
Fibrosis in Chronic Kidney Disease: Pathophysiology and Therapeutic Targets

 ,
,  ,
,  and
and
Abstract
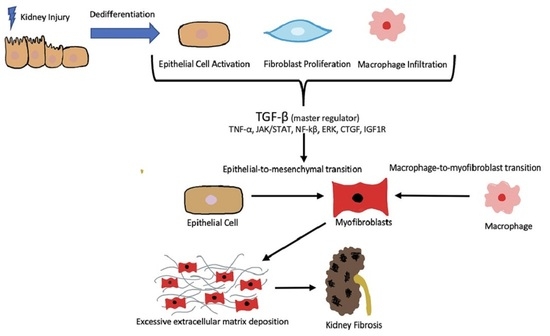
:
1. Introduction
2. Fibrosis in Chronic Kidney Disease: Overview
3. Clinical Assessment of Kidney Fibrosis
3.1. Kidney Biopsy
3.2. Imaging
3.2.1. Diffusion-Weighted Magnetic Resonance Imaging
3.2.2. Shear Wave Elastography
3.2.3. Biomarkers of Kidney Fibrosis
4. Pathophysiology of Fibrosis in the Setting of Chronic Kidney Disease
4.1. The Fibrotic Niche and the Profibrotic Microenvironment
4.2. Cellular Mechanisms—Dysregulation of TGF-B/Smad in Promoting Renal Fibrosis
4.3. Diabetes
4.4. Epithelial–Mesenchymal Transition and the Macrophage
4.5. ICAM-1-Induced Epithelial–Mesenchymal Transition
4.6. Serum Amyloid a and Chronic Kidney Disease
5. Nephroprotective Drugs in Current Use
5.1. Renin-Angiotensin Blockers
5.2. Mineralocorticoid Receptor Antagonists
5.3. Sodium/Glucose Cotransporter 2 Inhibitors
6. Antifibrotic Drugs in Clinical Trials
6.1. Pirfenidone
6.2. Silencing of MicroRNA-21 and Lademirsen
7. Future Treatments and Potential Approaches
7.1. Strategies Targeting TGF-β
7.2. RNA-Based Therapeutics
7.3. Utilizing Transcriptional Regulators Snail1 and Twist1 as Therapeutic Targets
7.4. Mesenchymal Stem Cells
7.5. Src Family Kinases
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.C.; Zhang, L.X. Prevalence and disease burden of chronic kidney disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Jafar, T.H.; Nitsch, D.; Neuen, B.L.; Perkovic, V. Chronic kidney disease. Lancet 2021, 398, 786–802. [Google Scholar] [CrossRef] [PubMed]
- Rhee, C.; Kovesdy, C. Spotlight on CKD deaths—Increasing mortality worldwide. Nat. Rev. Nephrol. 2015, 11, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Tang, S.C.; Kashihara, N.; Kim, Y.S.; Togtokh, A.; Yang, C.W.; Zhao, M.H.; ISN North and East Asia Regional Board. Disease burden and challenges of chronic kidney disease in North and East Asia. Kidney Int. 2018, 94, 22–25. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Escribá-Martí, G.; Cámara-Ramos, I.; Climent-Catalá, M.T.; Escudero-Quesada, V.; Salar-Ibáñez, L. Pharmaceutical care program for patients with chronic kidney disease in the community pharmacy: Detection of nephrotoxic drugs and dose adjustment. Viability study. PLoS ONE 2022, 17, e0278648. [Google Scholar] [CrossRef]
- Düsing, P.; Zietzer, A.; Goody, P.R.; Hosen, M.R.; Kurts, C.; Nickenig, G.; Jansen, F. Vascular pathologies in chronic kidney disease: Pathophysiological mechanisms and novel therapeutic approaches. J. Mol. Med. 2021, 99, 335–348. [Google Scholar] [CrossRef]
- Charles, C.; Ferris, A.H. Chronic Kidney Disease. Prim. Care 2020, 47, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Mone, P.; Jankauskas, S.S.; Gambardella, J.; Santulli, G. Chronic kidney disease: Definition, updated epidemiology, staging, and mechanisms of increased cardiovascular risk. J. Clin. Hypertens. 2021, 23, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Akchurin, O.M. Chronic Kidney Disease and Dietary Measures to Improve Outcomes. Pediatr. Clin. N. Am. 2019, 66, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Martínez-Arias, L.; Alonso-Montes, C.; Cannata, P.; Martín-Carro, B.; Fernández-Martín, J.L.; Naves-Díaz, M.; Carrillo-López, N.; Cannata-Andía, J.B. Fibrosis in Chronic Kidney Disease: Pathogenesis and Consequences. Int. J. Mol. Sci. 2021, 22, 408. [Google Scholar] [CrossRef] [PubMed]
- Vanhove, T.; Goldschmeding, R.; Kuypers, D. Kidney Fibrosis: Origins and Interventions. Transplantation 2017, 101, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Genovese, F.; Manresa, A.A.; Leeming, D.J.; Karsdal, M.A.; Boor, P. The extracellular matrix in the kidney: A source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair 2014, 7, 4. [Google Scholar] [CrossRef]
