

Is Endothelial Activation a Critical Event in Thrombotic Thrombocytopenic Purpura?
 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Endothelium
2.1. Endothelial Cells
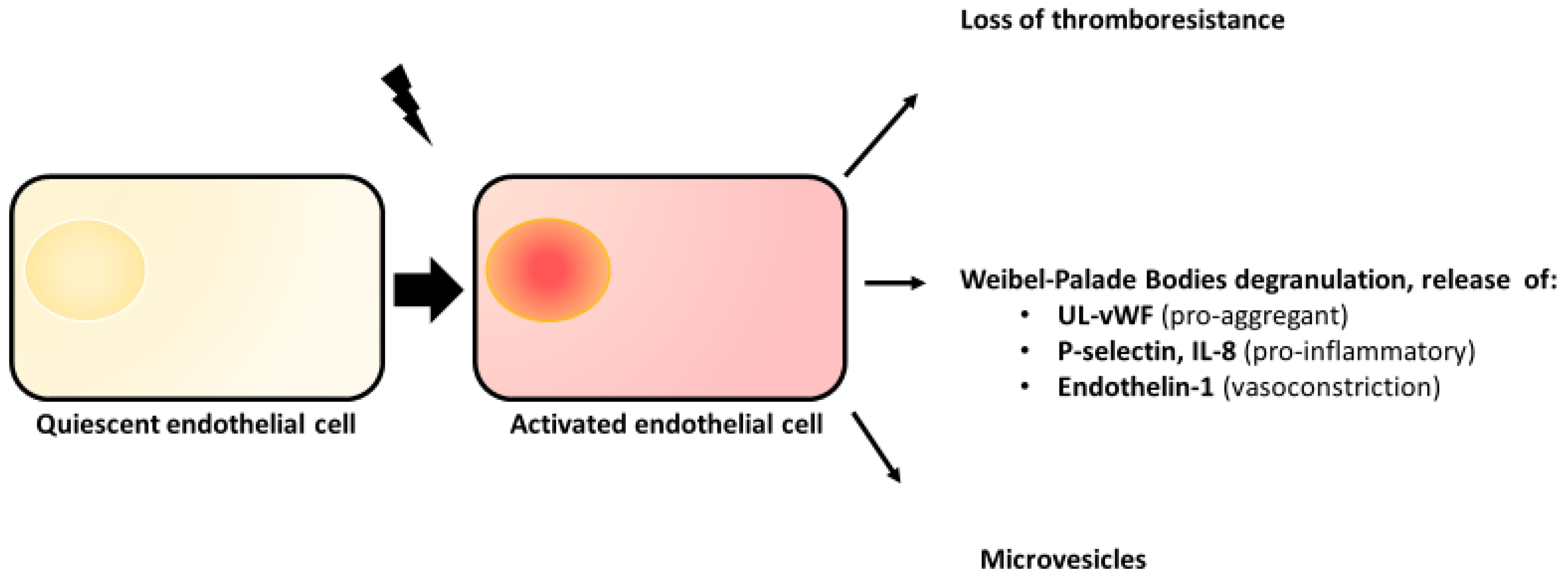
2.2. Endothelial Activation
2.3. Weibel–Palade Bodies
- –
- The Ca2+-mediated pathway: agonists, such as thrombin and histamine activate GqPCRs and GiPCRs, leading to the activation of phospholipase Cβ and the formation of inositol triphosphate (IP3). The fixation of IP3 on the endoplasmic reticulum membrane IP-3 receptor generates a Ca2+ intracellular influx which triggers the degranulation of the cortical pool of WPBs. It also increases vascular permeability involving VE-Cadherin phosphorylation and myosin light-chain phosphorylation and participates in the vesiculation process [33].
- –
- The cAMP-mediated pathway (cyclic adenosine monophosphate): other agonists, such as epinephrine and serotonin activate GsPCRs, inducing an increase in cAMP intracellular level. This results in the activation of the PKA (protein kinase A) that triggers a lower degranulation of WPB microtubular pool. In contrast to the Ca2+-mediated pathway, cAMP-mediated pathway is associated with a decrease in vascular permeability.
2.4. From Endothelial Activation to Endothelial Dysfunction
3. Evidence for Endothelial Activation in TTP
3.1. Clinical Points
3.2. Endothelial Exploration in Humans in TTP
3.3. In Vitro Experimental Data
3.4. In Vivo Experimental Data
4. What Are the Suspected Triggers for the Second Hit Hypothesis?
- –
- Some viruses, such as the Cytomegalovirus (CMV), have a tropism for ECs and can directly activate them [88].
- –
- The cytokines secreted in response to the infectious process, especially γ-interferon, TNFα and Interleukin-8, may act as CWP degranulation inductors [89]. Type 1 interferons (α and β) may induce TMA, as shown in a murine model [90]. Furthermore, γ-interferon and TNFα are cytokines that downregulate Adamts13 gene expression in the liver [91].
- –
- TLR-9 (Toll-like receptor-9) is a molecule belonging to innate immunity expressed on neutrophils and ECs which recognizes DNA present in bacteria and viruses. TLR-9 polymorphisms have been suspected to be a genetic risk factor of TTP crisis [87].
