


Immune and Hereditary Thrombotic Thrombocytopenic Purpura: Can ADAMTS13 Deficiency Alone Explain the Different Clinical Phenotypes?

 , , ,
, , , 
Abstract
:1. Introduction
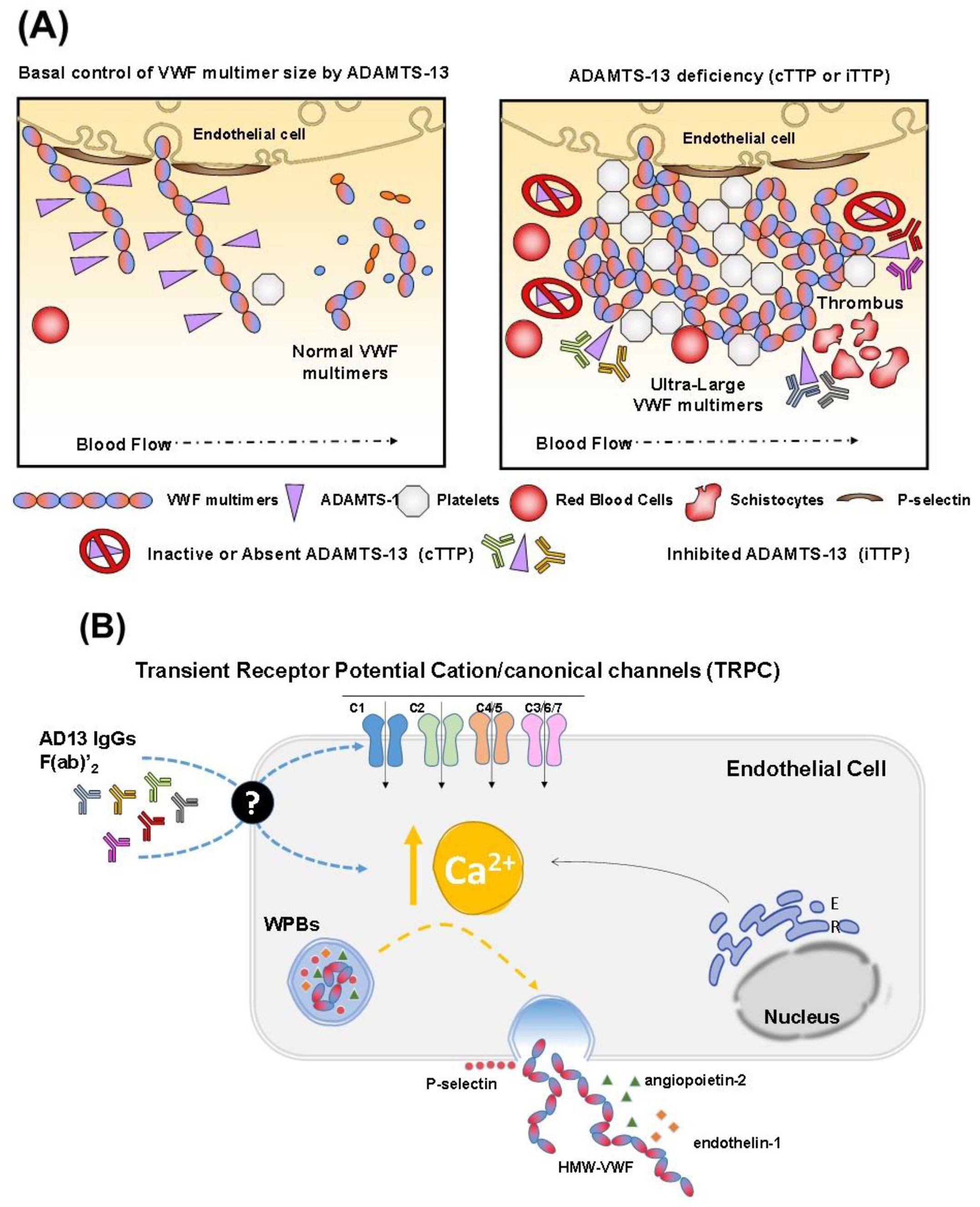
2. TTP Pathophysiology
3. Immune TTP (iTTP)
4. Clinical Symptoms of First Episodes and Relapse Incidence of iTTP
5. Clinical Symptoms of First Episodes and Relapse Incidence of Hereditary TTP
6. The Role of Anti-ADAMTS13 Antibodies in the Pathological Complications of iTTP
7. Effects on Therapies of the Different Pathogenetic Effectors of cTTP and iTTP
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Moake, J. Thrombotic microangiopathies: Multimers, metalloprotease, and beyond. Clin. Transl. Sci. 2009, 2, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Malgaj Vrecko, M.; Ales Rigler, A.; Veceric-Haler, Z. Coronavirus Disease 2019-Associated Thrombotic Microangiopathy: Literature Review. Int. J. Mol. Sci. 2022, 23, 11307. [Google Scholar] [CrossRef]
- Zheng, X.; Chung, D.; Takayama, T.K.; Majerus, E.M.; Sadler, J.E.; Fujikawa, K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J. Biol. Chem. 2001, 276, 41059–41063. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001, 413, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Furlan, M.; Robles, R.; Galbusera, M.; Remuzzi, G.; Kyrle, P.A.; Brenner, B.; Krause, M.; Scharrer, I.; Aumann, V.; Mittler, U.; et al. von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N. Engl. J. Med. 1998, 339, 1578–1584. [Google Scholar] [CrossRef]
- Tsai, H.M.; Lian, E.C. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N. Engl. J. Med. 1998, 339, 1585–1594. [Google Scholar] [CrossRef]
- Chung, D.W.; Fujikawa, K. Processing of von Willebrand factor by ADAMTS-13. Biochemistry 2002, 41, 11065–11070. [Google Scholar] [CrossRef]
- George, J.N. Clinical practice. Thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2006, 354, 1927–1935. [Google Scholar] [CrossRef]
- Roose, E.; Vidarsson, G.; Kangro, K.; Verhagen, O.; Mancini, I.; Desender, L.; Pareyn, I.; Vandeputte, N.; Vandenbulcke, A.; Vendramin, C.; et al. Anti-ADAMTS13 Autoantibodies against Cryptic Epitopes in Immune-Mediated Thrombotic Thrombocytopenic Purpura. Thromb. Haemost. 2018, 118, 1729–1742. [Google Scholar] [CrossRef]
- Roose, E.; Veyradier, A.; Vanhoorelbeke, K. Insights into ADAMTS13 structure: Impact on thrombotic thrombocytopenic purpura diagnosis and management. Curr. Opin. Hematol. 2020, 27, 320–326. [Google Scholar] [CrossRef]
- Roose, E.; Schelpe, A.S.; Tellier, E.; Sinkovits, G.; Joly, B.S.; Dekimpe, C.; Kaplanski, G.; Le Besnerais, M.; Mancini, I.; Falter, T.; et al. Open ADAMTS13, induced by antibodies, is a biomarker for subclinical immune-mediated thrombotic thrombocytopenic purpura. Blood 2020, 136, 353–361. [Google Scholar] [CrossRef]
