
Mitochondrial Metabolism in Major Depressive Disorder: From Early Diagnosis to Emerging Treatment Options

 , , , , and
, , , , and
Abstract
: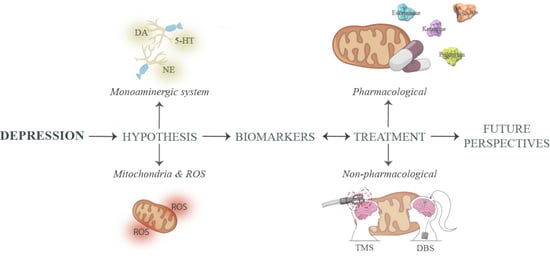
1. Introduction
1.1. Monoamine Deficiency
1.2. Mitochondrial Hypothesis
1.3. Mitochondrial Stress and Oxidative Damage in Depressive Disorders
2. Depression Biomarkers
2.1. Diagnosis Biomarkers
2.2. Prognostic Biomarkers
3. Treatment Options for Treatment-Resistant Depression
3.1. Pharmacological Treatment
3.1.1. Ketamine
Ketamine and Mitochondria
3.1.2. Esketamine
Esketamine and Mitochondria
3.1.3. Psilocybin
Psilocybin and Mitochondria
3.1.4. Anti-Inflammatory Drugs and Cytokine Inhibitors
Anti-Inflammatory Drugs and Mitochondria
3.2. Non-Pharmacological Treatment
3.2.1. Transcraneal Magnetic Stimulation
- Circular Coil: A round-shaped coil that allows for the induction of a homogeneous magnetic field with a preference for superficial cortical areas. This type of coil is often selected for more superficial or generalized stimulations in a broader brain region [126].
- Figure-Eight Coil: It has two loops forming a figure-eight structure, enabling greater focus of stimulation in the area where the two loops cross. This type of coil is usually chosen when aiming to direct magnetic pulses to specific brain regions, minimizing undesired stimulation in surrounding areas [126].
- 10–20 EEG System Method: Utilizes the International 10–20 EEG system to identify specific areas of the skull. Distances between electrodes on the scalp are measured to determine the coil’s location. Its use is common for routine clinical applications [127].
- Neuronavigation: This method employs medical images such as magnetic resonance imaging (MRI) or computed tomography (CT) to create a three-dimensional map of the brain. This real-time map guides the exact placement of the coil, allowing for precise positioning based on each individual’s brain anatomy. Its use is common for advanced clinical applications [128].
- Anatomical Methods: Points such as the central sulcus are used to orient the coil, with selected points depending on the stimulation goal. Stereotactic coordinates are employed to specify the three-dimensional position of the coil in relation to brain anatomy [126].
3.2.2. Deep Brain Stimulation
- -
- Subcallosal Cingulate Cortex (SCC): Associated with the regulation of negative mood states, with increased activity observed. It is interconnected with regions involved in emotion processing and motivation, with reciprocal pathways to other subcortical regions. The goal of DBS is to reduce hyperactivity and interrupt negative mood states. Studies have shown that remission rates are initially reduced but significantly increase in the long term [148].
- -
- Lateral Habenula (LHb): Implicated in negative mood states and found to be activated with reduced volume in patients with MDD [148].
- -
- Ventral Capsule and Ventral Striatum (VC and VS): Linked to mood regulation and reward. Due to varied outcomes in studies, a more extensive optimization phase is suggested.
- -
- Medial Forebrain Bundle (MFB): Associated with the reward circuitry. Various studies have shown that its modulation can produce significant changes in anhedonia both in the short and long term [149].
Non-Pharmacological Treatment Stimulation and Mitochondria
4. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Zvěřová, M.; Hroudová, J.; Fišar, Z.; Hansíková, H.; Kališová, L.; Kitzlerová, E.; Lambertová, A.; Raboch, J. Disturbances of Mitochondrial Parameters to Distinguish Patients with Depressive Episode of Bipolar Disorder and Major Depressive Disorder. Neuropsychiatr. Dis. Treat. 2019, 15, 233. [Google Scholar] [CrossRef]
- Ballard, E.D.; Yarrington, J.S.; Farmer, C.A.; Lener, M.S.; Kadriu, B.; Lally, N.; Williams, D.; Machado-Vieira, R.; Niciu, M.J.; Park, L.; et al. Parsing the Heterogeneity of Depression: An Exploratory Factor Analysis across Commonly Used Depression Rating Scales. J. Affect. Disord. 2018, 231, 51–57. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. The Molecular Neurobiology of Depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef]
- Pandarakalam, J.P. Challenges of Treatment-Resistant Depression. Psychiatr. Danub. 2018, 30, 273–284. [Google Scholar] [CrossRef]
- Henssler, J.; Alexander, D.; Schwarzer, G.; Bschor, T.; Baethge, C. Combining Antidepressants vs. Antidepressant Monotherapy for Treatment of Patients with Acute Depression: A Systematic Review and Meta-Analysis. JAMA Psychiatry 2022, 79, 300–312. [Google Scholar] [CrossRef]
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A Review of Recent Studies on Malondialdehyde as Toxic Molecule and Biological Marker of Oxidative Stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328. [Google Scholar] [CrossRef]
- Yang, B.; Chen, Y.; Shi, J. Reactive Oxygen Species (ROS)-Based Nanomedicine. Chem. Rev. 2019, 119, 4881–4985. [Google Scholar] [CrossRef] [PubMed]
- Massart, R.; Mongeau, R.; Lanfumey, L. Beyond the Monoaminergic Hypothesis: Neuroplasticity and Epigenetic Changes in a Transgenic Mouse Model of Depression. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2485–2494. [Google Scholar] [CrossRef]
