

Cancer Metabolism: Fasting Reset, the Keto-Paradox and Drugs for Undoing


{kind=link}
Abstract
:1. Introduction
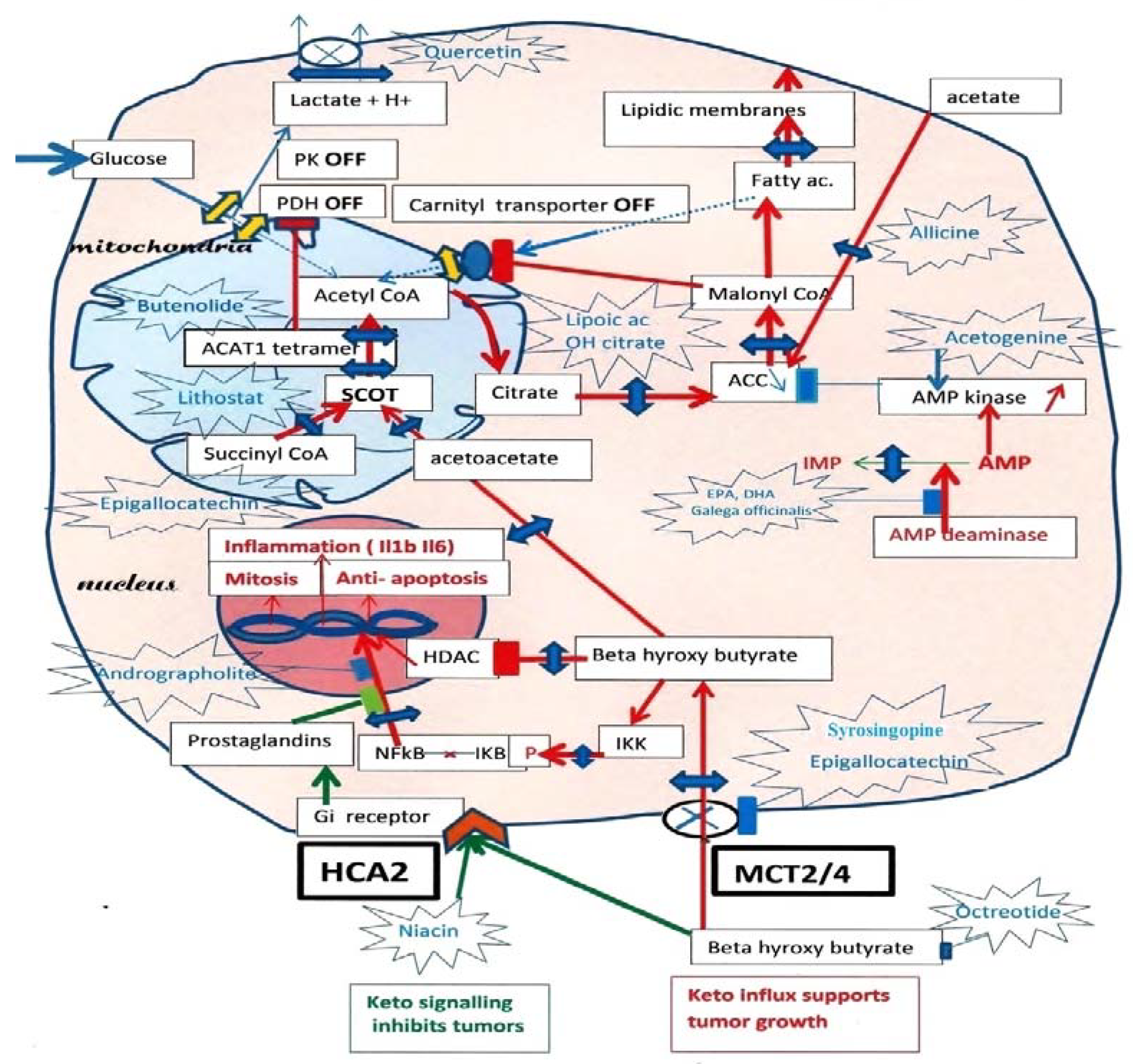
2. The Metabolic Vulnerability of Tumor Cells
2.1. Tumor Metabolism
2.2. An Essential Observation Illustrates the Ketolytic Dependency of Tumor Cells
2.3. What about Ketone Diets?
3. Metabolic Features of Ketolytic-Dependent Tumors
3.1. The Keto Diet: Is It Appropriate?
3.2. Why Is Pyruvate Kinase Phosphorylated?
3.3. How Do We Switch Back to Glycolysis?
3.3.1. Selection of the Acetyl-CoA Source
3.3.2. Role of AMP Deaminase
3.4. A Metabolic Hypothesis for How Cancer Forms?
4. How to Undo the Metabolic Advantage of Tumors
4.1. A Possible Reset of the Metabolic Rewiring Mechanism in Cancer
4.2. Cutting the Ketone Influx
4.3. Cutting the SCOT-ACAT1 Ketolytic Steps
4.4. Possible Compounds for Inhibiting SCOT
4.5. Compounds Affecting ACAT1
4.6. Compounds Acting down Stream of SCOT-ACAT1
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warburg, O. On the origin of cancer cell. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–370. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.; Eigenbrodt, E. The tumor metabolome. Anticancer Res. 2003, 23, 1149–1154. [Google Scholar] [PubMed]
- Eigenbrodt, E.; Gerbracht, U.; Mazurek, S.; Presek, P.; Friis, R. Carbohydrate metabolism and neoplasia: New Perspectives for diagnosis and therapy. In Biochemical and Molecular Aspects of Selected Cancers; Prestlow, T.G., Prestlow, T.P., Eds.; Academic Press: Cambridge, MA, USA, 1994; Volume 2, pp. 311–385. [Google Scholar]
- Schlichtholz, B.; Turyn, J.; Goyke, E.; Biernacki, M.; Jaskiewicz, K.; Sledzinski, Z.; Swierczynski, J. Enhanced citrate synthase activity in human pancreatic cancer. Pancreas 2005, 30, 99–104. [Google Scholar] [CrossRef]
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Invest. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [Green Version]
- Israël, M.; Schwartz, L. Tumor Cells are Vitally Dependent upon Ketolysis, Inhibition of Succinyl CoA:3- Oxoacid- CoA Transferase Should Block Them. OAJBS 2020, 2, 220–226. [Google Scholar] [CrossRef]
- Fan, J.; Lin, R.; Chen, D.; Xia, S.; Elf, S.E.; Liu, S.; Pan, Y.; Pan, Y.; Xu, H.; Qian, Z.; et al. Tetrameric Acetyl-CoA Acetyltransferase 1 is Important for tumor growth. Mol. Cell 2016, 64, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Israël, M. Genetic adaptation controlled by methylations and acetylation’s at the nuclear and cytosolic Levels: A hypothetical model. Neurochem. Res. 2003, 25, 631–635. [Google Scholar] [CrossRef]
- Halestrap, A.P. The Monocarboxylate Transporter Family—Structure and Functional Characterization. IUMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef]