- Scully, M. Inhibitory anti-ADAMTS 13 antibodies: Measurement and clinical application. Blood Rev. 2010, 24, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Froissart, A.; Buffet, M.; Veyradier, A.; Poullin, P.; Provot, F.; Malot, S.; Schwarzinger, M.; Galicier, L.; Vanhille, P.; Vernant, J.P.; et al. Efficacy and safety of first-line rituximab in severe, acquired thrombotic thrombocytopenic purpura with a suboptimal response to plasma exchange. Experience of the French Thrombotic Microangiopathies Reference Center. Crit. Care Med. 2012, 40, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Peyvandi, F.; Callewaert, F. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2016, 374, 2497–2498. [Google Scholar] [CrossRef]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knobl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Lancellotti, S.; Sacco, M.; Basso, M.; De Cristofaro, R. Mechanochemistry of von Willebrand factor. Biomol. Concepts 2019, 10, 194–208. [Google Scholar] [CrossRef]
- Shankaran, H.; Neelamegham, S. Hydrodynamic forces applied on intercellular bonds, soluble molecules, and cell-surface receptors. Biophys. J. 2004, 86, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Scheiflinger, F.; Knobl, P.; Trattner, B.; Plaimauer, B.; Mohr, G.; Dockal, M.; Dorner, F.; Rieger, M. Nonneutralizing IgM and IgG antibodies to von Willebrand factor-cleaving protease (ADAMTS-13) in a patient with thrombotic thrombocytopenic purpura. Blood 2003, 102, 3241–3243. [Google Scholar] [CrossRef]
- Shelat, S.G.; Smith, P.; Ai, J.; Zheng, X.L. Inhibitory autoantibodies against ADAMTS-13 in patients with thrombotic thrombocytopenic purpura bind ADAMTS-13 protease and may accelerate its clearance in vivo. J. Thromb. Haemost. 2006, 4, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Rieger, M.; Mannucci, P.M.; Kremer Hovinga, J.A.; Herzog, A.; Gerstenbauer, G.; Konetschny, C.; Zimmermann, K.; Scharrer, I.; Peyvandi, F.; Galbusera, M.; et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005, 106, 1262–1267. [Google Scholar] [CrossRef]
- Thomas, M.R.; de Groot, R.; Scully, M.A.; Crawley, J.T. Pathogenicity of Anti-ADAMTS13 Autoantibodies in Acquired Thrombotic Thrombocytopenic Purpura. EBioMedicine 2015, 2, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Soejima, K.; Matsumoto, M.; Kokame, K.; Yagi, H.; Ishizashi, H.; Maeda, H.; Nozaki, C.; Miyata, T.; Fujimura, Y.; Nakagaki, T. ADAMTS-13 cysteine-rich/spacer domains are functionally essential for von Willebrand factor cleavage. Blood 2003, 102, 3232–3237. [Google Scholar] [CrossRef] [PubMed]
- Luken, B.M.; Turenhout, E.A.; Hulstein, J.J.; Van Mourik, J.A.; Fijnheer, R.; Voorberg, J. The spacer domain of ADAMTS13 contains a major binding site for antibodies in patients with thrombotic thrombocytopenic purpura. Thromb. Haemost. 2005, 93, 267–274. [Google Scholar] [CrossRef]
