman, T.B.; Cunningham, L.L.; et al. Gene Therapy Restores Balance and Auditory Functions in a Mouse Model of Usher Syndrome. Mol. Ther. 2017, 25, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Dulon, D.; Papal, S.; Patni, P.; Cortese, M.; Vincent, P.F.Y.; Tertrais, M.; Emptoz, A.; Tlili, A.; Bouleau, Y.; Michel, V.; et al. Clarin-1 Gene Transfer Rescues Auditory Synaptopathy in Model of Usher Syndrome. J. Clin. Investig. 2018, 128, 3382–3401. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Meijer, E.J.; Ivanchenko, M.V.; Tenneson, K.; Emond, F.; Hanlon, K.S.; Indzhykulian, A.A.; Volak, A.; Karavitaki, K.D.; Tamvakologos, P.I.; et al. Gene Transfer with AAV9-PHP.B Rescues Hearing in a Mouse Model of Usher Syndrome 3A and Transduces Hair Cells in a Non-Human Primate. Mol. Ther. Methods Clin. Dev. 2019, 13, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Shubina-Oleinik, O.; Nist-Lund, C.; French, C.; Rockowitz, S.; Shearer, A.E.; Holt, J.R. Dual-Vector Gene Therapy Restores Cochlear Amplification and Auditory Sensitivity in a Mouse Model of DFNB16 Hearing Loss. Sci. Adv. 2021, 7, 7629. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zou, L.; Li, K.; Hou, H.; Hu, Q.; Liu, S.; Li, J.; Song, C.; Chen, J.; Wang, S.; et al. Template-Independent Genome Editing in the Pcdh15av−3j Mouse, a Model of Human DFNB23 Nonsyndromic Deafness. Cell Rep. 2022, 40, 111061. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Shubina-Oleinik, O.; Holt, J.R. Emerging Gene Therapies for Genetic Hearing Loss. JARO J. Assoc. Res. Otolaryngol. 2017, 18, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Martinez, J.; Patkaniowska, A.; Lendeckel, W.; Tuschl, T. Functional Anatomy of SiRNAs for Mediating Efficient RNAi in Drosophila Melanogaster Embryo Lysate. EMBO J. 2001, 20, 6877–6888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowdy, S.F. Overcoming Cellular Barriers for RNA Therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]
- Hannon, G.J.; Rossi, J.J. Unlocking the Potential of the Humangenome with RNA Interference. Nature 2004, 431, 371–378. [Google Scholar] [CrossRef]
- Nikitenko, N.A.; Speiseder, T.; Lam, E.; Rubtsov, P.M.; Tonaeva, K.D.; Borzenok, S.A.; Dobner, T.; Prassolov, V.S. Regulation of Human Adenovirus Replication by RNA Interference. Acta Nat. 2015, 7, 2015. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Fukushima, K.; Nishizaki, K.; Smith, R.J.H. In Vitro and in Vivo Suppression of GJB2 Expression by RNA Interference. Hum. Mol. Genet. 2005, 14, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Smith, L.E.; Bailey, R.A.; Compton, J.G.; Bale, S.J.; White, T.W.; Paul, D.L.; Richard, G.; White, T.W.; Smith, L.E.; et al. RAPID COMMUNICATION Functional Defects of Cx26 Resulting from a Heterozygous Missense Mutation in a Family with Dominant Deaf-Mutism and Palmoplantar Keratoderma. Hum. Genet. 1998, 103, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, F.; Lina-Granade, G.; Plauchu, H.; Bruzzone, R.; Chaib, H.; Levi-Acobas, F.; Weil, D.; Petit, C. Connexin 26 Gene Linked to a Dominant Deafness. Nature 1998, 393, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parryll, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 Mutations in Hereditary Non-Syndromic Sensorineural Deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef]