- Schwartz, L.; Abolhassani, M.; Guais, A.; Sanders, E.; Steyaert, J.M.; Campion, F.; Israël, M. A combination of alpha lipoic acid and calcium hydroxycitrate is efficient against mouse cancer models: Preliminary results. Oncol. Rep. 2010, 23, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, L.; Guais, A.; Israël, M.; Junod, B.; Steyaert, J.M.; Crespi, E.; Baronzio, G.; Abolhassani, M. Tumor regression with a combination of drugs interfering with tumor metabolism: Efficacy of hydroxycitrate, lipoic acid and capsaicin. Investig. New Drugs 2013, 31, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Focke, M.; Feld, A.; Lichtenthaler, K. Allicin a naturally occurring antibiotic from garlic, specifically Inhibits acetyl-CoA Synthetase. FEBS 1990, 261, 106–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, B.A.; Richardson, T.; Amundsen, C.H. Inhibition of Cholesterol biosynthesis and Acetyl-CoA synthetase by bovine Milk and Orotic acid. J. Dairy Sci. 1977, 60, 1846–1853. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Zhang, H.; He, L.; Liu, C.; Xu, Y.; Qian, P.-Y. Butenolide Inhibits Marine Fouling by altering the primary metabolism of three target organisms. ACS Chem. Biol. 2012, 7, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-L.; Zhang, G.; Sun, J.; Xu, Y.; Han, Z.; Liu, L.-L. Cochliomycin A inhibits the larval settlement of Amphibalanus amphitrite by activating the NO/cGMP pathway. Biofouling 2016, 32, 35–44. [Google Scholar] [CrossRef]
- Venturelli, S.; Belz, R.G.; Kämper, A.; Berger, A.; von Horn, K.; Wegner, A.; Böcker, A.; Zabulon, G.; Langenecker, T.; Kohlbacher, O.; et al. Plants release Precursors of Histone Deacetylase inhibitors to suppress growth of competitors. Plant Cell 2015, 27, 3175–3189. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.C.; Riseborough, J.A.; Flematti, G.R.; Stevens, J.; Ghisalberti, E.L.; Dixon, K.W.; Smith, S.M. Karrikins discovered in smoke trigger Arabidopsis seed germination by a mechanism requiring GibberellicAcid synthesis and light. Plant Physiol. 2009, 149, 863–873. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, M.; Miyajima, E.; Tsushima, K. Kinetics of the interactions of AMP Deaminase with Fatty acids. J. Biol. Chem. 1979, 254, 1521–1525. [Google Scholar] [CrossRef]
- Kashan, A. Biological roles and therapeutic potential of hydroxyl-carboxilic acid receptors. Front. Endocrinol. 2011, 2, 1–12. [Google Scholar]
- Okumura, S.; Konishi, Y.; Narukawa, M.; Sugiura, Y.; Yoshimoto, S.; Arai, Y.; Sato, S.; Yoshida, Y.; Tsuji, S.; Uemura, K.; et al. Gut bacteria Identified in colorectal cancer patients promote tumorigenesis via butyrate secretion. Nat. Commun. 2021, 12, 5674. [Google Scholar] [CrossRef]