- Cruz-Solbes, A.S.; Youker, K. Epithelial to Mesenchymal Transition (EMT) and Endothelial to Mesenchymal Transition (EndMT): Role and Implications in Kidney Fibrosis. Results Probl. Cell Differ. 2017, 60, 345–372. [Google Scholar] [CrossRef] [PubMed]
- Leung, G.; Kirpalani, A.; Szeto, S.G.; Deeb, M.; Foltz, W.; Simmons, C.A.; Yuen, D.A. Could MRI Be Used To Image Kidney Fibrosis? A Review of Recent Advances and Remaining Barriers. Clin. J. Amer. Soc. Nephrol. 2017, 12, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Palsson, R.; Kaze, A.D.; Chen, M.E.; Palacios, P.; Sabbisetti, V.; Betensky, R.A.; Steinman, T.I.; Thadhani, R.I.; McMahon, G.M.; et al. The Prognostic Value of Histopathologic Lesions in Native Kidney Biopsy Specimens: Results from the Boston Kidney Biopsy Cohort Study. J. Am. Soc. Nephrol. 2018, 29, 2213–2224. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Mimura, I.; Shoji, K.; Tanaka, T.; Nangaku, M. Hypoxia and fibrosis in chronic kidney disease: Crossing at pericytes. Kidney Int. Suppl. 2014, 4, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.V.; Li, L.P.; Thacker, J.M.; Li, W.; Hack, B.; Kohn, O.; Sprague, S.M. Cortical Perfusion and Tubular Function as Evaluated by Magnetic Resonance Imaging Correlates with Annual Loss in Renal Function in Moderate Chronic Kidney Disease. Am. J. Nephrol. 2019, 49, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Kanellis, J.; Hugo, C.; Truong, L.; Anderson, S.; Kerjaschki, D.; Schreiner, G.F.; Johnson, R.J. Role of the microvascular endothelium in progressive renal disease. J. Am. Soc. Nephrol. 2002, 13, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Fine, L.G.; Norman, J.T. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: From hypothesis to novel therapeutics. Kidney Int. 2008, 74, 867–872. [Google Scholar] [CrossRef]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ are mechanoregulators of TGF-β-Smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef]
- Chen, W.C.; Lin, H.H.; Tang, M.J. Regulation of Proximal Tubular Cell Differentiation and Proliferation in Primary Culture by Matrix Stiffness and ECM Components. Am. J. Physiol. Renal Physiol. 2014, 307, F695–F707. [Google Scholar] [CrossRef]
- Gewin, L. The many talents of transforming growth factor-β in the kidney. Curr. Opin. Nephrol. Hypertens. 2019, 28, 203–210. [Google Scholar] [CrossRef]
- Georges, P.C.; Hui, J.J.; Gombos, Z.; McCormick, M.E.; Wang, A.Y.; Uemura, M.; Mick, R.; Janmey, P.A.; Furth, E.E.; Wells, R.G. Increased stiffness of the rat liver precedes matrix deposition: Implications for fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1147–G1154. [Google Scholar] [CrossRef]
- Chade, A.R. Renal vascular structure and rarefaction. Compr. Physiol. 2013, 3, 817–831. [Google Scholar] [CrossRef]
- Jiang, K.; Ferguson, C.M.; Lerman, L.O. Noninvasive assessment of renal fibrosis by magnetic resonance imaging and ultrasound techniques. Transl. Res. 2019, 209, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Street, J.M.; Souza, A.C.; Alvarez-Prats, A.; Horino, T.; Hu, X.; Yuen, P.S.; Star, R.A. Automated quantification of renal fibrosis with Sirius Red and polarization contrast microscopy. Physiol. Rep. 2014, 2, e12088. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yang, X.; Zhang, M.; Zhou, W.; Jin, Y.; Zhou, H.; Zhou, Y.; Wang, Q.; Mou, S. Effects of receiving renal biopsy on the prognosis of chronic kidney disease patients with impaired renal function. BMC Nephrol. 2023, 24, 56. [Google Scholar] [CrossRef] [PubMed]