- Luken, B.M.; Turenhout, E.A.; Kaijen, P.H.; Greuter, M.J.; Pos, W.; van Mourik, J.A.; Fijnheer, R.; Voorberg, J. Amino acid regions 572-579 and 657-666 of the spacer domain of ADAMTS13 provide a common antigenic core required for binding of antibodies in patients with acquired TTP. Thromb. Haemost. 2006, 96, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Velasquez Pereira, L.C.; Roose, E.; Graca, N.A.G.; Sinkovits, G.; Kangro, K.; Joly, B.S.; Tellier, E.; Kaplanski, G.; Falter, T.; Von Auer, C.; et al. Immunogenic hotspots in the spacer domain of ADAMTS13 in immune-mediated thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2021, 19, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Roose, E.; Schelpe, A.S.; Joly, B.S.; Peetermans, M.; Verhamme, P.; Voorberg, J.; Greinacher, A.; Deckmyn, H.; De Meyer, S.F.; Coppo, P.; et al. An open conformation of ADAMTS-13 is a hallmark of acute acquired thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2018, 16, 378–388. [Google Scholar] [CrossRef]
- Halkidis, K.; Siegel, D.L.; Zheng, X.L. A human monoclonal antibody against the distal carboxyl terminus of ADAMTS-13 modulates its susceptibility to an inhibitor in thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2021, 19, 1888–1895. [Google Scholar] [CrossRef]
- Mariotte, E.; Azoulay, E.; Galicier, L.; Rondeau, E.; Zouiti, F.; Boisseau, P.; Poullin, P.; de Maistre, E.; Provot, F.; Delmas, Y.; et al. Epidemiology and pathophysiology of adulthood-onset thrombotic microangiopathy with severe ADAMTS13 deficiency (thrombotic thrombocytopenic purpura): A cross-sectional analysis of the French national registry for thrombotic microangiopathy. Lancet Haematol. 2016, 3, e237–e245. [Google Scholar] [CrossRef]
- Reese, J.A.; Muthurajah, D.S.; Kremer Hovinga, J.A.; Vesely, S.K.; Terrell, D.R.; George, J.N. Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: Comparison of incidence, demographic and clinical features. Pediatr. Blood Cancer 2013, 60, 1676–1682. [Google Scholar] [CrossRef]
- Martino, S.; Jamme, M.; Deligny, C.; Busson, M.; Loiseau, P.; Azoulay, E.; Galicier, L.; Pene, F.; Provot, F.; Dossier, A.; et al. Thrombotic Thrombocytopenic Purpura in Black People: Impact of Ethnicity on Survival and Genetic Risk Factors. PLoS ONE 2016, 11, e0156679. [Google Scholar] [CrossRef]
- Hrdinova, J.; D'Angelo, S.; Graca, N.A.G.; Ercig, B.; Vanhoorelbeke, K.; Veyradier, A.; Voorberg, J.; Coppo, P. Dissecting the pathophysiology of immune thrombotic thrombocytopenic purpura: Interplay between genes and environmental triggers. Haematologica 2018, 103, 1099–1109. [Google Scholar] [CrossRef]
- Coppo, P.; Busson, M.; Veyradier, A.; Wynckel, A.; Poullin, P.; Azoulay, E.; Galicier, L.; Loiseau, P.; French Reference Centre For Thrombotic, M. HLA-DRB1*11: A strong risk factor for acquired severe ADAMTS13 deficiency-related idiopathic thrombotic thrombocytopenic purpura in Caucasians. J. Thromb. Haemost. 2010, 8, 856–859. [Google Scholar] [CrossRef]