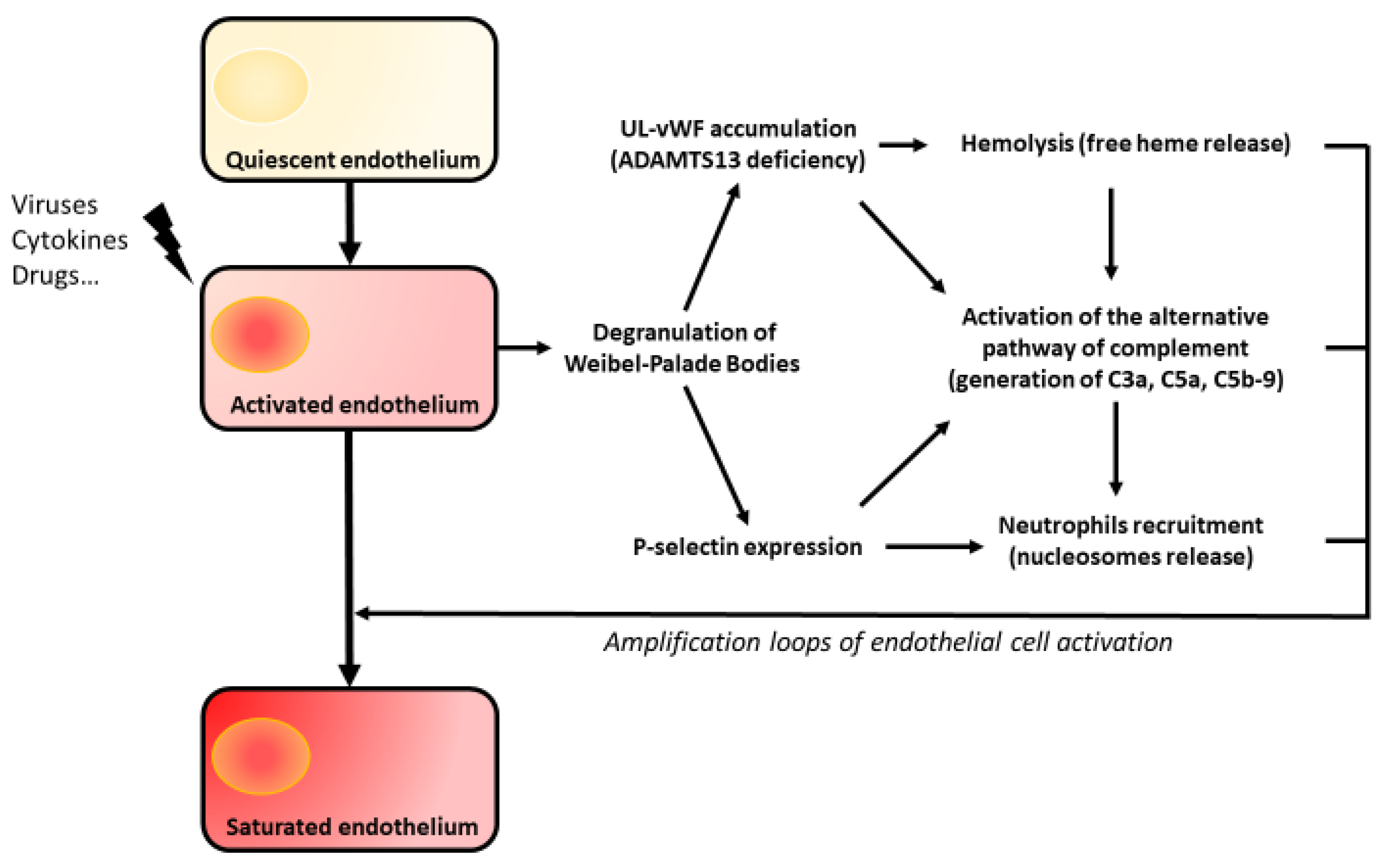
5. Amplification Loops of Endothelial Aggression
5.1. The Complement System
5.2. Hemolysis
5.3. Nucleosomes
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- George, J.N.; Nester, C.M. Syndromes of Thrombotic Microangiopathy. N. Engl. J. Med. 2014, 371, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moake, J.L.; Rudy, C.K.; Troll, J.H.; Weinstein, M.J.; Colannino, N.M.; Azocar, J.; Seder, R.H.; Hong, S.L.; Deykin, D. Unusually Large Plasma Factor VIII: Von Willebrand Factor Multimers in Chronic Relapsing Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 1982, 307, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, K.; Suzuki, H.; McMullen, B.; Chung, D. Purification of human von Willebrand factor–cleaving protease and its identification as a new member of the metalloproteinase family. Blood 2001, 98, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chung, D.; Takayama, T.K.; Majerus, E.M.; Sadler, J.E.; Fujikawa, K. Structure of von Willebrand Factor-cleaving Protease (ADAMTS13), a Metalloprotease Involved in Thrombotic Thrombocytopenic Purpura. J. Biol. Chem. 2001, 276, 41059–41063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotta, L.A.; Garagiola, I.; Palla, R.; Cairo, A.; Peyvandi, F. ADAMTS13 mutations and polymorphisms in congenital thrombotic thrombocytopenic purpura. Hum. Mutat. 2010, 31, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-M.; Lian, E.C.-Y. Antibodies to von Willebrand Factor–Cleaving Protease in Acute Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 1998, 339, 1585–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, M.; Yarranton, H.; Liesner, R.; Cavenagh, J.; Hunt, B.; Benjamin, S.; Bevan, D.; Mackie, I.; Machin, S. Regional UK TTP Registry: Correlation with laboratory ADAMTS 13 analysis and clinical features. Br. J. Haematol. 2008, 142, 819–826. [Google Scholar] [CrossRef]
- Kremer Hovinga, J.A.; Coppo, P.; Lämmle, B.; Moake, J.L.; Miyata, T.; Vanhoorelbeke, K. Thrombotic thrombocytopenic purpura. Nat. Rev. Dis. Prim. 2017, 3, 17020. [Google Scholar] [CrossRef] [Green Version]
- Reeves, H.M.; Maitta, R.W. Comparison of absolute immature platelet count to the PLASMIC score at presentation in predicting ADAMTS13 deficiency in suspected thrombotic thrombocytopenic purpura. Thromb. Res. 2022, 215, 30–36. [Google Scholar] [CrossRef]
- Zhu, M.-L.; Reeves, H.M.; Maitta, R.W. Immature platelet dynamics correlate with ADAMTS13 deficiency and predict therapy response in immune-mediated thrombotic thrombocytopenic purpura. Thromb. Res. 2021, 198, 72–78. [Google Scholar] [CrossRef]
- George, J.N. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010, 116, 4060–4069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppo, P. Treatment of autoimmune thrombotic thrombocytopenic purpura in the more severe forms. Transfus. Apher. Sci. 2017, 56, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Cataland, S.; Knöbl, P.; Wu, H.; Artoni, A.; Westwood, J.-P.; Mansouri Taleghani, M.; Jilma, B.; et al. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2016, 374, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Zeldin, R.K. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Poullin, P.; Bornet, C.; Veyradier, A.; Coppo, P. Caplacizumab to treat immune-mediated thrombotic thrombocytopenic purpura. Drugs Today 2019, 55, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Deford, C.C.; Reese, J.A.; Schwartz, L.H.; Perdue, J.J.; Kremer Hovinga, J.A.; Lämmle, B.; George, J.N. Multiple major morbidities and increased mortality during long-term follow-up after recovery from thrombotic thrombocytopenic purpura. Blood 2013, 122, 2023–2029. [Google Scholar] [CrossRef] [Green Version]
- Kremer Hovinga, J.A.; Vesely, S.K.; Terrell, D.R.; Lämmle, B.; George, J.N. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood 2010, 115, 1500–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggenenti, P.; Noris, M.; Remuzzi, G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001, 60, 831–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, R.J.; Nakagawa, T.; Johnson, R.J.; Thurman, J.M. The Role of Endothelial Cell Injury in Thrombotic Microangiopathy. Am. J. Kidney Dis. 2010, 56, 1168–1174. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.M. Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndrome. In Platelets; Elsevier: Amsterdam, The Netherlands, 2013; pp. 883–907. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780123878373000432 (accessed on 2 August 2019).