- Luciano, R.L.; Moeckel, G.W. Update on the Native Kidney Biopsy: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 73, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Nissen, C.J.; Moreno, V.; Davis, V.G.; Walker, P.D. Increasing Incidence of Inadequate Kidney Biopsy Samples Over Time: A 16-Year Retrospective Analysis From a Large National Renal Biopsy Laboratory. Kidney Int. Rep. 2021, 7, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Whittier, W.L.; Korbet, S.M. Timing of complications in percutaneous renal biopsy. J. Am. Soc. Nephrol. 2004, 15, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Brachemi, S.; Bollée, G. Renal biopsy practice: What is the gold standard? World J. Nephrol. 2014, 3, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Khosroshahi, H.T.; Abedi, B.; Daneshvar, S.; Sarbaz, Y.; Shakeri Bavil, A. Future of the Renal Biopsy: Time to Change the Conventional Modality Using Nanotechnology. Int. J. Biomed. Imaging 2017, 2017, 6141734. [Google Scholar] [CrossRef]
- Bülow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef]
- Zhan, T.; Lou, A. Comparison of outcomes of an 18-gauge vs 16-gauge ultrasound-guided percutaneous renal biopsy: A systematic review and meta-analysis. Ren. Fail. 2023, 45, 2257806. [Google Scholar] [CrossRef]
- Lim, C.Y.; Khay, S.L. Bleeding complications after percutaneous kidney biopsies—Nationwide experience from Brunei Darussalam. World J. Nephrol. 2023, 12, 147–158. [Google Scholar] [CrossRef]
- Ge, X.Y.; Lan, Z.K.; Lan, Q.Q.; Lin, H.S.; Wang, G.D.; Chen, J. Diagnostic accuracy of ultrasound-based multimodal radiomics modeling for fibrosis detection in chronic kidney disease. Eur. Radiol. 2023, 33, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Lan, Z.K.; Lan, Q.Q.; Lin, H.S.; Wang, G.D.; Chen, J. Usefulness of urinary biomarkers to estimate the interstitial fibrosis surface in diabetic nephropathy with normal kidney function. Nephrol. Dial. Transplant. 2022, 37, 2102–2110. [Google Scholar] [CrossRef]
- Srivastava, A.; Tomar, B.; Prajapati, S.; Gaikwad, A.B.; Mulay, S.R. Advanced non-invasive diagnostic techniques for visualization and estimation of kidney fibrosis. Drug Discov. Today 2021, 26, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Hysi, E.; Yuen, D.A. Imaging of renal fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, Y.; Liu, X.; Tang, X.; Xu, F.; Zhang, M.; Xie, G.; Zhang, L.; Li, X.; Liu, Z.H. Evaluation of renal fibrosis by mapping histology and magnetic resonance imaging. Kidney Dis. 2021, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, B.; Merville, P.; Soulabaille, B.; Taton, B.; Kaminski, H.; Visentin, J.; Vermorel, A.; Bouzgarrou, M.; Couzi, L.; Grenier, N. Magnetic Resonance Elastography as Surrogate Marker of Interstitial Fibrosis in Kidney Transplantation: A Prospective Study. Kidney360 2022, 3, 1924–1933. [Google Scholar] [CrossRef]
- Makvandi, K.; Hockings, P.D.; Jensen, G.; Unnerstall, T.; Leonhardt, H.; Jarl, L.V.; Englund, C.; Francis, S.; Sundgren, A.K.; Hulthe, J.; et al. Multiparametric magnetic resonance imaging allows non-invasive functional and structural evaluation of diabetic kidney disease. Clin. Kidney J. 2022, 15, 1387–1402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, L.J. Functional MRI as a Tool for Evaluating Interstitial Fibrosis and Prognosis in Kidney Disease. Kidney Dis. 2020, 6, 7–12. [Google Scholar] [CrossRef]
- Ahmed, S.; Bughio, S.; Hassan, M.; Lal, S.; Ali, M. Role of Ultrasound in the Diagnosis of Chronic Kidney Disease and its Correlation with Serum Creatinine Level. Cureus 2019, 11, e4241. [Google Scholar] [CrossRef]
- Singla, R.K.; Kadatz, M.; Rohling, R.; Nguan, C. Kidney Ultrasound for Nephrologists: A Review. Kidney Med. 2022, 4, 100464. [Google Scholar] [CrossRef]
- Fiorini, F.; Barozzi, L. The role of ultrasonography in the study of medical nephropathy. J. Ultrasound 2007, 10, 161–167. [Google Scholar] [CrossRef]
- Leong, S.S.; Wong, J.H.D.; Md Shah, M.N.; Vijayananthan, A.; Jalalonmuhali, M.; Ng, K.H. Shear wave elastography in the evaluation of renal parenchymal stiffness in patients with chronic kidney disease. Br. J. Radiol. 2018, 91, 20180235. [Google Scholar] [CrossRef]