- Scully, M.; Brown, J.; Patel, R.; McDonald, V.; Brown, C.J.; Machin, S. Human leukocyte antigen association in idiopathic thrombotic thrombocytopenic purpura: Evidence for an immunogenetic link. J. Thromb. Haemost. 2010, 8, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Mancini, I.; Giacomini, E.; Pontiggia, S.; Artoni, A.; Ferrari, B.; Pappalardo, E.; Gualtierotti, R.; Trisolini, S.M.; Capria, S.; Facchini, L.; et al. The HLA Variant rs6903608 Is Associated with Disease Onset and Relapse of Immune-Mediated Thrombotic Thrombocytopenic Purpura in Caucasians. J. Clin. Med. 2020, 9, 3379. [Google Scholar] [CrossRef] [PubMed]
- John, M.L.; Hitzler, W.; Scharrer, I. The role of human leukocyte antigens as predisposing and/or protective factors in patients with idiopathic thrombotic thrombocytopenic purpura. Ann. Hematol. 2012, 91, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Kuwana, M.; Tanaka, H.; Hosomichi, K.; Hasegawa, A.; Uyama, H.; Nishio, K.; Omae, T.; Hishizawa, M.; Matsui, M.; et al. HLA loci predisposing to immune TTP in Japanese: Potential role of the shared ADAMTS13 peptide bound to different HLA-DR. Blood 2020, 135, 2413–2419. [Google Scholar] [CrossRef]
- Jin, M.; Casper, T.C.; Cataland, S.R.; Kennedy, M.S.; Lin, S.; Li, Y.J.; Wu, H.M. Relationship between ADAMTS13 activity in clinical remission and the risk of TTP relapse. Br. J. Haematol. 2008, 141, 651–658. [Google Scholar] [CrossRef]
- Peyvandi, F.; Lavoretano, S.; Palla, R.; Feys, H.B.; Vanhoorelbeke, K.; Battaglioli, T.; Valsecchi, C.; Canciani, M.T.; Fabris, F.; Zver, S.; et al. ADAMTS13 and anti-ADAMTS13 antibodies as markers for recurrence of acquired thrombotic thrombocytopenic purpura during remission. Haematologica 2008, 93, 232–239. [Google Scholar] [CrossRef]
- Page, E.E.; Kremer Hovinga, J.A.; Terrell, D.R.; Vesely, S.K.; George, J.N. Clinical importance of ADAMTS13 activity during remission in patients with acquired thrombotic thrombocytopenic purpura. Blood 2016, 128, 2175–2178. [Google Scholar] [CrossRef]
- Miyata, T.; Fan, X. A second hit for TMA. Blood 2012, 120, 1152–1154. [Google Scholar] [CrossRef]
- Reti, M.; Farkas, P.; Csuka, D.; Razso, K.; Schlammadinger, A.; Udvardy, M.L.; Madach, K.; Domjan, G.; Bereczki, C.; Reusz, G.S.; et al. Complement activation in thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2012, 10, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Jay, L.; Lin, S.; Han, C.; Yang, S.; Cataland, S.R.; Masias, C. Interrelationship between ADAMTS13 activity, von Willebrand factor, and complement activation in remission from immune-mediated trhrombotic thrombocytopenic purpura. Br. J. Haematol. 2020, 189, e18–e20. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.C.; Yang, S.; Haven, S.; Holers, V.M.; Lundberg, A.S.; Wu, H.; Cataland, S.R. Complement activation and mortality during an acute episode of thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2013, 11, 1925–1927. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Sartain, S.; Moake, J. Ultralarge von Willebrand factor-induced platelet clumping and activation of the alternative complement pathway in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndromes. Hematol. Oncol. Clin. N. Am. 2015, 29, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Bettoni, S.; Galbusera, M.; Gastoldi, S.; Donadelli, R.; Tentori, C.; Sparta, G.; Bresin, E.; Mele, C.; Alberti, M.; Tortajada, A.; et al. Interaction between Multimeric von Willebrand Factor and Complement: A Fresh Look to the Pathophysiology of Microvascular Thrombosis. J. Immunol. 2017, 199, 1021–1040. [Google Scholar] [CrossRef]
- Turner, N.A.; Moake, J. Assembly and activation of alternative complement components on endothelial cell-anchored ultra-large von Willebrand factor links complement and hemostasis-thrombosis. PLoS ONE 2013, 8, e59372. [Google Scholar] [CrossRef]
- Feng, S.; Liang, X.; Cruz, M.A.; Vu, H.; Zhou, Z.; Pemmaraju, N.; Dong, J.F.; Kroll, M.H.; Afshar-Kharghan, V. The interaction between factor H and Von Willebrand factor. PLoS ONE 2013, 8, e73715. [Google Scholar] [CrossRef]
- Feng, S.; Liang, X.; Kroll, M.H.; Chung, D.W.; Afshar-Kharghan, V. von Willebrand factor is a cofactor in complement regulation. Blood 2015, 125, 1034–1037. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, D.; Cao, W.; Song, W.C.; Zheng, X.L. Synergistic effects of ADAMTS13 deficiency and complement activation in pathogenesis of thrombotic microangiopathy. Blood 2019, 134, 1095–1105. [Google Scholar] [CrossRef]
- Domingo-Gonzalez, A.; Regalado-Artamendi, I.; Martin-Rojas, R.M.; Perez-Rus, G.; Perez-Corral, A.; Diez-Martin, J.L.; Pascual-Izquierdo, C. Application of the French TMA Reference Center Score and the mortality in TTP Score in de novo and relapsed episodes of acquired thrombotic thrombocytopenic purpura at a tertiary care facility in Spain. J. Clin. Apher. 2021, 36, 420–428. [Google Scholar] [CrossRef]
- Adeyemi, A.; Razakariasa, F.; Chiorean, A.; de Passos Sousa, R. Epidemiology, treatment patterns, clinical outcomes, and disease burden among patients with immune-mediated thrombotic thrombocytopenic purpura in the United States. Res. Pract. Thromb. Haemost. 2022, 6, e12802. [Google Scholar] [CrossRef]
- von Krogh, A.S.; Quist-Paulsen, P.; Waage, A.; Langseth, O.O.; Thorstensen, K.; Brudevold, R.; Tjonnfjord, G.E.; Largiader, C.R.; Lammle, B.; Kremer Hovinga, J.A. High prevalence of hereditary thrombotic thrombocytopenic purpura in central Norway: From clinical observation to evidence. J. Thromb. Haemost. 2016, 14, 73–82. [Google Scholar] [CrossRef] [PubMed]
- van Dorland, H.A.; Taleghani, M.M.; Sakai, K.; Friedman, K.D.; George, J.N.; Hrachovinova, I.; Knobl, P.N.; von Krogh, A.S.; Schneppenheim, R.; Aebi-Huber, I.; et al. The International Hereditary Thrombotic Thrombocytopenic Purpura Registry: Key findings at enrollment until 2017. Haematologica 2019, 104, 2107–2115. [Google Scholar] [CrossRef]