- Jaffe, E.A. Cell biology of endothelial cells. Hum. Pathol. 1987, 18, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007, 100, 174–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturtzel, C. Endothelial Cells. Adv. Exp. Med. Biol. 2017, 1003, 71–91. [Google Scholar] [PubMed]
- Sabatier, F.; Camoin-Jau, L.; Anfosso, F.; Sampol, J.; Dignat-George, F. Circulating endothelial cells, microparticles and progenitors: Key players towards the definition of vascular competence. J. Cell Mol. Med. 2009, 13, 454–471. [Google Scholar] [CrossRef] [PubMed]
- Vallier, L.; Cointe, S.; Lacroix, R.; Bonifay, A.; Judicone, C.; Dignat-George, F.; Kwaan, H.C. Microparticles and Fibrinolysis. Semin. Thromb. Hemost. 2017, 43, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Rayes, J.; Frimat, M.; Fremeaux-Bacchi, V. Endothelial cells: Source, barrier, and target of defensive mediators. Immunol. Rev. 2016, 274, 307–329. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Pober, J.S.; Cotran, R.S. The role of endothelial cells in inflammation. Transplantation 1990, 50, 537–544. [Google Scholar] [CrossRef]
- Lowenstein, C.J.; Morrell, C.N.; Yamakuchi, M. Regulation of Weibel–Palade Body Exocytosis. Trends Cardiovasc. Med. 2005, 15, 302–308. [Google Scholar] [CrossRef]
- Mourik, M.; Eikenboom, J. Lifecycle of Weibel-Palade bodies. Hamostaseologie 2017, 37, 13–24. [Google Scholar] [CrossRef]
- Rondaij, M.G.; Bierings, R.; Kragt, A.; Van Mourik, J.A.; Voorberg, J. Dynamics and Plasticity of Weibel-Palade Bodies in Endothelial Cells. Arter. Thromb. Vasc. Biol. 2006, 26, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Tiruppathi, C.; Minshall, R.D.; Paria, B.C.; Vogel, S.M.; Malik, A.B. Role of Ca2+ signaling in the regulation of endothelial permeability. Vasc. Pharmacol. 2002, 39, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Ludmer, P.L.; Selwyn, A.P.; Shook, T.L.; Wayne, R.R.; Mudge, G.H.; Alexander, R.W.; Ganz, P. Paradoxical Vasoconstriction Induced by Acetylcholine in Atherosclerotic Coronary Arteries. N. Engl. J. Med. 1986, 315, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Poredos, P. Endothelial dysfunction in the pathogenesis of atherosclerosis. Int. Angiol. 2002, 21, 109–116. [Google Scholar] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [Green Version]
- Huynh, D.T.N.; Heo, K.-S. Therapeutic targets for endothelial dysfunction in vascular diseases. Arch. Pharmacal Res. 2019, 42, 848–861. [Google Scholar] [CrossRef]
- Moatti-Cohen, M.; Garrec, C.; Wolf, M.; Boisseau, P.; Galicier, L.; Azoulay, E.; Stepanian, A.; Delmas, Y.; Rondeau, E.; Bezieau, S.; et al. Unexpected frequency of Upshaw-Schulman syndrome in pregnancy-onset thrombotic thrombocytopenic purpura. Blood 2012, 119, 5888–5897. [Google Scholar] [CrossRef]
- Mariotte, E.; Azoulay, E.; Galicier, L.; Rondeau, E.; Zouiti, F.; Boisseau, P.; Poullin, P.; de Maistre, E.; Provôt, F.; Delmas, Y.; et al. Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): A cross-sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol. 2016, 3, e237–e245. [Google Scholar] [CrossRef]
- Page, E.E.; Hovinga, J.A.K.; Terrell, D.R.; Vesely, S.K.; George, J.N. Clinical importance of ADAMTS13 activity during remission in patients with acquired thrombotic thrombocytopenic purpura. Blood 2016, 128, 2175–2178. [Google Scholar] [CrossRef] [Green Version]
- George, J.N. Measuring ADAMTS13 activity in patients with suspected thrombotic thrombocytopenic purpura: When, how, and why? Transfusion 2015, 55, 11–13. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Lekakis, J.; Abraham, P.; Balbarini, A.; Blann, A.; Boulanger, C.M.; Cockcroft, J.; Cosentino, F.; Deanfield, J.; Gallino, A.; Ikonomidis, I.; et al. Methods for evaluating endothelial function: A position statement from the European Society of Cardiology Working Group on Peripheral Circulation. Eur. J. Cardiovasc. Prev. Rehabil. 2011, 18, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Horstman, L.L.; Jy, W.; Jimenez, J.J.; Ahn, Y.S. Endothelial microparticles as markers of endothelial dysfunction. Front. Biosci. 2004, 9, 1118–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Hanano, M.; Wada, K.; Tatewaki, W.; Niwano, H.; Shibata, A.; Tsubouchi, J.; Nakano, M.; Nakamura, T. Circulating thrombomodulin in thrombotic thrombocytopenic purpura. Am. J. Hematol. 1991, 38, 174–177. [Google Scholar] [CrossRef]
- Mori, Y.; Wada, H.; Okugawa, Y.; Tamaki, S.; Nakasaki, T.; Watanabe, R.; Gabazza, E.C.; Nishikawa, M.; Minami, N.; Shiku, H. Increased Plasma Thrombomodulin as a Vascular Endothelial Cell Marker in Patients with Thrombotic Thrombocytopenic Purpura and Hemolytic Uremic Syndrome. Clin. Appl. Thromb. 2001, 7, 5–9. [Google Scholar] [CrossRef]
- Chong, B.H.; Murray, B.; Berndt, M.C.; Dunlop, L.C.; Brighton, T.; Chesterman, C.N. Plasma P-selectin is increased in thrombotic consumptive platelet disorders. Blood 1994, 83, 1535–1541. [Google Scholar] [CrossRef] [Green Version]
- Katayama, M.; Handa, M.; Araki, Y.; Ambo, H.; Kawai, Y.; Watanabe, K.; Ikeda, Y. Soluble P-selectin is present in normal circulation and its plasma level is elevated in patients with thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome. Br. J. Haematol. 1993, 84, 702–710. [Google Scholar] [CrossRef]
- Wada, H.; Kaneko, T.; Ohiwa, M.; Tanigawa, M.; Hayashi, T.; Tamaki, S.; Minami, N.; Deguchi, K.; Suzuki, K.; Nakano, T.; et al. Increased levels of vascular endothelial cell markers in thrombotic thrombocytopenic purpura. Am. J. Hematol. 1993, 44, 101–105. [Google Scholar] [CrossRef]
- Widemann, A.; Pasero, C.; Arnaud, L.; Poullin, P.; Loundou, A.D.; Choukroun, G.; Sanderson, F.; Lacroix, R.; Sabatier, F.; Coppo, P.; et al. Circulating endothelial cells and progenitors as prognostic factors during autoimmune thrombotic thrombocytopenic purpura: Results of a prospective multicenter French study. J. Thromb. Haemost. 2014, 12, 1601–1609. [Google Scholar] [CrossRef]