- Dimitrieva-Posocco, O.; Wong, A.C.; Lundgren, P.; Golos, A.M.; Descamps, H.C.; Cramer, Z.; Tian, Y.; Yueh, B.; Eskiocak, O.; Egervari, G.; et al. Beta-Hydroxybutyrate suppresses colorectal cancer. Nat. Commun. 2022, 605, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menez, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [PubMed]
- Poff, A.M.; Ari, C.; Arnold, P.; Seyfried, T.N.; D’Agostino, D.P. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. J. Cancer 2014, 135, 1711–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor–stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [Green Version]
- Klement, R.J. Wilhelm Brünings’ forgotten contribution to the metabolic treatment of cancer utilizing hypoglycemia and very low carbohydrate (ketogenic) diet. J. Tradit. Complement. Med. 2019, 9, 192–200. [Google Scholar] [CrossRef]
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; Wernstedt Asterholm, I.; Benrick, A.; Briant, L.J.B.; Chibalina, M.V.; et al. Insulin inhibits glucagon release by SGTL2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Israël, M.; Schwartz, L. Inhibition of the ketolytic acetyl CoA supply to tumors could be their “Achilles heel”. Int. J. Cancer 2020, 147, 1755–1757. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism:cancer’s Achilles’ heel. Cancer Cell. 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Kobayashi, S.; Shimizu, M. Tea Polyphenols inhibit the Transport of Dietary Phenolic Acids Mediated by the Monocarboxylic Acid Transporter (MCT) in Intestinal Caco-2 Cell Monolayers). J. Agric. Food Chem. 2003, 51, 7296–7302. [Google Scholar] [CrossRef] [PubMed]
- Pournourmohammadi, S.; Grimaldi, M.; Stridh, M.H.; Lavallard, V.; Waagepetersen, H.S.; Wollheim, C.B.; Maechler, P. Epigallocatechin-3–gallate (EGC) activates AMPK through the inhibition of glutamatedehydrogenase in muscle and pancreatic Bcells: A potential beneficial effect in the pre-diabetic state. Int. J. Biochem. Cell Biol. 2017, 88, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.K.; Chang, P.H.; Kuo, W.H.; Chen, C.L.; Jeng, Y.M.; Chang, K.G.; Shew, J.-Y.; Hu, C.-M.; Lee, W.-H. Adipocytes promote Malignant growth of breast tumors with monocarboxylate transporter 2 expression via Beta-Hydroxybutyrate. Nat. Commun. 2017, 8, 14706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.-M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual inhibition of Lactate transporters MCT1and MCT4 is synthetic lethal with metformin, due to NAD+Depletion in cancer cells. Cell Rep. 2018, 25, 3047–3058. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.H.; Yen, T.L.; Hsu, C.Y.; Thomas, P.A.; Sheu, J.R.; Jayakumar, T. Multi-targeting Andrographolide, a novel NFkB Inhibitor, as a potential therapeutic agent for stroke. Int. J. Mol. Sci. 2017, 18, 1638. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, R.; Berg, E.; Tenenbaum, G.; Israël, M. Inhibition of SCOT and Ketolysis decreases tumorGrowth and inflammation in the Lewis cancer model. JJ. Oncol. Clin. Res. 2022, 3, 1–12. [Google Scholar]