- Lancellotti, S.; Peyvandi, F.; Pagliari, M.T.; Cairo, A.; Abdel-Azeim, S.; Chermak, E.; Lazzareschi, I.; Mastrangelo, S.; Cavallo, L.; Oliva, R.; et al. The D173G mutation in ADAMTS-13 causes a severe form of congenital thrombotic thrombocytopenic purpura. A clinical, biochemical and in silico study. Thromb. Haemost. 2016, 115, 51–62. [Google Scholar] [CrossRef]
- Alwan, F.; Vendramin, C.; Liesner, R.; Clark, A.; Lester, W.; Dutt, T.; Thomas, W.; Gooding, R.; Biss, T.; Watson, H.G.; et al. Characterization and treatment of congenital thrombotic thrombocytopenic purpura. Blood 2019, 133, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Kokame, K.; Matsumoto, M.; Soejima, K.; Yagi, H.; Ishizashi, H.; Funato, M.; Tamai, H.; Konno, M.; Kamide, K.; Kawano, Y.; et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc. Natl. Acad. Sci. USA 2002, 99, 11902–11907. [Google Scholar] [CrossRef]
- Plaimauer, B.; Fuhrmann, J.; Mohr, G.; Wernhart, W.; Bruno, K.; Ferrari, S.; Konetschny, C.; Antoine, G.; Rieger, M.; Scheiflinger, F. Modulation of ADAMTS13 secretion and specific activity by a combination of common amino acid polymorphisms and a missense mutation. Blood 2006, 107, 118–125. [Google Scholar] [CrossRef]
- Akiyama, M.; Kokame, K.; Miyata, T. ADAMTS13 P475S polymorphism causes a lowered enzymatic activity and urea lability in vitro. J. Thromb. Haemost. 2008, 6, 1830–1832. [Google Scholar] [CrossRef] [PubMed]
- Hager, H.B.; Andersen, M.T. A neonate presenting with jaundice, anemia, and thrombocytopenia. Blood 2018, 131, 1627. [Google Scholar] [CrossRef] [PubMed]
- Tarasco, E.; Butikofer, L.; Friedman, K.D.; George, J.N.; Hrachovinova, I.; Knobl, P.N.; Matsumoto, M.; von Krogh, A.S.; Aebi-Huber, I.; Cermakova, Z.; et al. Annual incidence and severity of acute episodes in hereditary thrombotic thrombocytopenic purpura. Blood 2021, 137, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, S.; Lammle, B.; Cataland, S.R. Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management. J. Clin. Med. 2021, 10, 536. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Mudde, G.C.; Rieger, M.; Veyradier, A.; Kremer Hovinga, J.A.; Scheiflinger, F. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2009, 7, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Sinkovits, G.; Szilagyi, A.; Farkas, P.; Inotai, D.; Szilvasi, A.; Tordai, A.; Razso, K.; Reti, M.; Prohaszka, Z. Concentration and Subclass Distribution of Anti-ADAMTS13 IgG Autoantibodies in Different Stages of Acquired Idiopathic Thrombotic Thrombocytopenic Purpura. Front. Immunol. 2018, 9, 1646. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daeron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Tellier, E.; Widemann, A.; Cauchois, R.; Faccini, J.; Lagarde, M.; Brun, M.; Robert, P.; Robert, S.; Bachelier, R.; Poullin, P.; et al. Immune thrombotic thrombocytopenic purpura plasmas induce calcium- and IgG-dependent endothelial activation: Correlations with disease severity. Haematologica 2023, 108, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Paule, R.; Leon, J.; Daugan, M.; Robe-Rybkine, T.; Poillerat, V.; Torset, C.; Fremeaux-Bacchi, V.; Dimitrov, J.D.; Roumenina, L.T. P-selectin drives complement attack on endothelium during intravascular hemolysis in TLR-4/heme-dependent manner. Proc. Natl. Acad. Sci. USA 2019, 116, 6280–6285. [Google Scholar] [CrossRef]