- Di Lisa, F.; Kaludercic, N.; Carpi, A.; Menabò, R.; Giorgio, M. Mitochondrial Pathways for ROS Formation and Myocardial Injury: The Relevance of P66Shc and Monoamine Oxidase. Basic Res. Cardiol. 2009, 104, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.W.Y.; Tsai, S.J.; Chen, T.J.; Lin, C.H.; Hong, C.J. Association Study of the Serotonin Transporter Promoter Polymorphism and Symptomatology and Antidepressant Response in Major Depressive Disorders. Mol. Psychiatry 2002, 7, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Pezawas, L.; Meyer-Lindenberg, A.; Drabant, E.M.; Verchinski, B.A.; Munoz, K.E.; Kolachana, B.S.; Egan, M.F.; Mattay, V.S.; Hariri, A.R.; Weinberger, D.R. 5-HTTLPR Polymorphism Impacts Human Cingulate-Amygdala Interactions: A Genetic Susceptibility Mechanism for Depression. Nat. Neurosci. 2005, 8, 828–834. [Google Scholar] [CrossRef]
- Jorm, A.F.; Henderson, A.S.; Jacomb, P.A.; Christensen, H.; Korten, A.E.; Rodgers, B.; Tan, X.; Easteal, S. Association of a Functional Polymorphism of the Monoamine Oxidase A Gene Promoter with Personality and Psychiatric Symptoms. Psychiatr. Genet. 2000, 10, 87–90. [Google Scholar] [CrossRef]
- Moncrieff, J.; Cooper, R.E.; Stockmann, T.; Amendola, S.; Hengartner, M.P.; Horowitz, M.A. The Serotonin Theory of Depression: A Systematic Umbrella Review of the Evidence. Mol. Psychiatry 2023, 28, 3243. [Google Scholar] [CrossRef]
- Domschke, K.; Dannlowski, U.; Ohrmann, P.; Lawford, B.; Bauer, J.; Kugel, H.; Heindel, W.; Young, R.; Morris, P.; Arolt, V.; et al. Cannabinoid Receptor 1 (CNR1) Gene: Impact on Antidepressant Treatment Response and Emotion Processing in Major Depression. Eur. Neuropsychopharmacol. 2008, 18, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, J.A.; Markovic, T.; Massaly, N.; Morón, J.A. Endogenous Opioid Systems Alterations in Pain and Opioid Use Disorder. Front. Syst. Neurosci. 2022, 16, 1014768. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovács, R. Mitochondria and Neuronal Activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed]
- Weckmann, K.; Labermaier, C.; Asara, J.M.; Müller, M.B.; Turck, C.W. Time-Dependent Metabolomic Profiling of Ketamine Drug Action Reveals Hippocampal Pathway Alterations and Biomarker Candidates. Transl. Psychiatry 2014, 4, e481. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Benatti, C.; Blom, J.M.C.; Caraci, F.; Tascedda, F. The Many Faces of Mitochondrial Dysfunction in Depression: From Pathology to Treatment. Front. Pharmacol. 2019, 10, 995. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lian, G. ROS and Diseases: Role in Metabolism and Energy Supply. Mol. Cell Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ruan, M.; Chen, J.; Fang, Y. Major Depressive Disorder: Advances in Neuroscience Research and Translational Applications. Neurosci. Bull. 2021, 37, 863–880. [Google Scholar] [CrossRef]
- Holper, L.; Ben-Shachar, D.; Mann, J. Multivariate Meta-Analyses of Mitochondrial Complex I and IV in Major Depressive Disorder, Bipolar Disorder, Schizophrenia, Alzheimer Disease, and Parkinson Disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef]
- Giménez-Palomo, A.; Dodd, S.; Anmella, G.; Carvalho, A.F.; Scaini, G.; Quevedo, J.; Pacchiarotti, I.; Vieta, E.; Berk, M. The Role of Mitochondria in Mood Disorders: From Physiology to Pathophysiology and to Treatment. Front. Psychiatry 2021, 12, 546801. [Google Scholar] [CrossRef]
- Karabatsiakis, A.; Böck, C.; Salinas-Manrique, J.; Kolassa, S.; Calzia, E.; Dietrich, D.E.; Kolassa, I.T. Mitochondrial Respiration in Peripheral Blood Mononuclear Cells Correlates with Depressive Subsymptoms and Severity of Major Depression. Transl. Psychiatry 2014, 4, e397. [Google Scholar] [CrossRef]
- Cikánková, T.; Fišar, Z.; Bakhouche, Y.; Ľupták, M.; Hroudová, J. In Vitro Effects of Antipsychotics on Mitochondrial Respiration. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 1209–1223. [Google Scholar] [CrossRef]
- Martins, M.R.; Petronilho, F.C.; Gomes, K.M.; Dal-Pizzol, F.; Streck, E.L.; Quevedo, J. Antipsychotic-Induced Oxidative Stress in Rat Brain. Neurotox. Res. 2008, 13, 63–69. [Google Scholar] [CrossRef]
- Dean, J.; Keshavan, M. The Neurobiology of Depression: An Integrated View. Asian J. Psychiatr. 2017, 27, 101–111. [Google Scholar] [CrossRef]
- Kotzaeroglou, A.; Tsamesidis, I. The Role of Equilibrium between Free Radicals and Antioxidants in Depression and Bipolar Disorder. Medicines 2022, 9, 57. [Google Scholar] [CrossRef]
- Maria Michel, T.; Pulschen, D.; Thome, J. The Role of Oxidative Stress in Depressive Disorders. Curr. Pharm. Des. 2012, 18, 5890–5899. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chai, Y.; Ding, J.H.; Sun, X.L.; Hu, G. Chronic Mild Stress Damages Mitochondrial Ultrastructure and Function in Mouse Brain. Neurosci. Lett. 2011, 488, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, U.; Dasgupta, A.; Rout, J.K.; Singh, O.P. Effects of Lithium Therapy on Na +−K +−ATPase Activity and Lipid Peroxidation in Bipolar Disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 37, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.F. Non-Motor Symptoms in Parkinson’s Disease. Park. Relat. Disord. 2016, 22, S119–S122. [Google Scholar] [CrossRef]
- Mora, C.; Zonca, V.; Riva, M.A.; Cattaneo, A. Blood Biomarkers and Treatment Response in Major Depression. Exp. Rev. Mol. Diagn. 2018, 18, 513–529. [Google Scholar] [CrossRef]