- Michaely, H.J.; Metzger, L.; Haneder, S.; Hansmann, J.; Schoenberg, S.O.; Attenberger, U.I. Renal BOLD-MRI does not reflect renal function in chronic kidney disease. Kidney Int. 2012, 81, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Andersen, U.B.; Haddock, B.; Asmar, A. Multiparametric magnetic resonance imaging: A robust tool to test pathogenesis and pathophysiology behind nephropathy in humans. Clin. Physiol. Funct. Imaging 2023, 43, 207–210. [Google Scholar] [CrossRef]
- Thiravit, S.; Suwanchatree, P.; Skulratanasak, P.; Thiravit, P.; Suvannarerg, V. Correlation between Apparent Diffusion Coefficient Values of the Renal Parenchyma and Estimated Glomerular Filtration Rates on 3-T Diffusion-Weighted Echo-Planar Magnetic Resonance Imaging. J. Comput. Assist. Tomogr. 2019, 43, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, C.M.; Eirin, A.; Abumoawad, A.; Saad, A.; Jiang, K.; Hedayat, A.F.; Misra, S.; Glockner, J.; Textor, S.C.; Lerman, L.O. Renal fibrosis detected by diffusion-weighted magnetic resonance imaging remains unchanged despite treatment in subjects with renovascular disease. Sci. Rep. 2020, 10, 16300. [Google Scholar] [CrossRef]
- Li, J.; An, C.; Kang, L.; Mitch, W.E.; Wang, Y. Recent Advances in Magnetic Resonance Imaging Assessment of Renal Fibrosis. Adv. Chronic Kidney Dis. 2017, 24, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Baliyan, V.; Das, C.J.; Sharma, R.; Gupta, A.K. Diffusion weighted imaging: Technique and applications. World J. Radiol. 2016, 8, 785–798. [Google Scholar] [CrossRef]
- De Perrot, T.; Sadjo Zoua, C.; Glessgen, C.G.; Botsikas, D.; Berchtold, L.; Salomir, R.; De Seigneux, S.; Thoeny, H.C.; Vallée, J.P. Diffusion-Weighted MRI in the Genitourinary System. J. Clin. Med. 2022, 11, 1921. [Google Scholar] [CrossRef]
- Togao, O.; Doi, S.; Kuro-o, M.; Masaki, T.; Yorioka, N.; Takahashi, M. Assessment of renal fibrosis with diffusion-weighted MR imaging: Study with murine model of unilateral ureteral obstruction. Radiology 2010, 255, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, Z.J.; Liu, M.; Zhu, J.; Zhang, X.; Zhang, T.; Li, S.; Li, Y. Assessment of renal fibrosis in chronic kidney disease using diffusion-weighted MRI. Clin. Radiol. 2014, 69, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Morrell, G.R.; Zhang, J.L.; Lee, V.S. Magnetic Resonance Imaging of the Fibrotic Kidney. J. Am. Soc. Nephrol. 2017, 28, 2564–2570. [Google Scholar] [CrossRef] [PubMed]
- Sigmund, E.E.; Vivier, P.H.; Sui, D.; Lamparello, N.A.; Tantillo, K.; Mikheev, A.; Rusinek, H.; Babb, J.S.; Storey, P.; Lee, V.S.; et al. Intravoxel incoherent motion and diffusion-tensor imaging in renal tissue under hydration and furosemide flow challenges. Radiology 2012, 263, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Caroli, A.; Schneider, M.; Friedli, I.; Ljimani, A.; De Seigneux, S.; Boor, P.; Gullapudi, L.; Kazmi, I.; Mendichovszky, I.A.; Notohamiprodjo, M.; et al. Diffusion-weighted magnetic resonance imaging to assess diffuse renal pathology: A systematic review and statement paper. Nephrol. Dial. Transplant. 2018, 33, ii29–ii40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Schwartz, M.; Küstner, T.; Martirosian, P.; Seith, F. Multiparametric Functional MRI of the Kidney: Current State and Future Trends with Deep Learning Approaches. Multiparametrische funktionelle Nierenbildgebung in der MRT: Aktueller Status und zukunftsweisende Entwicklungen mit Deep Learning. Rofo 2022, 194, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Ding, X.; Ding, Y.; Cao, B.; Fu, C.; Kuehn, B.; Benkert, T.; Grimm, R.; Nickel, D.; Zhou, J.; et al. Evaluation of interstitial fibrosis in chronic kidney disease by multiparametric functional MRI and histopathologic analysis. Eur. Radiol. 2022, 33, 4138–4147. [Google Scholar] [CrossRef]
- Mo, X.L.; Meng, H.Y.; Wu, Y.Y.; Wei, X.Y.; Li, Z.K.; Yang, S.Q. Shear Wave Elastography in the Evaluation of Renal Parenchymal Stiffness in Patients With Chronic Kidney Disease: A Meta-Analysis. J. Clin. Med. Res. 2022, 14, 95–105. [Google Scholar] [CrossRef]