- Frimat, M.; Tabarin, F.; Dimitrov, J.D.; Poitou, C.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013, 122, 282–292. [Google Scholar] [CrossRef]
- Cauchois, R.; Muller, R.; Lagarde, M.; Dignat-George, F.; Tellier, E.; Kaplanski, G. Is Endothelial Activation a Critical Event in Thrombotic Thrombocytopenic Purpura? J. Clin. Med. 2023, 12, 758. [Google Scholar] [CrossRef]
- Wu, X.W.; Li, Q.Z.; Lian, E.C. Plasma from a patient with thrombotic thrombocytopenic purpura induces endothelial cell apoptosis and platelet aggregation. Thromb. Res. 1999, 93, 79–87. [Google Scholar] [CrossRef]
- Jimenez, J.J.; Jy, W.; Mauro, L.M.; Horstman, L.L.; Ahn, Y.S. Elevated endothelial microparticles in thrombotic thrombocytopenic purpura: Findings from brain and renal microvascular cell culture and patients with active disease. Br. J. Haematol. 2001, 112, 81–90. [Google Scholar] [CrossRef]
- Widemann, A.; Pasero, C.; Arnaud, L.; Poullin, P.; Loundou, A.D.; Choukroun, G.; Sanderson, F.; Lacroix, R.; Sabatier, F.; Coppo, P.; et al. Circulating endothelial cells and progenitors as prognostic factors during autoimmune thrombotic thrombocytopenic purpura: Results of a prospective multicenter French study. J. Thromb. Haemost. 2014, 12, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Feys, H.B.; Roodt, J.; Vandeputte, N.; Pareyn, I.; Lamprecht, S.; van Rensburg, W.J.; Anderson, P.J.; Budde, U.; Louw, V.J.; Badenhorst, P.N.; et al. Thrombotic thrombocytopenic purpura directly linked with ADAMTS13 inhibition in the baboon (Papio ursinus). Blood 2010, 116, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.; Earley, S. Transient Receptor Potential Channels and Endothelial Cell Calcium Signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef]
- Birnbaumer, L. The TRPC class of ion channels: A critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 395–426. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.; Chan, W.Y.; Leung, P.C.; Kwan, H.Y.; Liu, C.; Huang, Y.; Michel, V.; Yew, D.T.; Yao, X. Expression of TRPC homologs in endothelial cells and smooth muscle layers of human arteries. Histochem. Cell Biol. 2004, 122, 553–561. [Google Scholar] [CrossRef]
- Graciaa, S.; Adeagbo, S.; Fong, G.; Rollins, M.; McElfresh, P.; Zerra, P.E.; Bennett, C.; Josephson, C.D.; Briones, M.; Fasano, R.M.; et al. Clinical features and neurological outcomes in pediatric immune-mediated thrombotic thrombocytopenic purpura: A report from a large pediatric hematology center. Pediatr. Blood Cancer 2022, 69, e29992. [Google Scholar] [CrossRef]