- Kobayashi, M.; Wada, H.; Wakita, Y.; Shimura, M.; Nakase, T.; Hiyoyama, K.; Nagaya, S.; Minami, N.; Nakano, T.; Shiku, H. Decreased plasma tissue factor pathway inhibitor levels in patients with thrombotic thrombocytopenic purpura. Thromb. Haemost. 1995, 73, 10–14. [Google Scholar]
- Glas-Greenwalt, P.; Hall, J.M.; Panke, T.W.; Kant, K.S.; Allen, C.M.; Pollak, V.E. Fibrinolysis in health and disease: Abnormal levels of plasminogen activator, plasminogen activator inhibitor, and protein C in thrombotic thrombocytopenic purpura. J. Lab. Clin. Med. 1986, 108, 415–422. [Google Scholar] [PubMed]
- Asada, Y.; Sumiyoshi, A.; Hayashi, T.; Suzumiya, J.; Kaketani, K. Immunohistochemistry of vascular lesion in thrombotic thrombocytopenic purpura, with special reference to factor VIII related antigen. Thromb. Res. 1985, 38, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Tersteeg, C.; de Maat, S.; De Meyer, S.F.; Smeets, M.W.; Barendrecht, A.D.; Roest, M.; Pasterkamp, G.; Fijnheer, R.; Vanhoorelbeke, K.; de Groot, P.G.; et al. Plasmin Cleavage of von Willebrand Factor as an Emergency Bypass for ADAMTS13 Deficiency in Thrombotic Microangiopathy. Circulation 2014, 129, 1320–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, E.C.-Y. Pathogenesis of Thrombotic Thrombocytopenic Purpura: ADAMTS13 Deficiency and Beyond. Semin. Thromb. Hemost. 2005, 31, 625–632. [Google Scholar] [CrossRef] [PubMed]
- van Mourik, J.A.; Boertjes, R.; Huisveld, I.A.; Fijnvandraat, K.; Pajkrt, D.; van Genderen, P.J.; Fijnheer, R. von Willebrand factor propeptide in vascular disorders: A tool to distinguish between acute and chronic endothelial cell perturbation. Blood 1999, 94, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Béranger, N.; Benghezal, S.; Savigny, S.; Capdenat, S.; Joly, B.S.; Coppo, P.; Stepanian, A.; Veyradier, A. Loss of von Willebrand factor high-molecular-weight multimers at acute phase is associated with detectable anti-ADAMTS13 IgG and neurological symptoms in acquired thrombotic thrombocytopenic purpura. Thromb. Res. 2019, 181, 29–35. [Google Scholar] [CrossRef]
- Jimenez, J.J.; Jy, W.; Mauro, L.M.; Horstman, L.L.; Ahn, Y.S. Elevated endothelial microparticles in thrombotic thrombocytopenic purpura: Findings from brain and renal microvascular cell culture and patients with active disease. Br. J. Haematol. 2001, 112, 81–90. [Google Scholar] [CrossRef]
- Jimenez, J.J.; Jy, W.; Mauro, L.M.; Horstman, L.L.; Soderland, C.; Ahn, Y.S. Endothelial microparticles released in thrombotic thrombocytopenic purpura express von Willebrand factor and markers of endothelial activation. Br. J. Haematol. 2003, 123, 896–902. [Google Scholar] [CrossRef] [Green Version]
- Lefevre, P.; George, F.; Durand, J.M.; Sampol, J. Detection of Circulating Endothelial Cells in Thrombotic Thrombocytopenic Purpura. Thromb. Haemost. 1993, 69, 522. [Google Scholar] [CrossRef]
- Dang, C.T.; Magid, M.S.; Weksler, B.; Chadburn, A.; Laurence, J. Enhanced endothelial cell apoptosis in splenic tissues of patients with thrombotic thrombocytopenic purpura. Blood 1999, 93, 1264–1270. [Google Scholar] [CrossRef]
- Jimenez, J.J.; Jy, W.; Mauro, L.M.; Horstman, L.L.; Soderland, C.; Ahn, Y.S. Response to Laurence. Br. J. Haematol. 2004, 125, 416–417. [Google Scholar] [CrossRef]
- Burns, E.R.; Zucker-Franklin, D. Pathologic effects of plasma from patients with thrombotic thrombocytopenic purpura on platelets and cultured vascular endothelial cells. Blood 1982, 60, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurence, J.; Mitra, D.; Steiner, M.; Staiano-Coico, L.; Jaffe, E. Plasma from patients with idiopathic and human immunodeficiency Virus-Associated thrombotic thrombocytopenic purpura induces apoptosis in microvascular endothelial cells. Transfus. Med. Rev. 1996, 10, 315. [Google Scholar] [CrossRef]
- Mitra, D.; Jaffe, E.A.; Weksler, B.; Hajjar, K.A.; Soderland, C.; Laurence, J. Thrombotic thrombocytopenic purpura and sporadic hemolytic-uremic syndrome plasmas induce apoptosis in restricted lineages of human microvascular endothelial cells. Blood 1997, 89, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Jy, W.; Jimenez, J.J.; Mauro, L.M.; Horstman, L.L.; Cheng, P.; Ahn, E.R.; Bidot, C.J.; Ahn, Y.S. Endothelial microparticles induce formation of platelet aggregates via a von Willebrand factor/ristocetin dependent pathway, rendering them resistant to dissociation. J. Thromb. Haemost. 2005, 3, 1301–1308. [Google Scholar] [CrossRef]
- Laurence, J. Endothelial cell activation and apoptosis in the thrombotic microangiopathies. Br. J. Haematol. 2004, 125, 415–416. [Google Scholar] [CrossRef]
- Tellier, E.; Widemann, A.; Cauchois, R.; Faccini, J.; Lagarde, M.; Brun, M.; Kaplanski, G. Immune thrombotic thrombocytopenic purpura plasmas induce calcium- and IgG-dependent endothelial activation: Correlations with disease severity. Haematologica 2022. [Google Scholar] [CrossRef]
- Motto, D.G.; Chauhan, A.K.; Zhu, G.; Homeister, J.; Lamb, C.B.; Desch, K.C.; Zhang, W.; Tsai, H.-M.; Wagner, D.D.; Ginsburg, D. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J. Clin. Investig. 2005, 115, 2752–2761. [Google Scholar] [CrossRef] [Green Version]
- Banno, F.; Kokame, K.; Okuda, T.; Honda, S.; Miyata, S.; Kato, H.; Tomiyama, Y.; Miyata, T. Complete deficiency in ADAMTS13 is prothrombotic, but it alone is not sufficient to cause thrombotic thrombocytopenic purpura. Blood 2006, 107, 3161–3166. [Google Scholar] [CrossRef]