- Desvignes, C.; Dabadie, A.; Aschero, A.; Ruocco, A.; Garaix, F.; Daniel, L.; Ferlicot, S.; Villes, V.; Loundou, A.D.; Gorincour, G.; et al. Technical feasibility and correlations between shear-wave elastography and histology in kidney fibrosis in children. Pediatr. Radiol. 2021, 51, 1879–1888. [Google Scholar] [CrossRef]
- Iyama, T.; Sugihara, T.; Takata, T.; Isomoto, H. Renal Ultrasound Elastography: A Review of the Previous Reports on Chronic Kidney Diseases. Appl. Sci. 2021, 11, 9677. [Google Scholar] [CrossRef]
- Lee, A.; Joo, D.J.; Han, W.K.; Jeong, H.J.; Oh, M.J.; Kim, Y.S.; Oh, Y.T. Renal tissue elasticity by acoustic radiation force impulse: A prospective study of healthy kidney donors. Medicine 2021, 100, e23561. [Google Scholar] [CrossRef] [PubMed]
- Samir, A.E.; Allegretti, A.S.; Zhu, Q.; Dhyani, M.; Anvari, A.; Sullivan, D.A.; Trottier, C.A.; Dougherty, S.; Williams, W.W.; Babitt, J.L.; et al. Shear wave elastography in chronic kidney disease: A pilot experience in native kidneys. BMC Nephrol. 2015, 16, 119. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, C.; Li, J.; Huo, H.; Song, D. Diagnostic Accuracy of Real-Time Shear Wave Elastography for Staging of Liver Fibrosis: A Meta-Analysis. Med. Sci. Monit. 2016, 22, 1349–1359. [Google Scholar] [CrossRef]
- Bob, F.; Grosu, I.; Sporea, I.; Bota, S.; Popescu, A.; Sirli, R.; Petrica, L.; Schiller, A. Is there a correlation between kidney shear wave velocity measured with VTQ and histological parameters in patients with chronic glomerulonephritis? A pilot study. Med. Ultrason. 2018, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.T.H.; Ooi, E.H.; Foo, J.J.; Ng, K.H.; Wong, J.H.D.; Leong, S.S. Shear Wave Elastography: A Review on the Confounding Factors and Their Potential Mitigation in Detecting Chronic Kidney Disease. Ultrasound Med. Biol. 2021, 47, 2033–2047. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Kim, S.I.; Choi, M.E. Therapeutic targets for treating fibrotic kidney diseases. Transl. Res. 2015, 165, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Akdeniz, A.; Weitz, S.; Usinger, W.R.; Molineaux, C.; Jones, S.E.; Langham, R.G.; Jerums, G. Urinary connective tissue growth factor excretion in patients with type 1 diabetes and nephropathy. Diabetes Care 2003, 26, 2632–2636. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, S.; Gao, H.; Yang, Y.; Liu, X.; Li, W. Factors associated with the progression of mesangial lesions in IgA nephropathy: A comparative analysis of renal re-biopsies. Front. Endocrinol. 2022, 13, 1004289. [Google Scholar] [CrossRef]
- Shukla, N.; Kumari, S.; Verma, P.; Kushwah, A.S.; Banarjee, M.; Sankhwar, S.N.; Srivastava, A.; Ansari, M.S.; Gautam, N.K. Genotypic Analysis of COL4A1 Gene in Diabetic Nephropathy and Type 2 Diabetes Mellitus Patients: A Comparative Genetic Study. DNA Cell Biol. 2023, 42, 541–547. [Google Scholar] [CrossRef]
- Neprasova, M.; Maixnerova, D.; Sparding, N.; Genovese, F.; Karsdal, M.A.; Koprivova, H.; Kollar, M.; Suchanek, M.; Hruskova, Z.; Tesar, V. Serum and Urine Biomarkers Related to Kidney Fibrosis Predict Kidney Outcome in Czech Patients with IgA Nephropathy. Int. J. Mol. Sci. 2023, 24, 2064. [Google Scholar] [CrossRef]
- Genovese, F.; Kring, D.; Rasmussen, G.; Karsdal, M.A.; Jesky, M.A.; Ferro, C.; Fenton, A.; Cockwell, P. Imbalanced turnover of collagen type III is associated with disease progression and mortality in high-risk chronic kidney disease patients. Clin. Kidney J. 2021, 14, 593–601. [Google Scholar] [CrossRef]
- Genovese, F.; Boor, P.; Papasotiriou, M.; Leeming, D.J.; Karsdal, M.A.; Floege, J. Turnover of type III collagen reflects disease severity and is associated with progression and microinflammation in patients with IgA nephropathy. Nephrol. Dial. Transpl. 2016, 31, 472–479. [Google Scholar] [CrossRef]