- Scully, M.; Knobl, P.; Kentouche, K.; Rice, L.; Windyga, J.; Schneppenheim, R.; Kremer Hovinga, J.A.; Kajiwara, M.; Fujimura, Y.; Maggiore, C.; et al. Recombinant ADAMTS-13: First-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood 2017, 130, 2055–2063. [Google Scholar] [CrossRef]
- Boothby, A.; Mazepa, M. Caplacizumab for congenital thrombotic thrombocytopenic purpura. Am. J. Hematol. 2022, 97, E420–E421. [Google Scholar] [CrossRef]


{kind=link}
| Clinical Form | Diagnostic Features |
|---|---|
| Thrombotic Thrombocytopenic Purpura (TTP) | ADAMTS13 deficiency |
| Infection-associated TMA | Shiga-toxin, Streptococcus pneumonia, Campylobacter jejuni, Cytomegalovirus, Human immunodeficiency virus, Parvovirus B19, Epstein–Barr virus, BK virus, Influenza |
| Complement-mediated hemolytic uremic syndrome (HUS) | Dysregulation of complement factors and their inhibitors |
| Secondary TMAs | Cancer, Transplantation, Antiphospholipid antibody Systemic lupus erythematosus syndrome, Scleroderma, Vasculitis/ glomerulonephritis |
| Disseminated intravascular coagulation | Sepsis, cancer |
| Drug-induced TMA | Calcineurin or mTOR inhibitors, Quinine, Interferon Vascular endothelial growth factor or proteasome inhibitors Estrogen/progesterone, Gemcitabine/ mitomycin C, Cocaine |
| Malignant hypertension-induced TMA | Extreme levels of blood pressure, severe headache, papilledema |
| Pregnancy-associated TMA | HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome, HUS, TTP |
| Metabolism-associated TMA | Cobalamin responsive methylmalonic acidemia, mutation of Diacylglycerolkinase epsilon |
| COVID-19 associated TMA | SARS-COV2 infection, evidence of microangiopathy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lancellotti, S.; Sacco, M.; Tardugno, M.; Ferretti, A.; De Cristofaro, R. Immune and Hereditary Thrombotic Thrombocytopenic Purpura: Can ADAMTS13 Deficiency Alone Explain the Different Clinical Phenotypes? J. Clin. Med. 2023, 12, 3111. https://doi.org/10.3390/jcm12093111
Lancellotti S, Sacco M, Tardugno M, Ferretti A, De Cristofaro R. Immune and Hereditary Thrombotic Thrombocytopenic Purpura: Can ADAMTS13 Deficiency Alone Explain the Different Clinical Phenotypes? Journal of Clinical Medicine. 2023; 12(9):3111. https://doi.org/10.3390/jcm12093111
Chicago/Turabian StyleLancellotti, Stefano, Monica Sacco, Maira Tardugno, Antonietta Ferretti, and Raimondo De Cristofaro. 2023. "Immune and Hereditary Thrombotic Thrombocytopenic Purpura: Can ADAMTS13 Deficiency Alone Explain the Different Clinical Phenotypes?" Journal of Clinical Medicine 12, no. 9: 3111. https://doi.org/10.3390/jcm12093111
APA StyleLancellotti, S., Sacco, M., Tardugno, M., Ferretti, A., & De Cristofaro, R. (2023). Immune and Hereditary Thrombotic Thrombocytopenic Purpura: Can ADAMTS13 Deficiency Alone Explain the Different Clinical Phenotypes? Journal of Clinical Medicine, 12(9), 3111. https://doi.org/10.3390/jcm12093111