- Uddin, M. Blood-Based Biomarkers in Depression: Emerging Themes in Clinical Research. Mol. Diagn. Ther. 2014, 18, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Buckley, T.M.; Schatzberg, A.F. A Pilot Study of the Phase Angle between Cortisol and Melatonin in Major Depression—A Potential Biomarker? J. Psychiatr. Res. 2010, 44, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Gururajan, A.; Clarke, G.; Dinan, T.G.; Cryan, J.F. Molecular Biomarkers of Depression. Neurosci. Biobehav. Rev. 2016, 64, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Fuchikami, M.; Morinobu, S.; Segawa, M.; Okamoto, Y.; Yamawaki, S.; Ozaki, N.; Inoue, T.; Kusumi, I.; Koyama, T.; Tsuchiyama, K.; et al. DNA Methylation Profiles of the Brain-Derived Neurotrophic Factor (BDNF) Gene as a Potent Diagnostic Biomarker in Major Depression. PLoS ONE 2011, 6, e23881. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Lin, Y.S.; Chen, C.C.; Bai, C.H.; Wu, S.R. Cytokines and Serotonin Transporter in Patients with Major Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; Von Döbeln, U.; Hagenfeldt, L.; Hällström, T. Alterations of Mitochondrial Function and Correlations with Personality Traits in Selected Major Depressive Disorder Patients. J. Affect. Disord. 2003, 76, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Hroudová, J.; Fišar, Z.; Kitzlerová, E.; Zvěřová, M.; Raboch, J. Mitochondrial Respiration in Blood Platelets of Depressive Patients. Mitochondrion 2013, 13, 795–800. [Google Scholar] [CrossRef]
- Chen, M.H.; Li, C.T.; Lin, W.C.; Hong, C.J.; Tu, P.C.; Bai, Y.M.; Cheng, C.M.; Su, T.P. Rapid Inflammation Modulation and Antidepressant Efficacy of a Low-Dose Ketamine Infusion in Treatment-Resistant Depression: A Randomized, Double-Blind Control Study. Psychiatry Res. 2018, 269, 207–211. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Gold, P.W.; Luckenbaugh, D.A.; Ballard, E.D.; Richards, E.M.; Henter, I.D.; De Sousa, R.T.; Niciu, M.J.; Yuan, P.; Zarate, C.A. The Role of Adipokines in the Rapid Antidepressant Effects of Ketamine. Mol. Psychiatry 2017, 22, 127–133. [Google Scholar] [CrossRef]
- Gkesoglou, T.; Bargiota, S.I.; Iordanidou, E.; Vasiliadis, M.; Bozikas, V.P.; Agorastos, A. Prognostic Significance of Blood-Based Baseline Biomarkers in Treatment-Resistant Depression: A Literature Review of Available Studies on Treatment Response. Brain Sci. 2022, 12, 940. [Google Scholar] [CrossRef]
- Maas, J.W.; Fawcett, J.A.; Dekirmenjian, H. Catecholamine Metabolism, Depressive Illness, and Drug Response. Arch. Gen. Psychiatry 1972, 26, 252–262. [Google Scholar] [CrossRef]
- Tsai, M.C.; Huang, T.L. Increased Activities of Both Superoxide Dismutase and Catalase Were Indicators of Acute Depressive Episodes in Patients with Major Depressive Disorder. Psychiatry Res. 2015, 235, 38–42. [Google Scholar] [CrossRef]
- Lopresti, A.L.; Maker, G.L.; Hood, S.D.; Drummond, P.D. A Review of Peripheral Biomarkers in Major Depression: The Potential of Inflammatory and Oxidative Stress Biomarkers. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 48, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant Effects of Ketamine in Depressed Patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Danyeli, L.V.; Gö, F.N.; Mrü, Z.; Sen, D.; Li, M.; Walter, M. Ketamine in Psychiatric Disorders. In NeuroPsychopharmacotherapy; Springer: Berlin/Heidelberg, Germany, 2021; pp. 1–44. [Google Scholar] [CrossRef]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A Randomized Trial of an N-Methyl-D-Aspartate Antagonist in Treatment-Resistant Major Depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, K.A.B.; Levitch, C.F.; Perez, A.M.; Brallier, J.W.; Parides, M.K.; Soleimani, L.; Feder, A.; Iosifescu, D.V.; Charney, D.S.; Murrough, J.W. A Randomized Controlled Trial of Intranasal Ketamine in Major Depressive Disorder. Biol. Psychiatry 2014, 76, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Bahji, A.; Vazquez, G.H.; Zarate, C.A. Comparative Efficacy of Racemic Ketamine and Esketamine for Depression: A Systematic Review and Meta-Analysis. J. Affect. Disord. 2021, 278, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.T.; Toprak, M.; Turner, M.S.; Levine, S.P.; Katz, R.B.; Sanacora, G. A Survey of the Clinical, Off-Label Use of Ketamine as a Treatment for Psychiatric Disorders. Am. J. Psychiatry 2017, 174, 695–696. [Google Scholar] [CrossRef] [PubMed]
- Vyklicky, V.; Korinek, M.; Smejkalova, T.; Balik, A.; Krausova, B.; Kaniakova, M.; Lichnerova, K.; Cerny, J.; Krusek, J.; Dittert, I.; et al. Structure, Function, and Pharmacology of NMDA Receptor Channels. Physiol. Res. 2014, 63, S191. [Google Scholar] [CrossRef]
- Zanos, P.; Gould, T.D. Mechanisms of Ketamine Action as an Antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef]
- Weckmann, K.; Deery, M.J.; Howard, J.A.; Feret, R.; Asara, J.M.; Dethloff, F.; Filiou, M.D.; Iannace, J.; Labermaier, C.; Maccarrone, G.; et al. Ketamine’s Antidepressant Effect Is Mediated by Energy Metabolism and Antioxidant Defense System. Sci. Rep. 2017, 7, 15788. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H. Ketamine Blocks Bursting in the Lateral Habenula to Rapidly Relieve Depression. Nature 2018, 554, 317–322. [Google Scholar] [CrossRef]
- Ma, S.; Chen, M.; Jiang, Y.; Xiang, X.; Wang, S.; Wu, Z.; Li, S.; Cui, Y.; Wang, J.; Zhu, Y.; et al. Sustained Antidepressant Effect of Ketamine through NMDAR Trapping in the LHb. Nature 2023, 622, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Rapid-Acting Antidepressant Ketamine, Its Metabolites and Other Candidates: A Historical Overview and Future Perspective. Psychiatry Clin. Neurosci. 2019, 73, 613–627. [Google Scholar] [CrossRef]