- Sontheimer, E.J.; Barrangou, R. The Bacterial Origins of the CRISPR Genome-Editing Revolution. Hum. Gene Ther. 2015, 26, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Dusko Ehrlich, S. Clustered Regularly Interspaced Short Palindrome Repeats (CRISPRs) Have Spacers of Extrachromosomal Origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Doudna, J.A. Applications of CRISPR Technologies in Research and Beyond. Nat. Biotechnol. 2016, 34, 933–941. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-CrRNA Ribonucleoprotein Complex Mediates Specific DNA Cleavage for Adaptive Immunity in Bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a Versatile Tool for Engineering Biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Di Stazio, M.; Foschi, N.; Athanasakis, E.; Gasparini, P.; d’Adamo, A.P. Systematic Analysis of Factors That Improve Homologous Direct Repair (HDR) Efficiency in CRISPR/Cas9 Technique. PLoS ONE 2021, 16, e0247603. [Google Scholar] [CrossRef] [PubMed]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient Introduction of Specific Homozygous and Heterozygous Mutations Using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.W.; Arbab, M.; Hsu, J.Y.; Worstell, D.; Culbertson, S.J.; Krabbe, O.; Cassa, C.A.; Liu, D.R.; Gifford, D.K.; Sherwood, R.I. Predictable and Precise Template-Free CRISPR Editing of Pathogenic Variants. Nature 2018, 563, 646–651. [Google Scholar] [CrossRef]
- Leenay, R.T.; Aghazadeh, A.; Hiatt, J.; Tse, D.; Roth, T.L.; Apathy, R.; Shifrut, E.; Hultquist, J.F.; Krogan, N.; Wu, Z.; et al. Large Dataset Enables Prediction of Repair after CRISPR–Cas9 Editing in Primary T Cells. Nat. Biotechnol. 2019, 37, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; de Angeli, P.; Páleníková, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the Mutations Generated by Repair of Cas9-Induced Double-Strand Breaks. Nat. Biotechnol. 2019, 37, 64–82. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of Double-Strand Breaks Induced by CRISPR–Cas9 Leads to Large Deletions and Complex Rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Lessard, S.; Francioli, L.; Alfoldi, J.; Tardif, J.C.; Ellinor, P.T.; MacArthur, D.G.; Lettre, G.; Orkin, S.H.; Canver, M.C. Human Genetic Variation Alters CRISPR-Cas9 on- and off-Targeting Specificity at Therapeutically Implicated Loci. Proc. Natl. Acad. Sci. USA 2017, 114, E11257–E11266. [Google Scholar] [CrossRef] [Green Version]
- Modarai, S.R.; Kanda, S.; Bloh, K.; Opdenaker, L.M.; Kmiec, E.B. Precise and Error-Prone CRISPR-Directed Gene Editing Activity in Human CD34+ Cells Varies Widely among Patient Samples. Gene Ther. 2021, 28, 105–113. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved Base Excision Repair Inhibition and Bacteriophage Mu Gam Protein Yields C:G-to-T:A Base Editors with Higher Efficiency and Product Purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Jo, D.H.; Cho, C.S.; Shin, J.H.; Seo, J.H.; Yu, G.; Gopalappa, R.; Kim, D.; Cho, S.R.; Kim, J.H.; et al. Application of Prime Editing to the Correction of Mutations and Phenotypes in Adult Mice with Liver and Eye Diseases. Nat. Biomed. Eng. 2022, 6, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Dong, X.; Cheng, H.; Zheng, C.; Chen, Z.; Rodríguez, T.C.; Liang, S.Q.; Xue, W.; Sontheimer, E.J. A Split Prime Editor with Untethered Reverse Transcriptase and Circular RNA Template. Nat. Biotechnol. 2022, 40, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.; Alexander, I.E.; Edelstein, M.L.; Abedi, M.R.; Wixon, J. Gene Therapy Clinical Trials Worldwide to 2012–An Update. J. Gene Med. 2013, 15, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Couture, L.A.; Gregory, R.J.; Graham, S.M.; Smith, A.E.; Welsh, M.J. Adenovirus-Mediated Gene Transfer Transiently Corrects the Chloride Transport Defect in Nasal Epithelia of Patients with Cystic Fibrosis. Cell 1993, 75, 207–216. [Google Scholar] [CrossRef]
- Kawamoto, K.; Oh, S.H.; Kanzaki, S.; Brown, N.; Raphael, Y. The Functional and Structural Outcome of Inner Ear Gene Transfer via the Vestibular and Cochlear Fluids in Mice. Mol. Ther. 2001, 4, 575–585. [Google Scholar] [CrossRef]