- Huang, J.; Motto, D.G.; Bundle, D.R.; Sadler, J.E. Shiga toxin B subunits induce VWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMTS13-deficient mice. Blood 2010, 116, 3653–3659. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.K.; Walsh, M.T.; Zhu, G.; Ginsburg, D.; Wagner, D.D.; Motto, D.G. The combined roles of ADAMTS13 and VWF in murine models of TTP, endotoxemia, and thrombosis. Blood 2008, 111, 3452–3457. [Google Scholar] [CrossRef] [PubMed]
- Feys, H.B.; Liu, F.; Dong, N.; Pareyn, I.; Vauterin, S.; Vandeputte, N.; Noppe, W.; Ruan, C.; Deckmyn, H.; Vanhoorelbeke, K. ADAMTS-13 plasma level determination uncovers antigen absence in acquired thrombotic thrombocytopenic purpura and ethnic differences. J. Thromb. Haemost. 2006, 4, 955–962. [Google Scholar] [CrossRef]
- Le Besnerais, M.; Favre, J.; Denis, C.V.; Mulder, P.; Martinet, J.; Nicol, L.; Benhamou, Y. Assessment of endothelial damage and cardiac injury in a mouse model mimicking thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2016, 14, 1917–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Abdelgawwad, M.S.; Di Zhang, L.X.; Wei, S.; Cao, W.; Zheng, X.L. Histone-induced thrombotic thrombocytopenic purpura in adamts13-/- zebrafish depends on von Willebrand factor. Haematologica 2019, 105, 1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, A.; Albánez, S.; Mewburn, J.; Nesbitt, K.; Gould, T.J.; Liaw, P.C.; James, P.D.; Swystun, L.L.; Lillicrap, D. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J. Thromb. Haemost. 2016, 14, 2274–2286. [Google Scholar] [CrossRef] [PubMed]
- Tersteeg, C.; Schiviz, A.; De Meyer, S.F.; Plaimauer, B.; Scheiflinger, F.; Rottensteiner, H.; Vanhoorelbeke, K. Potential for Recombinant ADAMTS13 as an Effective Therapy for Acquired Thrombotic Thrombocytopenic Purpura. Arter. Thromb. Vasc. Biol. 2015, 35, 2336–2342. [Google Scholar] [CrossRef] [Green Version]
- Deforche, L.; Tersteeg, C.; Roose, E.; Vandenbulcke, A.; Vandeputte, N.; Pareyn, I.; De Cock, E.; Rottensteiner, H.; Deckmyn, H.; De Meyer, S.F.; et al. Generation of Anti-Murine ADAMTS13 Antibodies and Their Application in a Mouse Model for Acquired Thrombotic Thrombocytopenic Purpura. PLoS ONE 2016, 11, e0160388. [Google Scholar] [CrossRef] [Green Version]
- Pickens, B.; Mao, Y.; Li, D.; Siegel, N.L.; Poncz, M.; Cines, U.B.; Zheng, X.L. Platelet-delivered ADAMTS13 inhibits arterial thrombosis and prevents thrombotic thrombocytopenic purpura in murine models. Blood 2015, 125, 3326–3334. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, Y.; Matsumoto, M.; Kokame, K.; Isonishi, A.; Soejima, K.; Akiyama, N.; Tomiyama, J.; Natori, K.; Kuranishi, Y.; Imamura, Y.; et al. Pregnancy-induced thrombocytopenia and TTP, and the risk of fetal death, in Upshaw-Schulman syndrome: A series of 15 pregnancies in 9 genotyped patients. Br. J. Haematol. 2009, 144, 742–754. [Google Scholar] [CrossRef]
- Falter, T.; Hovinga, J.A.K.; Lackner, K.; Füllemann, H.-G.; Lämmle, B.; Scharrer, I. Late onset and pregnancy-induced congenital thrombotic thrombocytopenic purpura. Hamostaseologie 2014, 34, 244–248. [Google Scholar] [CrossRef]
- Stirling, Y.; Woolf, L.; North, W.R.; Seghatchian, M.J.; Meade, T.W. Haemostasis in normal pregnancy. Thromb. Haemost. 1984, 52, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Furlan, M.; Robles, R.; Galbusera, M.; Remuzzi, G.; Kyrle, P.A.; Brenner, B.; Krause, M.; Scharrer, I.; Aumann, V.; Mittler, U.; et al. von Willebrand Factor–Cleaving Protease in Thrombotic Thrombocytopenic Purpura and the Hemolytic–Uremic Syndrome. N. Engl. J. Med. 1998, 339, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- George, J.N. The association of pregnancy with thrombotic thrombocytopenic purpura–hemolytic uremic syndrome. Curr. Opin. Hematol. 2003, 10, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Morioka, M.; Matsumoto, M.; Saito, M.; Kokame, K.; Miyata, T.; Fujimura, Y. A first bout of thrombotic thrombocytopenic purpura triggered by herpes simplex infection in a 45-year-old nulliparous female with Upshaw-Schulman syndrome. Blood Transfus. 2014, 12 (Suppl. 1), s153–s155. [Google Scholar] [PubMed]
- Bitzan, M.; Zieg, J. Influenza-associated thrombotic microangiopathies. Pediatr. Nephrol. 2018, 33, 2009–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgand, M.; Buffet, M.; Busson, M.; Loiseau, P.; Malot, S.; Amokrane, K.; Fortier, C.; London, J.; Bonmarchand, G.; Wynckel, A.; et al. High prevalence of infectious events in thrombotic thrombocytopenic purpura and genetic relationship with toll-like receptor 9 polymorphisms: Experience of the French Thrombotic Microangiopathies Reference Center. Transfusion 2014, 54, 389–397. [Google Scholar] [CrossRef]
- Popović, M.; Smiljanić, K.; Dobutović, B.; Syrovets, T.; Simmet, T.; Isenović, E.R. Human cytomegalovirus infection and atherothrombosis. J. Thromb. Thrombolysis 2012, 33, 160–172. [Google Scholar] [CrossRef]
- Bernardo, A.; Ball, C.; Nolasco, L.; Moake, J.F.; Dong, J.-F. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell–derived ultralarge von Willebrand factor multimers under flow. Blood 2004, 104, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, D.; McGlasson, S.; Jury, A.; Williams, J.; Scolding, N.; Bellamy, C.; Gunther, C.; Ritchie, D.; Gale, D.; Kanwar, Y.S.; et al. Type I interferon causes thrombotic microangiopathy by a dose-dependent toxic effect on the microvasculature. Blood 2016, 128, 2824–2833. [Google Scholar] [CrossRef]
- Cao, W.J.; Niiya, M.; Zheng, X.W.; Shang, D.Z. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J. Thromb. Haemost. 2008, 6, 1233–1235. [Google Scholar] [CrossRef] [Green Version]
- Malak, S.; Wolf, M.; Millot, G.A.; Mariotte, E.; Veyradier, A.; Meynard, J.-L.; Korach, J.-M.; Malot, S.; Bussel, A.; Azoulay, E.; et al. Human Immunodeficiency Virus-Associated Thrombotic Microangiopathies: Clinical Characteristics and Outcome According to ADAMTS13 Activity. Scand. J. Immunol. 2008, 68, 337–344. [Google Scholar] [CrossRef] [PubMed]