- Melchinger, H.; Calderon-Gutierrez, F.; Obeid, W.; Xu, L.; Shaw, M.M.; Luciano, R.L.; Kuperman, M.; Moeckel, G.W.; Kashgarian, M.; Wilson, F.P.; et al. Urine Uromodulin as a Biomarker of Kidney Tubulointerstitial Fibrosis. Clin. J. Am. Soc. Nephrol. 2022, 17, 1284–1292. [Google Scholar] [CrossRef]
- LaFavers, K.; Garimella, P.S. Uromodulin: More than a marker for chronic kidney disease progression. Curr. Opin. Nephrol. Hypertens. 2023, 32, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.G.; Puthumana, J.; Coca, S.G.; Gentry, M.; Parikh, C.R. Biomarkers for the detection of renal fibrosis and prediction of renal outcomes: A systematic review. BMC Nephrol. 2017, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Bagnasco, S.M.; Rosenberg, A.Z. Biomarkers of Chronic Renal Tubulointerstitial Injury. Histochem. Cytochem. 2019, 67, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Traykova-Brauch, M.; Schönig, K.; Greiner, O.; Miloud, T.; Jauch, A.; Bode, M.; Felsher, D.; Glick, A.; Kwiatowski, D.; Bujard, H.; et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat. Med. 2008, 14, 979–984. [Google Scholar] [CrossRef]
- Yuan, Q.; Ren, Q.; Li, L.; Tan, H.; Lu, M.; Tian, Y.; Huang, L.; Zhao, B.; Fu, H.; Hou, F.F.; et al. A Klotho-derived peptide protects against kidney fibrosis by targeting TGF-β signaling. Nat. Commun. 2022, 13, 438. [Google Scholar] [CrossRef]
- Kim, S.H.; Jin, J.A.; So, H.J.; Lee, S.H.; Kang, T.W.; Lee, J.U.; Choi, D.E.; Jeong, J.Y.; Chang, Y.K.; Choi, H.; et al. Urine-Derived Stem Cell-Secreted Klotho Plays a Crucial Role in the HK-2 Fibrosis Model by Inhibiting the TGF-β Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 5012. [Google Scholar] [CrossRef]
- Bullen, A.L.; Katz, R.; Jotwani, V.; Garimella, P.S.; Lee, A.K.; Estrella, M.M.; Shlipak, M.G.; Ix, J.H. Biomarkers of Kidney Tubule Health, CKD Progression, and Acute Kidney Injury in SPRINT (Systolic Blood Pressure Intervention Trial) Participants. Am. J. Kidney Dis. 2021, 78, 361–368.e1. [Google Scholar] [CrossRef]
- Puthumana, J.; Thiessen-Philbrook, H.; Xu, L.; Coca, S.G.; Garg, A.X.; Himmelfarb, J.; Bhatraju, P.K.; Ikizler, T.A.; Siew, E.D.; Ware, L.B.; et al. Biomarkers of inflammation and repair in kidney disease progression. J. Clin. Investig. 2021, 131, e139927. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Shimizu, A.; Masuda, Y.; Kuwahara, N.; Arai, T.; Kataoka, M.; Uchiyama, M.; Kaneko, T.; Akimoto, T.; Iino, Y.; et al. Involvement of matrix metalloproteinase-2 in the development of renal interstitial fibrosis in mouse obstructive nephropathy. Lab. Investig. 2012, 92, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Zhuang, S. New Insights Into the Role and Mechanism of Partial Epithelial-Mesenchymal Transition in Kidney Fibrosis. Front. Physiol. 2020, 11, 569322. [Google Scholar] [CrossRef]
- Kato, N.; Kosugi, T.; Sato, W.; Ishimoto, T.; Kojima, H.; Sato, Y.; Sakamoto, K.; Maruyama, S.; Yuzawa, Y.; Matsuo, S.; et al. Basigin/CD147 promotes renal fibrosis after unilateral ureteral obstruction. Am. J. Pathol. 2011, 178, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Tveitarås, M.K.; Skogstrand, T.; Leh, S.; Helle, F.; Iversen, B.M.; Chatziantoniou, C.; Reed, R.K.; Hultström, M. Matrix Metalloproteinase-2 Knockout and Heterozygote Mice Are Protected from Hydronephrosis and Kidney Fibrosis after Unilateral Ureteral Obstruction. PLoS ONE 2015, 10, e0143390. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhang, X.; Zhang, Y.; Li, L.; Chen, P. Role of MMP-2 and CD147 in kidney fibrosis. Open Life Sci. 2022, 17, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fu, H.; Liu, Y. The fibrogenic niche in kidney fibrosis: Components and mechanisms. Nat. Rev. Nephrol. 2022, 18, 545–557. [Google Scholar] [CrossRef]
- He, M.; Liu, Z.; Li, L.; Liu, Y. Cell-cell communication in kidney fibrosis. Nephrol. Dial. Transplant. 2023. [Google Scholar] [CrossRef]