- Luebke, A.E.; Foster, P.K.; Muller, C.D.; Peel, A.L. Cochlear Function and Transgene Expression in the Guinea Pig Cochlea, Using Adenovirus- and Adeno-Associated Virus-Directed Gene Transfer. Hum. Gene Ther. 2001, 12, 773–781. [Google Scholar] [CrossRef]
- Praetorius, M.; Baker, K.; Weich, C.; Plinkert, P.; Staecker, H. Hearing Preservation after Inner Ear Gene Therapy: The Effect of Vector and Surgical Approach. ORL J. Otorhinolaryngol. Relat. Spec. 2003, 65, 211–214. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and Problems with the Use of Viral Vectors for Gene Therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Apolonia, L.; Waddington, S.N.; Fernandes, C.; Ward, N.J.; Bouma, G.; Blundell, M.P.; Thrasher, A.J.; Collins, M.K.; Philpott, N.J. Stable Gene Transfer to Muscle Using Non-Integrating Lentiviral Vectors. Mol. Ther. 2007, 15, 1947–1954. [Google Scholar] [CrossRef]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral Vectors: A Look Back and Ahead on Gene Transfer Technology. New Microbiol. 2013, 36, 1–22. [Google Scholar] [PubMed]
- Hashimoto, T.; Gibbs, D.; Lillo, C.; Azarian, S.M.; Legacki, E.; Zhang, X.M.; Yang, X.J.; Williams, D.S. Lentiviral Gene Replacement Therapy of Retinas in a Mouse Model for Usher Syndrome Type 1B. Gene Ther. 2007, 14, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Zallocchi, M.; Binley, K.; Lad, Y.; Ellis, S.; Widdowson, P.; Iqball, S.; Scripps, V.; Kelleher, M.; Loader, J.; Miskin, J.; et al. EIAV-Based Retinal Gene Therapy in the Shaker1 Mouse Model for Usher Syndrome Type 1B: Development of UshStat. PLoS ONE 2014, 9, e94272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.J.; Mhatre, A.N.; Wareing, M.; Pettis, R.; Gao, W.-Q.; Zufferey, R.N.; Trono, D.; Lalwani, A.K. Transgene Expression in the Guinea Pig Cochlea Mediated by a Lentivirus-Derived Gene Transfer Vector. Hum. Gene Ther. 1999, 10, 1867–1873. [Google Scholar] [CrossRef]
- Parker, C.L.; Jacobs, T.M.; Huckaby, J.T.; Harit, D.; Lai, S.K. Efficient and Highly Specific Gene Transfer Using Mutated Lentiviral Vectors Redirected with Bispecific Antibodies. mBio 2020, 11, e02990-19. [Google Scholar] [CrossRef] [Green Version]
- Hastie, E.; Samulski, R.J. Adeno-Associated Virus at 50: A Golden Anniversary of Discovery, Research, and Gene Therapy Success–A Personal Perspective. Hum. Gene Ther. 2015, 26, 257–265. [Google Scholar]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [Green Version]
- Penaud-Budloo, M.; le Guiner, C.; Nowrouzi, A.; Toromanoff, A.; Chérel, Y.; Chenuaud, P.; Schmidt, M.; von Kalle, C.; Rolling, F.; Moullier, P.; et al. Adeno-Associated Virus Vector Genomes Persist as Episomal Chromatin in Primate Muscle. J. Virol. 2008, 82, 7875–7885. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-Associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.E.; Rolling, F.; Li, C.; Conrath, H.; Xiao, W.; Xiao, X.; Samulski, R.J. Cross-Packaging of a Single Adeno-Associated Virus (AAV) Type 2 Vector Genome into Multiple AAV Serotypes Enables Transduction with Broad Specificity. J. Virol. 2002, 76, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Stieger, K.; Lheriteau, E.; Moullier, P.; Rolling, F. AAV-Mediated Gene Therapy for Retinal Disorders in Large Animal Models. ILAR J. 2009, 50, 206–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hordeaux, J.; Wang, Q.; Katz, N.; Buza, E.L.; Bell, P.; Wilson, J.M. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol. Ther. 2018, 26, 664–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, P.S.; Lee, V.; Wei, Z.; Song, J.Y.; Casal, G.; Cronin, T.; Willett, K.; Huckfeldt, R.; Morgan, J.I.W.; Aleman, T.S.; et al. Evaluation of Dose and Safety of AAV7m8 and AAV8BP2 in the Non-Human Primate Retina. Hum. Gene Ther. 2017, 28, 154–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Nist-Lund, C.; Solanes, P.; Goldberg, H.; Wu, J.; Pan, B.; Schneider, B.L.; Holt, J.R. Efficient Viral Transduction in Mouse Inner Ear Hair Cells with Utricle Injection and AAV9-PHP.B. Hear Res. 2020, 394, 107882. [Google Scholar] [CrossRef] [PubMed]