- Pickart, C.M.; Jencks, W.P. Formation of stable anhydrides from CoA transferase and hydroxamic acids. J. Biol. Chem. 1979, 254, 9120–9129. [Google Scholar] [CrossRef]
- Gonçalves, J.M.; Barcellos Silva, C.M.; Rivero, E.R.C.; Cordeiro, M.M.R. Inhibition of Cancer stem cells promoted by Pimozide. Clin. Exp. Pharmacol. Physiol. 2019, 46, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Al Batran, R.; Gopal, K.; Capozzi, M.E.; Chahade, J.J.; Saleme, B.; Tabatabaei-Dakhili, S.A.; Greenwell, A.A.; Niu, J.; Almutairi, M.; Byrne, N.J.; et al. Pimozide Alleviates Hyperglycemia in Diet- Induced Obesity by inhibiting Skeletal Muscle Ketoneoxidation. Cell Metab. 2020, 31, 909–919. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Ketone body utilization drives tumor growth and metastasis. Cell Cycle 2012, 11, 3964–3971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, D.; Saha, S. Hydroxamic acid–A novel molecule for anticancer therapy. J. Adv. Pharm. Technol. Res. 2012, 3, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Rothemberg, M.L.; Nelson, R.; Hande, K.R. New drugs on the horizon: Matrix MetalloproteinaseInhibitors. Stem. Cells 1999, 17, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.R.; Bogdan, A.R.; Miyazawa, M.; Hashimoto, K. Siderophores in iron metabolism: From Mechanism to therapy. Trends Mol. Med. 2016, 22, 1077–1090. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.N.; Bergin, I.; Naik, M.; Hampton, A.; Allen, R.; Kunkel, S.L.; Rush, H.; Varani, J. A multi-mineral natural product inhibits liver tumor formation in C57BL mice. Biol. Trace Elem. Res. 2012, 147, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Konishi, H.; Fujiya, M.; Tanaka, H.; Ueno, N.; Moriichi, K.; Ikuta, K.; Akutsu, H.; Tanabe, H.; Kohgo, Y. Probiotic-derived ferrichrome Inhibits colon cancer progression via JNK-mediated apoptosis. Nat. Commun. 2016, 7, 12365. [Google Scholar] [CrossRef]
- Blatt, J.; Stitely, S. Antineuroblastoma activity of desferoxamine in human cell lines. Cancer Res. 1987, 47, 1749–1750. [Google Scholar]
- Reid, R.T.; Live, D.H.; Faulkner, D.J.; Butler, A. A siderophore from a marine bacterium with an Exceptional ferric ion affinity constant. Nature 1993, 336, 455–458. [Google Scholar] [CrossRef]