- Huang, R.; Fu, P.; Ma, L. Kidney fibrosis: From mechanisms to therapeutic medicines. Signal Transduct. Target. Ther. 2023, 8, 129. [Google Scholar] [CrossRef]
- Li, L.; He, M.; Tang, X.; Huang, J.; Li, J.; Hong, X.; Fu, H.; Liu, Y. Proteomic landscape of the extracellular matrix in the fibrotic kidney. Kidney Int. 2023, 103, 1063–1076. [Google Scholar] [CrossRef]
- Fu, H.; Tian, Y.; Zhou, L.; Zhou, D.; Tan, R.J.; Stolz, D.B.; Liu, Y. Tenascin-C Is a Major Component of the Fibrogenic Niche in Kidney Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 785–801. [Google Scholar] [CrossRef]
- Yin, Q.; Liu, H. Connective Tissue Growth Factor and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 365–380. [Google Scholar] [CrossRef]
- Li, L.; Liao, J.; Yuan, Q.; Hong, X.; Li, J.; Peng, Y.; He, M.; Zhu, H.; Zhu, M.; Hou, F.F.; et al. Fibrillin-1-enriched microenvironment drives endothelial injury and vascular rarefaction in chronic kidney disease. Sci. Adv. 2021, 7, eabc7170. [Google Scholar] [CrossRef]
- Wu, D.; Wu, J.; Liu, H.; Shi, S.; Wang, L.; Huang, Y.; Yu, X.; Lei, Z.; Ouyang, T.; Shen, J.; et al. A biomimetic renal fibrosis progression model on-chip evaluates anti-fibrotic effects longitudinally in a dynamic fibrogenic niche. Lab Chip 2023, 23, 4708–4725. [Google Scholar] [CrossRef]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [PubMed]
- Vega, G.; Alarcón, S.; San Martín, R. The cellular and signalling alterations conducted by TGF-β contributing to renal fibrosis. Cytokine 2016, 88, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, N.; Kang, J. SMAD regulatory networks construct a balanced immune system. Immunology 2013, 139, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yue, S.; Fang, J.; Zeng, J.; Chen, S.; Tian, J.; Nie, S.; Liu, X.; Ding, H. MicroRNA-10a/b inhibit TGF-β/Smad-induced renal fibrosis by targeting TGF-β receptor 1 in diabetic kidney disease. Mol. Ther. Nucleic Acids 2022, 28, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Hinz, B. TGF-β1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef]
- Wu, W.; Wang, X.; Yu, X.; Lan, H.Y. Smad3 Signatures in Renal Inflammation and Fibrosis. Int. J. Biol. Sci. 2022, 18, 2795–2806. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, T.; Lu, D.W.; Zhao, H.; Feng, Y.L.; Chen, H.; Chen, D.Q.; Vaziri, N.D.; Zhao, Y.Y. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 2018, 101, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M. Inflammatory Mediators and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.H. Mesangial cells and renal fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 165–194. [Google Scholar] [CrossRef]
- Ebefors, K.; Bergwall, L.; Nyström, J. The Glomerulus According to the Mesangium. Front. Med. 2022, 8, 740527. [Google Scholar] [CrossRef]
- Isaka, Y.; Nakamura, H.; Mizui, M.; Takabatake, Y.; Horio, M.; Kawachi, H.; Shimizu, F.; Imai, E.; Hori, M. DNAzyme for TGF-beta suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 2004, 66, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Gilham, D.; Wasiak, S.; Rakai, B.D.; Fu, L.; Tsujikawa, L.M.; Sarsons, C.D.; Carestia, A.; Lebioda, K.; Johansson, J.O.; Sweeney, M.; et al. Apabetalone Downregulates Fibrotic, Inflammatory and Calcific Processes in Renal Mesangial Cells and Patients with Renal Impairment. Biomedicines 2023, 11, 1663. [Google Scholar] [CrossRef]
- Li, H.Y.; Oh, Y.S.; Choi, J.W.; Jung, J.Y.; Jun, H.S. Blocking lysophosphatidic acid receptor 1 signaling inhibits diabetic nephropathy in db/db mice. Kidney Int. 2017, 91, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; McCue, P.; Dunn, S.R. Diabetic kidney disease in the db/db mouse. Am. J. Physiol. Renal Physiol. 2003, 284, F1138–F1144. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nam, G.Y.; Seo, E.; Jun, H.S. Inhibition of ChREBP ubiquitination via the ROS/Akt-dependent downregulation of Smurf2 contributes to lysophosphatidic acid-induced fibrosis in renal mesangial cells. J. Biomed. Sci. 2022, 29, 31. [Google Scholar] [CrossRef] [PubMed]