- Ivanchenko, M.V.; Hanlon, K.S.; Devine, M.K.; Tenneson, K.; Emond, F.; Lafond, J.F.; Kenna, M.A.; Corey, D.P.; Maguire, C.A. Preclinical Testing of AAV9-PHP.B for Transgene Expression in the Non-Human Primate Cochlea. Hear Res. 2020, 394, 107930. [Google Scholar] [CrossRef]
- Ivanchenko, M.V.; Hanlon, K.S.; Hathaway, D.M.; Klein, A.J.; Peters, C.W.; Li, Y.; Tamvakologos, P.I.; Nammour, J.; Maguire, C.A.; Corey, D.P. AAV-S: A Versatile Capsid Variant for Transduction of Mouse and Primate Inner Ear. Mol. Ther. Methods Clin. Dev. 2021, 21, 382–398. [Google Scholar] [CrossRef]
- Andres-Mateos, E.; Landegger, L.D.; Unzu, C.; Phillips, J.; Lin, B.M.; Dewyer, N.A.; Sanmiguel, J.; Nicolaou, F.; Valero, M.D.; Bourdeu, K.I.; et al. Choice of Vector and Surgical Approach Enables Efficient Cochlear Gene Transfer in Nonhuman Primate. Nat. Commun. 2022, 13, 1359. [Google Scholar] [CrossRef]
- Ranum, P.T.; Tecedor, L.; Keiser, M.S.; Chen, Y.H.; Leib, D.E.; Liu, X.; Davidson, B.L. Cochlear Transduction via Cerebrospinal Fluid Delivery of AAV in Non-Human Primates. Mol. Ther. 2023. [Google Scholar] [CrossRef]
- Landegger, L.D.; Pan, B.; Askew, C.; Wassmer, S.J.; Gluck, S.D.; Galvin, A.; Taylor, R.; Forge, A.; Stankovic, K.M.; Holt, J.R.; et al. A Synthetic AAV Vector Enables Safe and Efficient Gene Transfer to the Mammalian Inner Ear. Nat. Biotechnol. 2017, 35, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omichi, R.; Yoshimura, H.; Shibata, S.B.; Vandenberghe, L.H.; Smith, R.J.H. Hair Cell Transduction Efficiency of Single- and Dual-AAV Serotypes in Adult Murine Cochleae. Mol. Methods Clin. Dev. 2020, 17, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Huang, M.; Shu, Y.; Ruprecht, A.; Wang, H.; Tang, Y.; Vandenberghe, L.H.; Wang, Q.; Gao, G.; Kong, W.J.; et al. Delivery of Adeno-Associated Virus Vectors in Adult Mammalian Inner-Ear Cell Subtypes Without Auditory Dysfunction. Hum. Gene Ther. 2018, 29, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Geng, R.; Omar, A.; Gopal, S.R.; Chen, D.H.C.; Stepanyan, R.; Basch, M.L.; Dinculescu, A.; Furness, D.N.; Saperstein, D.; Hauswirth, W.; et al. Modeling and Preventing Progressive Hearing Loss in Usher Syndrome III. Sci. Rep. 2017, 7, 13480. [Google Scholar] [CrossRef] [Green Version]
- Isgrig, K.; McDougald, D.S.; Zhu, J.; Wang, H.J.; Bennett, J.; Chien, W.W. AAV2.7m8 Is a Powerful Viral Vector for Inner Ear Gene Therapy. Nat. Commun. 2019, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, T.; Kamiya, K.; Gotoh, S.; Sugitani, Y.; Suzuki, M.; Noda, T.; Minowa, O.; Ikeda, K. Perinatal GJB2 Gene Transfer Rescues Hearing in a Mouse Model of Hereditary Deafness. Hum. Mol. Genet. 2015, 24, 3651–3661. [Google Scholar] [CrossRef] [Green Version]
- Chien, W.W.; McDougald, D.S.; Roy, S.; Fitzgerald, T.S.; Cunningham, L.L. Cochlear Gene Transfer Mediated by Adeno-Associated Virus: Comparison of Two Surgical Approaches. Laryngoscope 2015, 125, 2557–2564. [Google Scholar] [CrossRef]
- Xia, L.; Yin, S.; Wang, J. Inner Ear Gene Transfection in Neonatal Mice Using Adeno-Associated Viral Vector: A Comparison of Two Approaches. PLoS ONE 2012, 7, e43218. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Liu, Y.; Qu, T.; Peng, Z.; Xie, J.; Wang, G.; Gong, S. Cochleovestibular Gene Transfer in Neonatal Mice by Canalostomy. Neuroreport 2017, 28, 682–688. [Google Scholar] [CrossRef]
- Gu, X.; Chai, R.; Guo, L.; Dong, B.; Li, W.; Shu, Y.; Huang, X.; Li, H. Transduction of Adeno-Associated Virus Vectors Targeting Hair Cells and Supporting Cells in the Neonatal Mouse Cochlea. Front. Cell. Neurosci. 2019, 13, 1–16. [Google Scholar] [CrossRef]