- Kishimoto, T.; Chawla, J.M.; Hagi, K.; Zarate, C.A.; Kane, J.M.; Bauer, M.; Correll, C.U. Single-Dose Infusion Ketamine and Non-Ketamine N-Methyl-d-Aspartate Receptor Antagonists for Unipolar and Bipolar Depression: A Meta-Analysis of Efficacy, Safety and Time Trajectories. Psychol. Med. 2016, 46, 1459–1472. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Chandra, J.; Sheriff, S.; Malinow, R. Opioid System Is Necessary but Not Sufficient for Antidepressive Actions of Ketamine in Rodents. Proc. Natl. Acad. Sci. USA 2020, 117, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Newport, D.J.; Carpenter, L.L.; McDonald, W.M.; Potash, J.B.; Tohen, M.; Nemeroff, C.B. Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. Am. J. Psychiatry 2015, 172, 950–966. [Google Scholar] [CrossRef] [PubMed]
- Bobo, W.V.; Voort, J.L.V.; Croarkin, P.E.; Leung, J.G.; Tye, S.J.; Frye, M.A. Ketamine for treatment-resistant unipolar and bipolar major depression: Critical review and implications for clinical practice. Depress. Anxiety 2016, 33, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hackett, M.; Carter, G.; Loo, C.; Gálvez, V.; Glozier, N.; Glue, P.; Lapidus, K.; McGirr, A.; Somogyi, A.A.; et al. Effects of Low-Dose and Very Low-Dose Ketamine among Patients with Major Depression: A Systematic Review and Meta-Analysis. Int. J. Neuropsychopharmacol. 2016, 19, pyv124. [Google Scholar] [CrossRef] [PubMed]
- Sanacora, G.; Heimer, H.; Hartman, D.; Mathew, S.J.; Frye, M.; Nemeroff, C.; Robinson Beale, R. Balancing the Promise and Risks of Ketamine Treatment for Mood Disorders. Neuropsychopharmacology 2017, 42, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Venâncio, C.; Félix, L.; Almeida, V.; Coutinho, J.; Antunes, L.; Peixoto, F.; Summavielle, T. Acute Ketamine Impairs Mitochondrial Function and Promotes Superoxide Dismutase Activity in the Rat Brain. Anesth. Analg. 2015, 120, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Moda-Sava, R.N.; Murdock, M.H.; Parekh, P.K.; Fetcho, R.N.; Huang, B.S.; Huynh, T.N.; Witztum, J.; Shaver, D.C.; Rosenthal, D.L.; Alway, E.J.; et al. Sustained Rescue of Prefrontal Circuit Dysfunction by Antidepressant-Induced Spine Formation. Science 2019, 364, eaat8078. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 342896. [Google Scholar] [CrossRef]
- Jonkman, K.; van Rijnsoever, E.; Olofsen, E.; Aarts, L.; Sarton, E.; van Velzen, M.; Niesters, M.; Dahan, A. Esketamine Counters Opioid-Induced Respiratory Depression. Br. J. Anaesth. 2018, 120, 1117–1127. [Google Scholar] [CrossRef]
- Gastaldon, C.; Papola, D.; Ostuzzi, G.; Barbui, C. Esketamine for Treatment Resistant Depression: A Trick of Smoke and Mirrors? Epidemiol. Psychiatr. Sci. 2020, 29, e79. [Google Scholar] [CrossRef] [PubMed]
- Janssen Announces, U.S. FDA Approval of SPRAVATOTM (Esketamine) CIII Nasal Spray for Adults with Treatment-Resistant Depression (TRD) Who Have Cycled through Multiple Treatments without Relief. Available online: https://www.jnj.com/media-center/press-releases/janssen-announces-u-s-fda-approval-of-spravatotm-esketamine-ciii-nasal-spray-for-adults-with-treatment-resistant-depression-trd-who-have-cycled-through-multiple-treatments-without-relief (accessed on 12 March 2024).
- Daly, E.J.; Trivedi, M.H.; Janik, A.; Li, H.; Zhang, Y.; Li, X.; Lane, R.; Lim, P.; Duca, A.R.; Hough, D.; et al. Efficacy of Esketamine Nasal Spray Plus Oral Antidepressant Treatment for Relapse Prevention in Patients with Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA Psychiatry 2019, 76, 893–903. [Google Scholar] [CrossRef]
- Salahudeen, M.S.; Wright, C.M.; Peterson, G.M. Esketamine: New Hope for the Treatment of Treatment-Resistant Depression? A Narrative Review. Ther. Adv. Drug Saf. 2020, 11, 2042098620937899. [Google Scholar] [CrossRef]
- Dang, Y.-H.; Ma, X.-C.; Zhang, J.-C.; Ren, Q.; Wu, J.; Gao, C.-G.; Hashimoto, K. Targeting of NMDA Receptors in the Treatment of Major Depression. Curr. Pharm. Des. 2014, 20, 5151–5159. [Google Scholar] [CrossRef]
- Saad, Z.; Hibar, D.; Fedgchin, M.; Popova, V.; Furey, M.L.; Singh, J.B.; Kolb, H.; Drevets, W.C.; Chen, G. Effects of Mu-Opiate Receptor Gene Polymorphism Rs1799971 (A118G) on the Antidepressant and Dissociation Responses in Esketamine Nasal Spray Clinical Trials. Int. J. Neuropsychopharmacol. 2020, 23, 549. [Google Scholar] [CrossRef] [PubMed]
- Jelen, L.A.; Stone, J.M.; Young, A.H.; Mehta, M.A. The Opioid System in Depression. Neurosci. Biobehav. Rev. 2022, 140, 104800. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Da-Silva, L.; Carlson, P.T.; Silva-Costa, L.C.; Martins-De-Souza, D.; De Almeida, V. Molecular Mechanisms Associated with Antidepressant Treatment on Major Depression. Complex. Psychiatry 2021, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Amasi-Hartoonian, N.; Pariante, C.M.; Cattaneo, A.; Sforzini, L. Understanding Treatment-Resistant Depression Using “Omics” Techniques: A Systematic Review. J. Affect. Disord. 2022, 318, 423–455. [Google Scholar] [CrossRef] [PubMed]
- Stultz, D.J.; Stanley, N.; Gills, T.; Osburn, S.; Burns, T.; Moomaw, S.; Pawlowska-Wajswol, S.; Walton, R. Three Months of Treatment with Esketamine: Effects on Depression, Insomnia, and Weight. Prim. Care Companion CNS Disord. 2020, 22, 26334. [Google Scholar] [CrossRef] [PubMed]