- del Arco, A.; Martinez, M.A.; Peña, J.M.; Gamallo, C.; González, J.J.; Barbado, F.J.; Vazquez, J.J. Thrombotic thrombocytopenic purpura associated with human immunodeficiency virus infection: Demonstration of p24 antigen in endothelial cells. Clin. Infect. Dis. 1993, 17, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, H.A.; Darnahal, M.; Vaezi, M.; Haghighi, S. COVID-19 associated thrombotic thrombocytopenic purpura (TTP); A case series and mini-review. Int. Immunopharmacol. 2021, 93, 107397. [Google Scholar] [CrossRef]
- Ward, S.E.; Curley, G.F.; Lavin, M.; Fogarty, H.; Karampini, E.; McEvoy, N.L.; Clarke, J.; Boylan, M.; Alalqam, R.; Worrall, A.P.; et al. Von Willebrand factor propeptide in severe coronavirus disease 2019 (COVID-19): Evidence of acute and sustained endothelial cell activation. Br. J. Haematol. 2021, 192, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Enjyoji, K.; Schmaier, A.A. Vasculopathy in COVID-19. Blood 2022, 140, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Shyy, J.Y. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- McCracken, I.R.; Saginc, G.; He, L.; Huseynov, A.; Daniels, A.; Fletcher, S.; Randi, A.M. Lack of Evidence of Angiotensin-Converting Enzyme 2 Expression and Replicative Infection by SARS-CoV-2 in Human Endothelial Cells. Circulation 2021, 143, 865–868. [Google Scholar] [CrossRef]
- Zakarija, A.; Kwaan, H.C.; Moake, J.L.; Bandarenko, N.; Pandey, D.K.; McKoy, J.M.; Yarnold, P.R.; Raisch, D.W.; Winters, J.L.; Raife, T.J.; et al. Ticlopidine- and clopidogrel-associated thrombotic thrombocytopenic purpura (TTP): Review of clinical, laboratory, epidemiological, and pharmacovigilance findings (1989–2008). Kidney Int. 2009, 75, S20–S24. [Google Scholar] [CrossRef] [Green Version]
- Mauro, M.; Zlatopolskiy, A.; Raife, T.J.; Laurence, J. Thienopyridine-linked thrombotic microangiopathy: Association with endothelial cell apoptosis and activation of MAP kinase signalling cascades. Br. J. Haematol. 2004, 124, 200–210. [Google Scholar] [CrossRef]
- Coppo, P.; Busson, M.; Veyradier, A.; Wynckel, A.; Poullin, P.; Azoulay, E.; Galicier, L.; Loiseau, P. HLA-DRB1*11: A strong risk factor for acquired severe ADAMTS13 deficiency-related idiopathic thrombotic thrombocytopenic purpura in Caucasians. J. Thromb. Haemost. 2010, 8, 856–859. [Google Scholar] [CrossRef]
- Scully, M.; Brown, J.; Patel, R.; Mcdonald, V.; Brown, C.J.; Machin, S. Human leukocyte antigen association in idiopathic thrombotic thrombocytopenic purpura: Evidence for an immunogenetic link. J. Thromb. Haemost. 2010, 8, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Sorvillo, N.; van Haren, S.; Kaijen, P.H.; Brinke, A.T.; Fijnheer, R.; Meijer, A.B.; Voorberg, J. Preferential HLA-DRB1*11–dependent presentation of CUB2-derived peptides by ADAMTS13-pulsed dendritic cells. Blood 2013, 121, 3502–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbij, F.C.; Turksma, A.W.; de Heij, F.; Kaijen, P.; Lardy, N.; Fijnheer, R.; Sorvillo, N.; Brinke, A.T.; Voorberg, J. CD4+ T cells from patients with acquired thrombotic thrombocytopenic purpura recognize CUB2 domain-derived peptides. Blood 2016, 127, 1606–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praprotnik, S.; Blank, M.; Levy, Y.; Tavor, S.; Boffa, M.-C.; Weksler, B.; Eldor, A.; Shoenfeld, Y. Anti-endothelial cell antibodies from patients with thrombotic thrombocytopenic purpura specifically activate small vessel endothelial cells. Int. Immunol. 2001, 13, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.F.; Wang, H.; Hornstein, A.; Hogarth, M.; Mody, M.; Garvey, M.B.; Blanchette, V.; Rock, G.; Freedman, J. Characterization of platelet glycoproteins and platelet/endothelial cell antibodies in patients with thrombotic thrombocytopenic purpura. Br. J. Haematol. 1999, 107, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.N.; Rock, G.; Jamieson, G.A. Anti-CD36 antibodies in thrombotic thrombocytopenic purpura. Br. J. Haematol. 1994, 88, 816–825. [Google Scholar] [CrossRef]
- Raife, T.J.; Atkinson, B.; Aster, R.H.; McFarland, J.G.; Gottschall, J.L. Minimal evidence of platelet and endothelial cell reactive antibodies in thrombotic thrombocytopenic purpura. Am. J. Hematol. 1999, 62, 82–87. [Google Scholar] [CrossRef]
- Drachenberg, C.B.; Papadimitriou, J.C. Endothelial injury in renal antibody-mediated allograft rejection: A schematic view based on pathogenesis. Transplantation 2013, 95, 1073–1083. [Google Scholar] [CrossRef]
- Rena, G.; Hack, B.K.; Minto, A.W.; Cunningham, P.N.; Alexander, J.J.; Haasb, M.; Quigg, R.J. A Complement-Dependent Model of Thrombotic Thrombocytopenic Purpura Induced by Antibodies Reactive with Endothelial Cells. Clin. Immunol. 2002, 103, 43–53. [Google Scholar] [CrossRef]
- Chang, J.C.; Shipstone, A.; Llenado-Lee, M.A. Postoperative thrombotic thrombocytopenic purpura following cardiovascular surgeries. Am. J. Hematol. 1996, 53, 11–17. [Google Scholar] [CrossRef]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Namal Rathnayaka, R.; Ranathunga, P.A.N.; Kularatne, S.A. Thrombotic Microangiopathy, Hemolytic Uremic Syndrome, and Thrombotic Thrombocytopenic Purpura Following Hump-nosed Pit Viper (Genus: Hypnale) Envenoming in Sri Lanka. Wilderness Environ. Med. 2019, 30, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.H.; Tan, N.H.; Sim, S.M.; Fung, S.Y.; Gnanathasan, C.A. Proteomic investigation of Sri Lankan hump-nosed pit viper (Hypnale hypnale) venom. Toxicon 2015, 93, 164–170. [Google Scholar] [CrossRef]