- Yoshimura, H.; Shibata, S.B.; Ranum, P.T.; Smith, R.J.H. Enhanced Viral-Mediated Cochlear Gene Delivery in Adult Mice by Combining Canal Fenestration with Round Window Membrane Inoculation. Sci. Rep. 2018, 8, 2980. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Hashimoto, K.; Xiao, R.; Vandenberghe, L.H.; Liberman, M.C. Cochlear Gene Therapy with Ancestral AAV in Adult Mice: Complete Transduction of Inner Hair Cells without Cochlear Dysfunction. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Chu, C.; Qi, J.; Li, W.; You, D.; Li, K.; Chen, X.; Zhao, W.; Cheng, C.; Liu, X.; et al. AAV-Ie Enables Safe and Efficient Gene Transfer to Inner Ear Cells. Nat. Commun. 2019, 10, 3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyka, F.M.; Boye, S.L.; Chiodo, V.A.; Hauswirth, W.W.; Boye, S.E. Dual Adeno-Associated Virus Vectors Result in Efficient in Vitro and in Vivo Expression of an Oversized Gene, MYO7A. Hum. Gene Ther. Methods 2014, 25, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapani, I.; Colella, P.; Sommella, A.; Iodice, C.; Cesi, G.; de Simone, S.; Marrocco, E.; Rossi, S.; Giunti, M.; Palfi, A.; et al. Effective Delivery of Large Genes to the Retina by Dual AAV Vectors. EMBO Mol. Med. 2014, 6, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Mcclements, M.E.; Maclaren, R.E. Adeno-Associated Virus (AAV) Dual Vector Strategies for Gene Therapy Encoding Large Transgenes. Yale J. Biol. Med. 2017, 90, 611–623. [Google Scholar]
- Koo, T.; Popplewell, L.; Athanasopoulos, T.; Dickson, G. Triple Trans-Splicing Adeno-Associated Virus Vectors Capable of Transferring the Coding Sequence for Full-Length Dystrophin Protein into Dystrophic Mice. Hum. Gene Ther. 2014, 25, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Ahmed, Z.M.; Riazuddin, S.; Bhinder, M.A.; Shahzad, M.; Husnain, T.; Riazuddin, S.; Griffith, A.J.; Friedman, T.B. Identities and Frequencies of Mutations of the Otoferlin Gene (OTOF) Causing DFNB9 Deafness in Pakistan. Clin. Genet. 2009, 75, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ballesteros, M.; Reynoso, R.; Olarte, M.; Villamar, M.; Morera, C.; Santarelli, R.; Arslan, E.; Medá, C.; Curet, C.; Völter, C.; et al. A Multicenter Study on the Prevalence and Spectrum of Mutations in the Otoferlin Gene (OTOF) in Subjects with Nonsyndromic Hearing Impairment and Auditory Neuropathy. Hum. Mutat. 2008, 29, 823–831. [Google Scholar] [CrossRef]
- Yasunaga, S.; Grati, M.; Cohen-Salmon, M.; El-Amraoui, A.; Mustapha, M.; Salem, N.; El-Zir, E.; Loiselet, J.; Petit, C. A Mutation in OTOF, Encoding Otoferlin, a FER-1-like Protein, Causes DFNB9, a Nonsyndromic Form of Deafness. Nat. Genet. 1999, 21, 363–369. [Google Scholar] [CrossRef]
- Yasunaga, S.; Grati, M.; Chardenoux, S.; Smith, T.N.; Friedman, T.B.; Lalwani, A.K.; Wilcox, E.R.; Petit, C. OTOF Encodes Multiple Long and Short Isoforms: Genetic Evidence That the Long Ones Underlie Recessive Deafness DFNB9. Am. J. Hum. Genet. 2000, 67, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Shi, X. Pathophysiology of the Cochlear Intrastrial Fluid-Blood Barrier (Review). Hear Res. 2016, 338, 52–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, S.B.; Yoshimura, H.; Ranum, P.T.; Goodwin, A.T.; Smith, R.J.H. Intravenous RAAV2/9 Injection for Murine Cochlear Gene Delivery. Sci. Rep. 2017, 7, 9609. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.A.; Li, Q.; Yang, J.; Goddard, J.C.; Fekete, D.M.; Lang, H. Adeno-Associated Virus-Mediated Gene Delivery into the Scala Media of the Normal and Deafened Adult Mouse Ear. Gene Ther. 2011, 18, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salt, A.N.; Hirose, K. Communication Pathways to and from the Inner Ear and Their Contributions to Drug Delivery. Hear Res. 2018, 362, 25–37. [Google Scholar] [CrossRef]