- Nikayin, S.; Murphy, E.; Krystal, J.H.; Wilkinson, S.T. Long-Term Safety of Ketamine and Esketamine in Treatment of Depression. Exp. Opin. Drug Saf. 2022, 21, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, J.; Li, X.; Wang, T.; Xu, Z.; Xu, X.; Zhou, X.; Chen, G. Adverse Effects of Esketamine for the Treatment of Major Depression Disorder: Findings from Randomized Controlled Trials. Psychiatr. Q. 2022, 93, 81–95. [Google Scholar] [CrossRef]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-Targeted Antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef]
- Krauss, S. Mitochondria: Structure and Role in Respiration. In Encyclopedia of Life Sciences; Nature Publishing Group: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Guan, X.; Yan, Q.; Wang, D.; Du, G.; Zhou, J. IGF-1 Signaling Regulates Mitochondrial Remodeling during Myogenic Differentiation. Nutrients 2022, 14, 1249. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z. Bin Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Davis, A.K.; Barrett, F.S.; May, D.G.; Cosimano, M.P.; Sepeda, N.D.; Johnson, M.W.; Finan, P.H.; Griffiths, R.R. Effects of Psilocybin-Assisted Therapy on Major Depressive Disorder: A Randomized Clinical Trial. JAMA Psychiatry 2021, 78, 1. [Google Scholar] [CrossRef]
- Aleksandrova, L.R.; Phillips, A.G. Neuroplasticity as a Convergent Mechanism of Ketamine and Classical Psychedelics. Trends Pharmacol. Sci. 2021, 42, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Haridy, R. Australia to Prescribe MDMA and Psilocybin for PTSD and Depression in World First. Nature 2023, 619, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, T.; Preller, K.H.; Kraehenmann, R.; Vollenweider, F.X. Modulatory Effect of the 5-HT1A Agonist Buspirone and the Mixed Non-Hallucinogenic 5-HT1A/2A Agonist Ergotamine on Psilocybin-Induced Psychedelic Experience. Eur. Neuropsychopharmacol. 2016, 26, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, R.G.; Osório, F.L.; Crippa, J.A.S.; Hallak, J.E.C. Classical Hallucinogens and Neuroimaging: A Systematic Review of Human Studies: Hallucinogens and Neuroimaging. Neurosci. Biobehav. Rev. 2016, 71, 715–728. [Google Scholar] [CrossRef] [PubMed]
- López-Giménez, J.F.; González-Maeso, J. Hallucinogens and Serotonin 5-HT2A Receptor-Mediated Signaling Pathways. Curr. Top. Behav. Neurosci. 2018, 36, 45. [Google Scholar] [CrossRef] [PubMed]
- Smausz, R.; Neill, J.; Gigg, J. Neural Mechanisms Underlying Psilocybin’s Therapeutic—The Need for Preclinical in Vivo. J. Psychopharmacol. 2022, 36, 781. [Google Scholar] [CrossRef] [PubMed]
- Golden, C.T.; Chadderton, P. Psilocybin Reduces Low Frequency Oscillatory Power and Neuronal Phase-Locking in the Anterior Cingulate Cortex of Awake Rodents. Sci. Rep. 2022, 12, 12702. [Google Scholar] [CrossRef]
- Gattuso, J.J.; Perkins, D.; Ruffell, S.; Lawrence, A.J.; Hoyer, D.; Jacobson, L.H.; Timmermann, C.; Castle, D.; Rossell, S.L.; Downey, L.A.; et al. Default Mode Network Modulation by Psychedelics: A Systematic Review. Int. J. Neuropsychopharmacol. 2023, 26, 155. [Google Scholar] [CrossRef]
- Siegel, J.S.; Subramanian, S.; Perry, D.; Kay, B.; Gordon, E.; Laumann, T.; Reneau, R.; Gratton, C.; Horan, C.; Metcalf, N.; et al. Psilocybin Desynchronizes Brain Networks. medRxiv 2023. [Google Scholar] [CrossRef]
- Buckner, R.L. The Brain’s Default Network: Origins and Implications for the Study of Psychosis. Dialogues Clin. Neurosci. 2013, 15, 351. [Google Scholar] [CrossRef]
- Kelly, J.R.; Gillan, C.M.; Prenderville, J.; Kelly, C.; Harkin, A.; Clarke, G.; O’Keane, V. Psychedelic Therapy’s Transdiagnostic Effects: A Research Domain Criteria (RDoC) Perspective. Front. Psychiatry 2021, 12, 800072. [Google Scholar] [CrossRef]
- Zamani, A.; Carhart-Harris, R.; Christoff, K. Prefrontal Contributions to the Stability and Variability of Thought and Conscious Experience. Neuropsychopharmacology 2022, 47, 329. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.X.; Kometer, M. The Neurobiology of Psychedelic Drugs: Implications for the Treatment of Mood Disorders. Nat. Rev. Neurosci. 2010, 11, 642–651. [Google Scholar] [CrossRef]
- Ly, C.; Greb, A.C.; Cameron, L.P.; Wong, J.M.; Barragan, E.V.; Wilson, P.C.; Burbach, K.F.; Soltanzadeh Zarandi, S.; Sood, A.; Paddy, M.R.; et al. Psychedelics Promote Structural and Functional Neural Plasticity. Cell Rep. 2018, 23, 3170. [Google Scholar] [CrossRef]
- Dwyer, J.M.; Duman, R.S. Activation of MTOR and Synaptogenesis: Role in the Actions of Rapid-Acting Antidepressants. Biol. Psychiatry 2013, 73, 1189. [Google Scholar] [CrossRef] [PubMed]
- Moliner, R.; Girych, M.; Brunello, C.A.; Kovaleva, V.; Biojone, C.; Enkavi, G.; Antenucci, L.; Kot, E.F.; Goncharuk, S.A.; Kaurinkoski, K.; et al. Psychedelics Promote Plasticity by Directly Binding to BDNF Receptor TrkB. Nat. Neurosci. 2023, 26, 1032–1041. [Google Scholar] [CrossRef]
- Cavarra, M.; Falzone, A.; Ramaekers, J.G.; Kuypers, K.P.C.; Mento, C. Psychedelic-Assisted Psychotherapy—A Systematic Review of Associated Psychological Interventions. Front. Psychol. 2022, 13, 887255. [Google Scholar] [CrossRef]
- Banushi, B.; Polito, V. A Comprehensive Review of the Current Status of the Cellular Neurobiology of Psychedelics. Biology 2023, 12, 1380. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Garg, N.J. Sirtuin Control of Mitochondrial Dysfunction, Oxidative Stress, and Inflammation in Chagas Disease Models. Front. Cell Infect. Microbiol. 2021, 11, 693051. [Google Scholar] [CrossRef]