- Vanuopadath, M.; Sajeev, N.; Murali, A.R.; Sudish, N.; Kangosseri, N.; Sebastian, I.R.; Jain, N.D.; Pal, A.; Raveendran, D.; Nair, B.G.; et al. Mass spectrometry-assisted venom profiling of Hypnale hypnale found in the Western Ghats of India incorporating de novo sequencing approaches. Int. J. Biol. Macromol. 2018, 118, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhouri, F.; Zuber, J.; Frémeaux-Bacchi, V.; Loirat, C. Haemolytic uraemic syndrome. Lancet 2017, 390, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Orth, D.; Khan, A.B.; Naim, A.; Grif, K.; Brockmeyer, J.; Karch, H.; Joannidis, M.; Clark, S.J.; Day, A.J.; Fidanzi, S.; et al. Shiga Toxin Activates Complement and Binds Factor H: Evidence for an Active Role of Complement in Hemolytic Uremic Syndrome. J. Immunol. 2009, 182, 6394–6400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, T.M.; Pruette, C.; Loeffler, L.F.; Weidemann, D.; Strouse, J.J.; Gavriilaki, E.; Brodsky, R.A. Typical Hus: Evidence of Acute Phase Complement Activation from a Daycare Outbreak. J. Clin. Exp. Nephrol. 2016, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Burwick, R.M.; Fichorova, R.N.; Dawood, H.Y.; Yamamoto, H.S.; Feinberg, B.B. Urinary excretion of C5b-9 in severe preeclampsia: Tipping the balance of complement activation in pregnancy. Hypertension 2013, 62, 1040–1045. [Google Scholar] [CrossRef] [Green Version]
- Jodele, S.; Licht, C.; Goebel, J.; Dixon, B.; Zhang, K.; Sivakumaran, T.A.; Davies, S.M.; Pluthero, F.; Lu, L.; Laskin, B.L. Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood 2013, 122, 2003–2007. [Google Scholar] [CrossRef]
- Jodele, S.; Zhang, K.; Zou, F.; Laskin, B.; Dandoy, C.; Myers, K.C.; Lane, A.; Meller, J.; Medvedovic, M.; Chen, J.; et al. The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood 2016, 127, 989–996. [Google Scholar] [CrossRef] [PubMed]
- de Fontbrune, F.S.; Galambrun, C.; Sirvent, A.; Huynh, A.; Faguer, S.; Nguyen, S.; de Latour, R.P. Use of Eculizumab in Patients with Allogeneic Stem Cell Transplant-Associated Thrombotic Microangiopathy: A Study From the SFGM-TC. Transplantation 2015, 99, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Westwood, J.-P.; Langley, K.; Heelas, E.; Machin, S.J.; Scully, M. Complement and cytokine response in acute Thrombotic Thrombocytopenic Purpura. Br. J. Haematol. 2014, 164, 858–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettoni, S.; Galbusera, M.; Gastoldi, S.; Donadelli, R.; Tentori, C.; Spartà, G.; Bresin, E.; Mele, C.; Alberti, M.; Tortajada, A.; et al. Interaction between Multimeric von Willebrand Factor and Complement: A Fresh Look to the Pathophysiology of Microvascular Thrombosis. J. Immunol. 2017, 199, 1021–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.C.; Yang, S.; Haven, S.; Holers, V.M.; Lundberg, A.S.; Wu, H.; Cataland, S.R. Complement activation and mortality during an acute episode of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2013, 11, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Cataland, S.R.; Holers, V.M.; Geyer, S.; Yang, S.; Wu, H.M. Biomarkers of terminal complement activation confirm the diagnosis of aHUS and differentiate aHUS from TTP. Blood 2014, 123, 3733–3738. [Google Scholar] [CrossRef] [Green Version]
- Tati, R.; Kristoffersson, A.-C.; Ståhl, A.-L.; Rebetz, J.; Wang, L.; Licht, C.; Motto, D.; Karpman, D. Complement Activation Associated with ADAMTS13 Deficiency in Human and Murine Thrombotic Microangiopathy. J. Immunol. 2013, 191, 2184–2193. [Google Scholar] [CrossRef] [Green Version]
- Sinkovits, G.; Farkas, P.; Csuka, D.; Rázsó, K.; Réti, M.; Radványi, G.; Demeter, J.; Prohászka, Z.; Mikes, B. Carboxiterminal pro-endothelin-1 as an endothelial cell biomarker in thrombotic thrombocytopenic purpura. Thromb. Haemost. 2016, 115, 1034–1043. [Google Scholar] [CrossRef]
- Turner, N.; Sartain, S.; Moake, J. Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes. Hematol. Clin. North Am. 2015, 29, 509–524. [Google Scholar] [CrossRef]
- Feng, S.; Liang, X.; Kroll, M.H.; Chung, D.W.; Afshar-Kharghan, V. von Willebrand factor is a cofactor in complement regulation. Blood 2015, 125, 1034–1037. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Zhang, D.; Cao, W.; Song, W.-C.; Zheng, X.L. Synergistic effects of ADAMTS13 deficiency and complement activation in pathogenesis of thrombotic microangiopathy. Blood 2019, 134, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Lorant, D.E.; Topham, M.K.; Whatley, R.E.; McEver, R.P.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. Inflammatory roles of P-selectin. J. Clin. Investig. 1993, 92, 559–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative Pathway Activation of Complement by Shiga Toxin Promotes Exuberant C3a Formation That Triggers Microvascular Thrombosis. J. Immunol. 2011, 187, 172–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foreman, K.E.; Vaporciyan, A.A.; Bonish, B.K.; Jones, M.L.; Johnson, K.J.; Glovsky, M.M.; Eddy, S.M.; A Ward, P. C5a-induced expression of P-selectin in endothelial cells. J. Clin. Investig. 1994, 94, 1147–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedesco, F.; Pausa, M.; Nardon, E.; Introna, M.; Mantovani, A.; Dobrina, A. The Cytolytically Inactive Terminal Complement Complex Activates Endothelial Cells to Express Adhesion Molecules and Tissue Factor Procoagulant Activity. J. Exp. Med. 1997, 185, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Torres, M.P.; Casiraghi, F.; Galbusera, M.; Macconi, D.; Gastoldi, S.; Todeschini, M.; Porrati, F.; Belotti, D.; Pogliani, E.M.; Remuzzi, G.; et al. Complement activation: The missing link between ADAMTS-13 deficiency and microvascular thrombosis of thrombotic microangiopathies. Thromb. Haemost. 2005, 93, 443–452. [Google Scholar] [CrossRef] [Green Version]