- Yamasoba, T.; Yagi, M.; Roessler, B.J.; Miller, J.M.; Raphael, Y. Inner Ear Transgene Expression after Adenoviral Vector Inoculation in the Endolymphatic Sac. Hum. Gene Ther. 1999, 10, 769–774. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, R.; Dai, C.; Steyger, P.S.; Yongfu, Y.Y. Comparison of Gentamicin Distribution in the Inner Ear Following Administration via the Endolymphatic Sac or Round Window. Laryngoscope 2010, 120, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Colletti, V.; Mandalà, M.; Carner, M.; Barillari, M.; Cerini, R.; Pozzi Mucelli, R.; Colletti, L. Evidence of Gadolinium Distribution from the Endolymphatic Sac to the Endolymphatic Compartments of the Human Inner Ear. Audiol. Neurotol. 2010, 15, 353–363. [Google Scholar] [CrossRef]
- García, M.D.L.F.; Segura, C.D.L.L.; Lesser, J.C.C.; Pianese, C.P. Endolymphatic Sac Surgery for Ménière’s Disease–Current Opinion and Literature Review. Int. Arch. Otorhinolaryngol. 2017, 21, 179–183. [Google Scholar]
- Dai, C.; Lehar, M.; Sun, D.Q.; Rvt, L.S.; Carey, J.P.; MacLachlan, T.; Brough, D.; Staecker, H.; della Santina, A.M.; Hullar, T.E.; et al. Rhesus Cochlear and Vestibular Functions Are Preserved After Inner Ear Injection of Saline Volume Sufficient for Gene Therapy Delivery. JARO J. Assoc. Res. Otolaryngol. 2017, 18, 601–617. [Google Scholar] [CrossRef]
- Jero, J.; Mhatre, A.N.; Tseng, C.J.; Stern, R.E.; Coling, D.E.; Goldstein, J.A.; Hong, K.; Zheng, W.W.; Hoque, A.T.M.S.; Lalwani, A.K. Cochlear Gene Delivery through an Intact Round Window Membrane in Mouse. Hum. Gene Ther. 2001, 12, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, Z.; Fan, L.; Landry, T.; Brown, J.; Yu, Z.; Yin, S.; Wang, J. Ultrasound-Microbubble Cavitation Facilitates Adeno-Associated Virus Mediated Cochlear Gene Transfection across the Round-Window Membrane. Bioeng. Transl. Med. 2021, 6, e10189. [Google Scholar] [CrossRef] [PubMed]
- Hitier, M.; Zhang, M.; Labrousse, M.; Barbier, C.; Patron, V.; Moreau, S. Persistent Stapedial Arteries in Human: From Phylogeny to Surgical Consequences. Surg. Radiol. Anat. 2013, 35, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Thorne, M.; Salt, A.N.; DeMott, J.E.; Henson, M.M.; Henson, O.W.; Gewalt, S.L. Cochlear Fluid Space Dimensions for Six Species Derived from Reconstructions of Three-Dimensional Magnetic Resonance Images. Laryngoscope 1999, 109, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.C.; Gosselin-Ildari, A.D. Cochlear Labyrinth Volume and Hearing Abilities in Primates. Anat. Rec. 2009, 292, 765–776. [Google Scholar] [CrossRef]
- Stöver, T.; Yagi, M.; Raphael, Y. Transduction of the Contralateral Ear after Adenovirus-Mediated Cochlear Gene Transfer. Gene Ther. 2000, 7, 377–383. [Google Scholar] [CrossRef]
- Burton, J.A.; Valero, M.D.; Hackett, T.A.; Ramachandran, R. The Use of Nonhuman Primates in Studies of Noise Injury and Treatment. J. Acoust. Soc. Am. 2019, 146, 3770–3789. [Google Scholar] [CrossRef] [Green Version]
- Manrique-Huarte, R.; de Linera-Alperi, M.A.; Parilli, D.; Rodriguez, J.A.; Borro, D.; Dueck, W.F.; Smyth, D.; Salt, A.; Manrique, M. Inner Ear Drug Delivery through a Cochlear Implant: Pharmacokinetics in a Macaque Experimental Model. Hear Res. 2021, 404, 108228. [Google Scholar] [CrossRef]
- Litovsky, R. Development of the Auditory System, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 129. [Google Scholar]
- Lim, R.; Brichta, A.M. Anatomical and Physiological Development of the Human Inner Ear. Hear Res. 2016, 338, 9–21. [Google Scholar] [CrossRef]
- Pedreira, D.A.L. Advances in Fetal Surgery. Einstein 2016, 14, 110–112. [Google Scholar] [CrossRef] [Green Version]