- Fanibunda, S.E.; Deb, S.; Maniyadath, B.; Tiwari, P.; Ghai, U.; Gupta, S.; Figueiredo, D.; Weisstaub, N.; Gingrich, J.A.; Vaidya, A.D.B.; et al. Serotonin Regulates Mitochondrial Biogenesis and Function in Rodent Cortical Neurons via the 5-HT2A Receptor and SIRT1–PGC-1α Axis. Proc. Natl. Acad. Sci. USA 2019, 166, 11028–11037. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Owens, G.C.; Crossin, K.L.; Edelman, D.B. Serotonin Stimulates Mitochondrial Transport in Hippocampal Neurons. Mol. Cell Neurosci. 2007, 36, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Kohler, O.; Krogh, J.; Mors, O.; Eriksen Benros, M. Inflammation in Depression and the Potential for Anti-Inflammatory Treatment. Curr. Neuropharmacol. 2016, 14, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. 2011, 10, 655. [Google Scholar]
- Smith, C.J.; Zhang, Y.; Koboldt, C.M.; Muhammad, J.; Zweifel, B.S.; Shaffer, A.; Talley, J.J.; Masferrer, J.L.; Seibert, K.; Isakson, P.C. Pharmacological Analysis of Cyclooxygenase-1 in Inflammation. Proc. Natl. Acad. Sci. USA 1998, 95, 13313. [Google Scholar] [CrossRef] [PubMed]
- Sethi, R.; Gómez-Coronado, N.; Walker, A.J.; Robertson, O.D.; Agustini, B.; Berk, M.; Dodd, S. Neurobiology and Therapeutic Potential of Cyclooxygenase-2 (COX-2) Inhibitors for Inflammation in Neuropsychiatric Disorders. Front Psychiatry 2019, 10, 605. [Google Scholar] [CrossRef]
- Novakova, I.; Subileau, E.A.; Toegel, S.; Gruber, D.; Lachmann, B.; Urban, E.; Chesne, C.; Noe, C.R.; Neuhaus, W. Transport Rankings of Non-Steroidal Antiinflammatory Drugs across Blood-Brain Barrier In Vitro Models. PLoS ONE 2014, 9, e86806. [Google Scholar] [CrossRef]
- Iyengar, R.L.; Gandhi, S.; Aneja, A.; Thorpe, K.; Razzouk, L.; Greenberg, J.; Mosovich, S.; Farkouh, M.E. NSAIDs Are Associated with Lower Depression Scores in Patients with Osteoarthritis. Am. J. Med. 2013, 126, 1017.e11–1017.e18. [Google Scholar] [CrossRef]
- Köhler-Forsberg, O.; Lydholm, C.N.; Hjorthøj, C.; Nordentoft, M.; Mors, O.; Benros, M.E. Efficacy of Anti-Inflammatory Treatment on Major Depressive Disorder or Depressive Symptoms: Meta-Analysis of Clinical Trials. Acta Psychiatr. Scand. 2019, 139, 404–419. [Google Scholar] [CrossRef]
- Müller, N.; Schwarz, M.J. The Immune-Mediated Alteration of Serotonin and Glutamate: Towards an Integrated View of Depression. Mol. Psychiatry 2007, 12, 988–1001. [Google Scholar] [CrossRef]
- He, Y.; Han, Y.; Liao, X.; Zou, M.; Wang, Y. Biology of Cyclooxygenase-2: An Application in Depression Therapeutics. Front. Psychiatry 2022, 13, 1037588. [Google Scholar] [CrossRef]
- Larrea, A.; Elexpe, A.; Díez-Martín, E.; Torrecilla, M.; Astigarraga, E.; Barreda-Gómez, G. Neuroinflammation in the Evolution of Motor Function in Stroke and Trauma Patients: Treatment and Potential Biomarkers. Curr. Issues Mol. Biol. 2023, 45, 8552–8585. [Google Scholar] [CrossRef]
- Harsanyi, S.; Kupcova, I.; Danisovic, L.; Klein, M. Selected Biomarkers of Depression: What Are the Effects of Cytokines and Inflammation? Int. J. Mol. Sci. 2023, 24, 578. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732. [Google Scholar] [CrossRef]
- Uzzan, S.; Azab, A.N. Anti-TNF-α Compounds as a Treatment for Depression. Molecules 2021, 26, 2368. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Górniak, B.; Limphaibool, N.; Puszczewicz, M. Cytokine Secretion and the Risk of Depression Development in Patients with Connective Tissue Diseases. Psychiatry Clin. Neurosci. 2019, 73, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603. [Google Scholar] [CrossRef]
- Calvo-Rodríguez, M.; Núñez, L.; Villalobos, C. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Neuroprotection in the Elderly: A View from the Mitochondria. Neural Regen. Res. 2015, 10, 1371. [Google Scholar] [CrossRef]
- Elexpe, A.; Sánchez-Sánchez, L.; Tolentino-Cortez, T.; Astigarraga, E.; Torrecilla, M.; Barreda-Gómez, G. Analysis of Mitochondrial Function in Cell Membranes as Indicator of Tissue Vulnerability to Drugs in Humans. Biomedicines 2022, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Uppala, R.; Dudiak, B.; Beck, M.E.; Bharathi, S.S.; Zhang, Y.; Stolz, D.B.; Goetzman, E.S. Aspirin Increases Mitochondrial Fatty Acid Oxidation. Biochem. Biophys. Res. Commun. 2017, 482, 346. [Google Scholar] [CrossRef]
- Somaa, F.A.; de Graaf, T.A.; Sack, A.T. Transcranial Magnetic Stimulation in the Treatment of Neurological Diseases. Front. Neurol. 2022, 13, 793253. [Google Scholar] [CrossRef]
- Hallett, M. Transcranial Magnetic Stimulation and the Human Brain. Nature 2000, 406, 147–150. [Google Scholar] [CrossRef]
- Groppa, S.; Oliviero, A.; Eisen, A.; Quartarone, A.; Cohen, L.G.; Mall, V.; Kaelin-Lang, A.; Mima, T.; Rossi, S.; Thickbroom, G.W.; et al. A Practical Guide to Diagnostic Transcranial Magnetic Stimulation: Report of an IFCN Committee. Clin. Neurophysiol. 2012, 123, 858. [Google Scholar] [CrossRef]
- Herwig, U.; Satrapi, P.; Schönfeldt-Lecuona, C. Using the International 10-20 EEG System for Positioning of Transcranial Magnetic Stimulation. Brain Topogr. 2003, 16, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Khoshnevisan, A.; Allahabadi, N.S. Neuronavigation: Principles, Clinical Applications and Potential Pitfalls. Iran. J. Psychiatry 2012, 7, 97. [Google Scholar]