- Muri, E.M.F.; Nieto, M.J.; Sindelarand, R.D.; Williamson, J.S. Hydroxamic Acids as Pharmacological Agents. Curr. Med. Chem. 2002, 9, 1631–1653. [Google Scholar] [CrossRef]
- Williamson, D.H.; Bates, M.W.; Page, M.A.; Krebs, H.A. Activities of enzymes involved in acetoacetate utilization in adult mammalian tissues. Biochem. J. 1971, 121, 41–47. [Google Scholar] [CrossRef]
- Mi, L.; Zhang, Y.; Su, A.; Tang, M.; Xing, Z.; He, T.; Wu, W.; Li, Z. Halofuginone for cancer treatment: A Systematic review of efficacy and molecular mechanisms. J. Funct. Foods 2022, 98, 105237. [Google Scholar] [CrossRef]
- Lee, I.-K.; Han, M.-S.; Lee, M.-S.; Kinm, Y.-S.; Yun, B.-S. Styrylpyrones from the medicinal fungus Phellinusbaumii and their antioxidant properties. Bioorg. Med. Chem. Lett. 2010, 20, 5459–5461. [Google Scholar] [CrossRef] [PubMed]
- Eliaguov, J.; Gopal, N.; Dixon, A.; Choudhury, M.; Konno, S. Potentiation of Anticancer Effect by Combination of Mushroom extract and Grapeseed extract on human Bladder cancer cells. Cancer Sci. Res. 2021, 4, 1–12. [Google Scholar] [CrossRef]
- Fiorillo, M.; Peiris-Pagès, M.; Sanchez-Alvarez, R.; Bartella, L.; Di Donna, L.; Dolce, V.; Sindona, G.; Sotgia, F.; Cappello, A.R.; Lissanti, M.P. Bergamot natural products eradicate cancer stem cells (CSCs) by targeting mevalonate, Rho-GDI-Signaling and mitochondria metabolism. BBA-Bioenerg. 2018, 1859, 984–996. [Google Scholar] [CrossRef]
- Liu, R.; Liu, P.; Bi, H.; Ling, J.; Zhang, H.; Zhang, M.; Hu, Y.; Chiao, P.J.; Huang, P.; Liu, J. Malignant transformation by oncogenic K-ras requires IDH2-mediated reductive carboxylation to promote glutamine utilization. Cancer Commun. 2022. Epub ahead of print. [Google Scholar] [CrossRef]
- Meneses da Silva, E.L.; Roblot, F.; Mahuteau, J.; Cavé, A. Coriadienin, the first Annonaceous Acetogenin with two double bonds isolated from Annona coriaceae. J. Nat. Prod. 1996, 59, 528–530. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Israël, M.; Berg, E.; Tenenbaum, G. Cancer Metabolism: Fasting Reset, the Keto-Paradox and Drugs for Undoing. J. Clin. Med. 2023, 12, 1589. https://doi.org/10.3390/jcm12041589
Israël M, Berg E, Tenenbaum G. Cancer Metabolism: Fasting Reset, the Keto-Paradox and Drugs for Undoing. Journal of Clinical Medicine. 2023; 12(4):1589. https://doi.org/10.3390/jcm12041589
Chicago/Turabian StyleIsraël, Maurice, Eric Berg, and Guy Tenenbaum. 2023. "Cancer Metabolism: Fasting Reset, the Keto-Paradox and Drugs for Undoing" Journal of Clinical Medicine 12, no. 4: 1589. https://doi.org/10.3390/jcm12041589
APA StyleIsraël, M., Berg, E., & Tenenbaum, G. (2023). Cancer Metabolism: Fasting Reset, the Keto-Paradox and Drugs for Undoing. Journal of Clinical Medicine, 12(4), 1589. https://doi.org/10.3390/jcm12041589