- Michalski, N.; Goutman, J.D.; Auclair, S.M.; de Monvel, J.B.; Tertrais, M.; Emptoz, A.; Parrin, A.; Nouaille, S.; Guillon, M.; Sachse, M.; et al. Otoferlin Acts as a Ca2+ Sensor for Vesicle Fusion and Vesicle Pool Replenishment at Auditory Hair Cell Ribbon Synapses. Elife 2017, 6, e31013. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Heon, E.; Hauswirth, W.W. Improvement and Decline in Vision with Gene Therapy in Childhood Blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bainbridge, J.W.B.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-Term Effect of Gene Therapy on Leber’s Congenital Amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichel, F.F.; Dauletbekov, D.L.; Klein, R.; Peters, T.; Ochakovski, G.A.; Seitz, I.P.; Wilhelm, B.; Ueffing, M.; Biel, M.; Wissinger, B.; et al. AAV8 Can Induce Innate and Adaptive Immune Response in the Primate Eye. Mol. Ther. 2017, 25, 2648–2660. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, A.C.; Tuddenham, E.G.D.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia, B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Hareendran, S.; Balakrishnan, B.; Sen, D.; Kumar, S.; Srivastava, A.; Jayandharan, G.R. Adeno-Associated Virus (AAV) Vectors in Gene Therapy: Immune Challenges and Strategies to Circumvent Them. Rev. Med. Virol. 2013, 23, 399–413. [Google Scholar] [CrossRef]
- Manno, C.S.; Arruda, V.R.; Pierce, G.F.; Glader, B.; Ragni, M.; Rasko, J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful Transduction of Liver in Hemophilia by AAV-Factor IX and Limitations Imposed by the Host Immune Response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Immune Responses to AAV Vectors: Overcoming Barriers to Successful Gene Therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Masat, E.; Pavani, G.; Mingozzi, F. Humoral Immunity to AAV Vectors in Gene Therapy: Challenges and Potential Solutions. Discov. Med. 2013, 15, 379–389. [Google Scholar]

| Gene (Deafness) | Mouse Model | Stage | Approach | Vector | Strategy | Results | References |
|---|---|---|---|---|---|---|---|
| VGLUT3 (DFNA25) | Vglut3-/- | Mature | RW | AAV2/1 | Replacement | Improvement in hearing to near-normal ABR thresholds | Akil et al., 2012 [8] |
| Neonatal | RW/Co | AAV2/1 | Replacement | ||||
| GJB6 (DFNB1) | Gjb6-/- | In utero | Otocyst | - | Replacement | Improvement of hearing (thresholds: 50 dB) | Miwa et al., 2013 [16] |
| Gjb6-/- | Neonatal | PSCC | BAAV | Replacement | Protein production without hearing improvement | Crispino et al., 2017 [17] | |
| GJB2 (DFNB1) | Foxg1-cCx26KO | Neonatal | Co | AAV2/1 | Replacement | Protein production without hearing improvement | Yu et al., 2014 [18] |
| MSRB3 (DFNB74) | MsrB3-/- | In utero | Otocyst | AAV2/1 | Replacement | Improvement in hearing to near-normal ABR thresholds | M.-A. Kim et al., 2015 [12] |
| TMC1 (DFNB7/11) | Tmc1Δ/Δ | Neonatal | RW | AAV2/1 | Replacement | Partial improvement of hearing (thresholds: 90 dB) | Askew et al., 2015 [19] |
| Tmc1Δ/Δ | AAV2/Anc80L65 | Replacement | Partial improvement of hearing (thresholds: 60 dB) Improvement of auditory cortex responses | Nist-Lund et al., 2019 [20] | |||
| Tmc1Y182C/Y182C | Neonatal | NR | AAV2/Anc80L65 | Base editing | Partial and transient improvement of hearing (thresholds: 90 dB) | Yeh et al., 2020 [21] | |
| TMC1 (DFNA36) | Tmc1Bth/+ | Mature | RW + PSCC fenestration | AAV2/9 | Regulation (miRNA) | Prevention of the progression of deafness | Yoshimura et al., 2019 [22] |
| Tmc1Bth/+ | Neonatal | Co | Liposome | Gene editing (CRISPR-Cas9) | Prevention of the progression of deafness | Gao et al., 2017 [23] | |
| Tmc1Bth/+ | Neonatal | Intracochlear | AAV2/Anc80L65 | Gene editing (CRISPR-Cas9) | Prevention of the progression of deafness up to one year after treatment | György et al., 2019 [9] | |