- del Conde, I.; Crúz, M.A.; Zhang, H.; López, J.A.; Afshar-Kharghan, V. Platelet activation leads to activation and propagation of the complement system. J. Exp. Med. 2005, 201, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Chapin, J.; Weksler, B.; Magro, C.; Laurence, J. Eculizumab in the treatment of refractory idiopathic thrombotic thrombocytopenic purpura. Br. J. Haematol. 2012, 157, 772–774. [Google Scholar] [CrossRef]
- Tsai, E.; Chapin, J.; Laurence, J.C.; Tsai, H.-M. Use of eculizumab in the treatment of a case of refractory, ADAMTS13-deficient thrombotic thrombocytopenic purpura: Additional data and clinical follow-up. Br. J. Haematol. 2013, 162, 558–559. [Google Scholar] [CrossRef]
- Pecoraro, C.; Ferretti, A.V.S.; Rurali, E.; Galbusera, M.; Noris, M.; Remuzzi, G. Treatment of Congenital Thrombotic Thrombocytopenic Purpura with Eculizumab. Am. J. Kidney Dis. 2015, 66, 1067–1070. [Google Scholar] [CrossRef]
- Vigna, E.; Petrungaro, A.; Perri, A.; Terzi, D.; Recchia, A.G.; Mendicino, F.; La Russa, A.; Bossio, S.; De Stefano, L.; Zinno, F.; et al. Efficacy of eculizumab in severe ADAMTS13-deficient thrombotic thrombocytopenic purpura (TTP) refractory to standard therapies. Transfus. Apher. Sci. 2018, 57, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: A novel mechanism of human disease. JAMA 2005, 293, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin Therapy Improves Cardiovascular Function by Preventing Heme-Induced Endothelial Toxicity in Mouse Models of Hemolytic Diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, K.; Morrell, C.N.; Cambien, B.; Yang, S.-X.; Yamakuchi, M.; Bao, C.; Hara, M.R.; Quick, R.A.; Cao, W.; O’Rourke, B.; et al. Nitric Oxide Regulates Exocytosis by S-Nitrosylation of N-ethylmaleimide-Sensitive Factor. Cell 2003, 115, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studt, J.-D.; Hovinga, J.A.K.; Antoine, G.; Hermann, M.; Rieger, M.; Scheiflinger, F.; Lämmle, B. Fatal congenital thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibitor: In vitro inhibition of ADAMTS13 activity by hemoglobin. Blood 2005, 105, 542–544. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Paule, R.; Leon, J.; Daugan, M.; Robe-Rybkine, T.; Poillerat, V.; Roumenina, L.T. P-selectin drives complement attack on endothelium during intravascular hemolysis in TLR-4/heme-dependent manner. Proc. Natl. Acad. Sci. USA 2019, 116, 6280–6285. [Google Scholar] [CrossRef] [Green Version]
- May, O.; Merle, N.S.; Grunenwald, A.; Gnemmi, V.; Leon, J.; Payet, C.; Robe-Rybkine, T.; Paule, R.; Delguste, F.; Satchell, S.C.; et al. Heme Drives Susceptibility of Glomerular Endothelium to Complement Overactivation Due to Inefficient Upregulation of Heme Oxygenase-1. Front. Immunol. 2018, 9, 3008. [Google Scholar] [CrossRef]
- Frimat, M.; Tabarin, F.; Dimitrov, J.; Poitou, C.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013, 122, 282–292. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones induce rapid and profound thrombocytopenia in mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Hovinga, J.A.K.; Schatzberg, D.; Wagner, D.D.; Lämmle, B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood 2012, 120, 1157–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, M.V.; Mejias, M.P.; Sabbione, F.; Fernandez-Brando, R.J.; Santiago, A.P.; Amaral, M.M.; Exeni, R.; Trevani, A.S.; Palermo, M.S. Induction of Neutrophil Extracellular Traps in Shiga Toxin-Associated Hemolytic Uremic Syndrome. J. Innate Immun. 2016, 8, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Gloude, N.J.; Khandelwal, P.; Luebbering, N.; Lounder, D.T.; Jodele, S.; Alder, M.N.; Lane, A.; Wilkey, A.; Lake, K.E.; Litts, B.; et al. Circulating dsDNA, endothelial injury, and complement activation in thrombotic microangiopathy and GVHD. Blood 2017, 130, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Yuen, J.; Pluthero, F.G.; Douda, D.N.; Riedl, M.; Cherry, A.; Ulanova, M.; Kahr, W.H.A.; Palaniyar, N.; Licht, C. NETosing Neutrophils Activate Complement Both on Their Own NETs and Bacteria via Alternative and Non-alternative Pathways. Front. Immunol. 2016, 7, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, T.; Fan, X. A second hit for TMA. Blood 2012, 120, 1152–1154. [Google Scholar] [CrossRef] [Green Version]
- Padilla, A.; Moake, J.L.; Bernardo, A.; Ball, C.; Wang, Y.; Arya, M.; Nolasco, L.; Turner, N.; Berndt, M.C.; Anvari, B.; et al. P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood 2004, 103, 2150–2156. [Google Scholar] [CrossRef] [Green Version]
- El-Mansi, S.; Nightingale, T.D. Emerging mechanisms to modulate VWF release from endothelial cells. Int. J. Biochem. Cell Biol. 2021, 131, 105900. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cauchois, R.; Muller, R.; Lagarde, M.; Dignat-George, F.; Tellier, E.; Kaplanski, G. Is Endothelial Activation a Critical Event in Thrombotic Thrombocytopenic Purpura? J. Clin. Med. 2023, 12, 758. https://doi.org/10.3390/jcm12030758
Cauchois R, Muller R, Lagarde M, Dignat-George F, Tellier E, Kaplanski G. Is Endothelial Activation a Critical Event in Thrombotic Thrombocytopenic Purpura? Journal of Clinical Medicine. 2023; 12(3):758. https://doi.org/10.3390/jcm12030758
Chicago/Turabian StyleCauchois, Raphael, Romain Muller, Marie Lagarde, Françoise Dignat-George, Edwige Tellier, and Gilles Kaplanski. 2023. "Is Endothelial Activation a Critical Event in Thrombotic Thrombocytopenic Purpura?" Journal of Clinical Medicine 12, no. 3: 758. https://doi.org/10.3390/jcm12030758
APA StyleCauchois, R., Muller, R., Lagarde, M., Dignat-George, F., Tellier, E., & Kaplanski, G. (2023). Is Endothelial Activation a Critical Event in Thrombotic Thrombocytopenic Purpura? Journal of Clinical Medicine, 12(3), 758. https://doi.org/10.3390/jcm12030758