- Bogdanov, M.; Schwabe, L. Transcranial Stimulation of the Dorsolateral Prefrontal Cortex Prevents Stress-Induced Working Memory Deficits. J. Neurosci. 2016, 36, 1429. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, J.; Guo, W. Emotional Roles of Mono-Aminergic Neurotransmitters in Major Depressive Disorder and Anxiety Disorders. Front. Psychol. 2018, 9, 412042. [Google Scholar] [CrossRef] [PubMed]
- Lev-Ran, S.; Shamay-Tsoory, S.G.; Zangen, A.; Levkovitz, Y. Transcranial Magnetic Stimulation of the Ventromedial Prefrontal Cortex Impairs Theory of Mind Learning. Eur. Psychiatry 2012, 27, 285–289. [Google Scholar] [CrossRef]
- Boes, A.D.; Uitermarkt, B.D.; Albazron, F.M.; Lan, M.J.; Liston, C.; Pascual-Leone, A.; Dubin, M.J.; Fox, M.D. Rostral Anterior Cingulate Cortex Is a Structural Correlate of Repetitive TMS Treatment Response in Depression. Brain Stimul. 2018, 11, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.J.; Ebmeier, K.P.; Glabus, M.F.; Goodwin, G.M. Cortical Grey Matter Reductions Associated with Treatment-Resistant Chronic Unipolar Depression. Controlled Magnetic Resonance Imaging Study. Br. J. Psychiatry 1998, 172, 527–532. [Google Scholar] [CrossRef]
- Schiena, G.; Franco, G.; Boscutti, A.; Delvecchio, G.; Maggioni, E.; Brambilla, P. Connectivity Changes in Major Depressive Disorder after RTMS: A Review of Functional and Structural Connectivity Data. Epidemiol. Psychiatr. Sci. 2021, 30, e59. [Google Scholar] [CrossRef]
- Du, L.; Liu, H.; Du, W.; Chao, F.; Zhang, L.; Wang, K.; Huang, C.; Gao, Y.; Tang, Y. Stimulated Left DLPFC-Nucleus Accumbens Functional Connectivity Predicts the Anti-Depression and Anti-Anxiety Effects of RTMS for Depression. Transl. Psychiatry 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Chib, V.S.; Yun, K.; Takahashi, H.; Shimojo, S. Noninvasive Remote Activation of the Ventral Midbrain by Transcranial Direct Current Stimulation of Prefrontal Cortex. Transl. Psychiatry 2013, 3, e268. [Google Scholar] [CrossRef] [PubMed]
- Siebner, H.R.; Funke, K.; Aberra, A.S.; Antal, A.; Bestmann, S.; Chen, R.; Classen, J.; Davare, M.; Di Lazzaro, V.; Fox, P.T.; et al. Transcranial Magnetic Stimulation of the Brain: What Is Stimulated?—A Consensus and Critical Position Paper. Clin. Neurophysiol. 2022, 140, 59. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.; Hamid, U.; Jamil, A.; Zainab, A.Z.; Yousuf, T.; Habib, S.; Tariq, S.M.; Ali, F. Transcranial Magnetic Stimulation as a Therapeutic Option for Neurologic and Psychiatric Illnesses. Cureus 2018, 10, e3456. [Google Scholar] [CrossRef] [PubMed]
- Baeken, C.; De Raedt, R. Neurobiological Mechanisms of Repetitive Transcranial Magnetic Stimulation on the Underlying Neuro Circuitry in Unipolar Depression. Dialogues Clin. Neurosci. 2011, 13, 139. [Google Scholar] [CrossRef] [PubMed]
- Kricheldorff, J.; Göke, K.; Kiebs, M.; Kasten, F.H.; Herrmann, C.S.; Witt, K.; Hurlemann, R. Evidence of Neuroplastic Changes after Transcranial Magnetic, Electric, and Deep Brain Stimulation. Brain Sci. 2022, 12, 929. [Google Scholar] [CrossRef]
- Wang, H.Y.; Crupi, D.; Liu, J.; Stucky, A.; Cruciata, G.; di Rocco, A.; Friedman, E.; Quartarone, A.; Ghilardi, M.F. Repetitive Transcranial Magnetic Stimulation Enhances BDNF–TrkB Signaling in Both Brain and Lymphocyte. J. Neurosci. 2011, 31, 11044. [Google Scholar] [CrossRef]
- Pandey, G.N.; Sharma, A.; Rizavi, H.S.; Ren, X. Dysregulation of Protein Kinase C in Adult Depression and Suicide: Evidence from Postmortem Brain Studies. Int. J. Neuropsychopharmacol. 2021, 24, 400. [Google Scholar] [CrossRef]
- Huerta, P.T.; Volpe, B.T. Transcranial Magnetic Stimulation, Synaptic Plasticity and Network Oscillations. J. Neuroeng. Rehabil. 2009, 6, 7. [Google Scholar] [CrossRef]
- Bliss, T.V.P.; Cooke, S.F. Long-Term Potentiation and Long-Term Depression: A Clinical Perspective. Clinics 2011, 66, 3. [Google Scholar] [CrossRef]
- Lozano, A.M.; Lipsman, N.; Bergman, H.; Brown, P.; Chabardes, S.; Chang, J.W.; Matthews, K.; McIntyre, C.C.; Schlaepfer, T.E.; Schulder, M.; et al. Deep Brain Stimulation: Current Challenges and Future Directions. Nat. Rev. Neurol. 2019, 15, 148. [Google Scholar] [CrossRef]
- Torres, C.V.; Lozano, A.M. Deep Brain Stimulation in the Treatment of Therapy-Refractory Depression. Rev. Neurol. 2008, 47, 477–482. [Google Scholar] [CrossRef]
- Ranzcp. Deep Brain Stimulation (DBS) in Psychiatric Practice; Ranzcp: Melbourne, Australia, 2022. [Google Scholar]
- Sartorius, A.; Kiening, K.L.; Kirsch, P.; von Gall, C.C.; Haberkorn, U.; Unterberg, A.W.; Henn, F.A.; Meyer-Lindenberg, A. Remission of Major Depression Under Deep Brain Stimulation of the Lateral Habenula in a Therapy-Refractory Patient. Biol. Psychiatry 2010, 67, e9–e11. [Google Scholar] [CrossRef] [PubMed]