| PJVK (DFNB59) | Pjvk-/- | Neonatal | RW | AAV2/8 | Replacement | Improvement in hearing to near-normal thresholds | Delmaghani et al., 2015 [10] |
| LHFPL5 (DFNB67) | Lhflp5-/- | Neonatal | RW | Exo-AAV2/1 | Replacement | Partial improvement of hearing (ABR thresholds: 80 dB) Partial improvement of vestibular function | György et al., 2017 [24] |
| OTOF (DFNB9) | Otof-/- | P6-P7 | RW | AAV2/6 | Replacement | Partial improvement of hearing (thresholds: 70 to 90 dB) | Al Moyed et al., 2019 [25] |
| Otof-/- | P10-P30 | RW | AAV2quadY-F | Replacement | Improvement in hearing to near-normal thresholds | Akil et al., 2019 [13] | |
| KCNQ1 (Jervell Lange-Nielsen) | Kcnq1-/- | Neonatal | RW | AAV2/1 | Replacement | Prevention of cochlear morphological abnormalities Improvement in hearing to near-normal thresholds | Chang et al., 2015 [26] |
| USH1C (Usher type 1C) | Ush1c c.216G > A | Neonatal | RW | Anc80L65 | Replacement | Complete restoration of balance Partial improvement of hearing (thresholds: 50 dB) | Pan et al., 2017 [27] |
| Neonatal | IP | - | Regulation (ASO) | Partial improvement of hearing (thresholds: 50 dB) | Lentz et al., 2013 [28] | ||
| Complete restoration of balance | Vijayakumar et al., 2017 [29] | ||||||
| Mature | Partial restoration of balance | ||||||
| Neonatal | RW/ITI | - | Regulation (ASO) | Complete restoration of balance Improvement in hearing to near-normal thresholds | Lentz et al., 2020 [11] | ||
| Mature | ITI | Partial improvement in hearing Significant improvement in balance function | |||||
| USH1G (Usher type 1G) | Ush1g-/- mice | Neonatal | RW | AAV2/8 | Replacement | Complete restoration of balance Partial improvement of hearing (thresholds: 50 dB) | Emptoz et al., 2017 [7] |
| WHRN (Usher type 2D) | Whrnwi/wi | Neonatal | RW | AAV2/8 | Replacement | Restoration of stereocilium structure and prevention of cell degeneration without improvement of hearing | Chien et al., 2015 [30] |
| Neonatal | PSCC | AAV2/8 | Replacement | Improvement of balance and hearing (thresholds: 90 dB) | Isgrig et al., 2017 [31] | ||
| CLRN (Usher type 3) | cClrn1KO | Neonatal | RW | AAV2/8 | Replacement | Preservation of synaptic morphology with a slight improvement in hearing | Dulon et al., 2018 [32] |
| Clrn-/- | Neonatal | PSCC | AAV9.PHP.B | Replacement | Partial improvement of hearing (thresholds: 40 dB for low frequencies) | György et al., 2019 [33] | |
| STRC (DFNB16) | Strc-/- | Neonatal | Utricle | AAV9.PHP.B | Replacement | Partial improvement of hearing (thresholds: 40 db) with restoration of DPOAE | Shubina-Oleinik et al., 2021 [34] |
| PCDH15 (DFNB23) | Pcdh15av3j | Neonatal | Co | AAV2/9 | Gene editing (CRISPR-Cas9) | Almost complete restoration of balance Partial improvement of hearing (thresholds: 90 dB) | Liu et al., 2022 [35] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lahlou, G.; Calvet, C.; Giorgi, M.; Lecomte, M.-J.; Safieddine, S. Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases. J. Clin. Med. 2023, 12, 1046. https://doi.org/10.3390/jcm12031046
Lahlou G, Calvet C, Giorgi M, Lecomte M-J, Safieddine S. Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases. Journal of Clinical Medicine. 2023; 12(3):1046. https://doi.org/10.3390/jcm12031046
Chicago/Turabian StyleLahlou, Ghizlene, Charlotte Calvet, Marie Giorgi, Marie-José Lecomte, and Saaid Safieddine. 2023. "Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases" Journal of Clinical Medicine 12, no. 3: 1046. https://doi.org/10.3390/jcm12031046
APA StyleLahlou, G., Calvet, C., Giorgi, M., Lecomte, M. -J., & Safieddine, S. (2023). Towards the Clinical Application of Gene Therapy for Genetic Inner Ear Diseases. Journal of Clinical Medicine, 12(3), 1046. https://doi.org/10.3390/jcm12031046