- Fenoy, A.J.; Schulz, P.; Selvaraj, S.; Burrows, C.; Spiker, D.; Cao, B.; Zunta-Soares, G.; Gajwani, P.; Quevedo, J.; Soares, J. Deep Brain Stimulation of the Medial Forebrain Bundle: Distinctive Responses in Resistant Depression. J. Affect. Disord. 2016, 203, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Khairuddin, S.; Ngo, F.Y.; Lim, W.L.; Aquili, L.; Khan, N.A.; Fung, M.L.; Chan, Y.S.; Temel, Y.; Lim, L.W. A Decade of Progress in Deep Brain Stimulation of the Subcallosal Cingulate for the Treatment of Depression. J. Clin. Med. 2020, 9, 3260. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, J.; Li, X.N.; Liang, J.; Song, L.; Wu, Y.; Liu, Z.; Sun, B.; Li, W.G. Neuronal and Synaptic Adaptations Underlying the Benefits of Deep Brain Stimulation for Parkinson’s Disease. Transl. Neurodegener. 2023, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, S.M.D.D.; Oostra, E.; Postma, T.S.; van der Werf, Y.D.; van den Heuvel, O.A. Repetitive Transcranial MagneticStimulation–Induced Neuroplasticity and the Treatment of Psychiatric Disorders: State of the Evidence and Future Opportunities. Biol Psychiatry 2024, 95, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Ji, Y.S.; Sun, X.L.; Liu, X.H.; Chen, Z.Y. Brain-Derived Neurotrophic Factor (BDNF)-Induced Mitochondrial Motility Arrest and Presynaptic Docking Contribute to BDNF-Enhanced Synaptic Transmission. J. Biol. Chem. 2014, 289, 1227. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, H.; Du, X.; Zhou, J.; Yuan, L.; Ren, H.; Yang, X.; Zhang, G.; Chen, X. Circulating Brain-Derived Neurotrophic Factor, Antioxidant Enzymes Activities, and Mitochondrial DNA in Bipolar Disorder: An Exploratory Report. Front. Psychiatry 2020, 11, 514658. [Google Scholar] [CrossRef]
- Toepp, S.L.; Turco, C.V.; Locke, M.B.; Nicolini, C.; Ravi, R.; Nelson, A.J. The Impact of Glucose on Corticospinal and Intracortical Excitability. Brain Sci. 2019, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Koy, A.; Cirak, S.; Gonzalez, V.; Becker, K.; Roujeau, T.; Milesi, C.; Baleine, J.; Cambonie, G.; Boularan, A.; Greco, F.; et al. Deep Brain Stimulation Is Effective in Pediatric Patients with GNAO1 Associated Severe Hyperkinesia. J. Neurol. Sci. 2018, 391, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, Z.; Su, Y.; Kang, L.; Geng, D.; Wang, Y.; Luan, F.; Wang, M.; Cui, H. Magnetic Stimulation Modulates Structural Synaptic Plasticity and Regulates BDNF–TrkB Signal Pathway in Cultured Hippocampal Neurons. Neurochem. Int. 2013, 62, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt Mediates Mitochondrial Protection in Cardiomyocytes through Phosphorylation of Mitochondrial Hexokinase-II. Cell Death Differ. 2007, 15, 521–529. [Google Scholar] [CrossRef]
- Yoshimura, R.; Hori, H.; Ikenouchi-Sugita, A.; Umene-Nakano, W.; Ueda, N.; Nakamura, J. Higher Plasma Interleukin-6 (IL-6) Level Is Associated with SSRI- or SNRI-Refractory Depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 722–726. [Google Scholar] [CrossRef]
- Yavi, M.; Lee, H.; Henter, I.D.; Park, L.T.; Zarate, C.A. Ketamine Treatment for Depression: A Review. Discover Mental Health 2022, 2, 9. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, Y.; Zhou, H.; Lu, H.; Zhang, Y.; Hua, J.; Liao, X. Esketamine Is Neuroprotective against Traumatic Brain Injury through Its Modulation of Autophagy and Oxidative Stress via AMPK/MTOR-Dependent TFEB Nuclear Translocation. Exp. Neurol. 2023, 366, 114436. [Google Scholar] [CrossRef]
- Morishita, T.; Fayad, S.M.; Higuchi, M.A.; Nestor, K.A.; Foote, K.D. Deep Brain Stimulation for Treatment-Resistant Depression: Systematic Review of Clinical Outcomes. Neurotherapeutics 2014, 11, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A. Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Area | Localization | Function | |
|---|---|---|---|
| Ventromedial Prefrontal Cortex | Subcortical region | Involved in the evaluation of rewards and punishments | [131] |
| Anterior Cingulate Gyrus | Frontal lobe | Involved in the regulation of emotional and stress responses | [132] |
| Dorsolateral Parietal Lobe | Parietal lobe | Involved in attention and working memory | [133] |
| Posterior Cingulate Cortex | Posterior region | Involved in the regulation of pain and emotion responses | [134] |
| Nucleus Accumbens | Subcortical region | Involved in the reward system, related to anhedonia | [135] |
| Ventral Tegmental Area | Brainstem | Associated with dopamine release; involved in the regulation of motivation and pleasure | [136] |
| Mayor Depressive Disorder | |
|---|---|
| Strengths | Opportunities |
|
|
| Weaknesses | Threats |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larrea, A.; Sánchez-Sánchez, L.; Diez-Martin, E.; Elexpe, A.; Torrecilla, M.; Astigarraga, E.; Barreda-Gómez, G. Mitochondrial Metabolism in Major Depressive Disorder: From Early Diagnosis to Emerging Treatment Options. J. Clin. Med. 2024, 13, 1727. https://doi.org/10.3390/jcm13061727
Larrea A, Sánchez-Sánchez L, Diez-Martin E, Elexpe A, Torrecilla M, Astigarraga E, Barreda-Gómez G. Mitochondrial Metabolism in Major Depressive Disorder: From Early Diagnosis to Emerging Treatment Options. Journal of Clinical Medicine. 2024; 13(6):1727. https://doi.org/10.3390/jcm13061727
Chicago/Turabian StyleLarrea, Ane, Laura Sánchez-Sánchez, Eguzkiñe Diez-Martin, Ane Elexpe, María Torrecilla, Egoitz Astigarraga, and Gabriel Barreda-Gómez. 2024. "Mitochondrial Metabolism in Major Depressive Disorder: From Early Diagnosis to Emerging Treatment Options" Journal of Clinical Medicine 13, no. 6: 1727. https://doi.org/10.3390/jcm13061727
APA StyleLarrea, A., Sánchez-Sánchez, L., Diez-Martin, E., Elexpe, A., Torrecilla, M., Astigarraga, E., & Barreda-Gómez, G. (2024). Mitochondrial Metabolism in Major Depressive Disorder: From Early Diagnosis to Emerging Treatment Options. Journal of Clinical Medicine, 13(6), 1727. https://doi.org/10.3390/jcm13061727