- Afkarian, M.; Zelnick, L.R.; Hall, Y.N.; Heagerty, P.J.; Tuttle, K.; Weiss, N.S.; de Boer, I.H. Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988–2014. JAMA 2016, 316, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wei, C.; Zhang, W.; Schlondorff, D.; Wu, J.; Cai, M.; He, W.; Baron, M.H.; Chuang, P.Y.; Liu, Z.; et al. Gene expression profiles of glomerular endothelial cells support their role in the glomerulopathy of diabetic mice. Kidney Int. 2018, 94, 326–345. [Google Scholar] [CrossRef]
- Liu, J.J.; Pek, S.L.T.; Ang, K.; Tavintharan, S.; Lim, S.C.; SMART2D study. Plasma leucine-rich alpha-2-glycoprotein 1 predicts rapid eGFR decline and albuminuria progression in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2017, 102, 3683–3691. [Google Scholar] [CrossRef]
- Hong, Q.; Zhang, L.; Fu, J.; Verghese, D.A.; Chauhan, K.; Nadkarni, G.N.; Li, Z.; Ju, W.; Kretzler, M.; Cai, G.Y.; et al. LRG1 Promotes Diabetic Kidney Disease Progression by Enhancing TGF-β-Induced Angiogenesis. J. Am. Soc. Nephrol. 2019, 30, 546–562. [Google Scholar] [CrossRef]
- Hong, Q.; Cai, H.; Zhang, L.; Li, Z.; Zhong, F.; Ni, Z.; Cai, G.; Chen, X.M.; He, J.C.; Lee, K. Modulation of transforming growth factor-β-induced kidney fibrosis by leucine-rich ⍺-2 glycoprotein-1. Kidney Int. 2022, 101, 299–314. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, S.; Han, T.; Cai, L.; Zhong, S.; Yang, X.; Zhang, S.; Li, Y.; Liu, K.; Zhou, B.; et al. Continuous genetic monitoring of transient mesenchymal gene activities in distal tubule and collecting duct epithelial cells during renal fibrosis. J. Cell. Biochem. 2024. [Google Scholar] [CrossRef] [PubMed]
- Rastaldi, M.P.; Ferrario, F.; Giardino, L.; Dell’Antonio, G.; Grillo, C.; Grillo, P.; Strutz, F.; Müller, G.A.; Colasanti, G.; D’Amico, G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002, 62, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Park, C.H.; Yoo, T.H. TGF-β Inhibitors for Therapeutic Management of Kidney Fibrosis. Pharmaceuticals 2022, 15, 1485. [Google Scholar] [CrossRef]
- Cohen, C.; Mhaidly, R.; Croizer, H.; Kieffer, Y.; Leclere, R.; Vincent-Salomon, A.; Robley, C.; Anglicheau, D.; Rabant, M.; Sannier, A.; et al. WNT-dependent interaction between inflammatory fibroblasts and FOLR2+ macrophages promotes fibrosis in chronic kidney disease. Nat. Commun. 2024, 15, 743. [Google Scholar] [CrossRef]
- Chen, Z.; Dong, F.; Lu, J.; Wei, L.; Tian, L.; Ge, H.; Zou, Y.; Ma, X.; Yang, Y.; Zhou, L.; et al. Polarized M2c macrophages have a promoting effect on the epithelial-to-mesenchymal transition of human renal tubular epithelial cells. Immunobiology 2018, 223, 826–833. [Google Scholar] [CrossRef]
- Cho, H.S.; Kim, J.H.; Jang, H.N.; Lee, T.W.; Jung, M.H.; Kim, T.H.; Chang, S.H.; Park, D.J. Alpha-lipoic acid ameliorates the epithelial mesenchymal transition induced by unilateral ureteral obstruction in mice. Sci. Rep. 2017, 7, 46065. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bai, Y.; Fu, C.; Liu, W.; Diao, Z. Exosomes from high glucose-treated macrophages promote epithelial-mesenchymal transition of renal tubular epithelial cells via long non-coding RNAs. BMC Nephrol. 2023, 24, 24. [Google Scholar] [CrossRef]
- Duffield, J.S.; Tipping, P.G.; Kipari, T.; Cailhier, J.F.; Clay, S.; Lang, R.; Bonventre, J.V.; Hughes, J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am. J. Pathol. 2005, 167, 1207–1219. [Google Scholar] [CrossRef]
- Yu, C.C.; Chien, C.T.; Chang, T.C. M2 Macrophage polarization modulates epithelial-mesenchymal transition in cisplatin-induced tubulointerstitial fibrosis. Biomedicine 2016, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Jiang, C.C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Liu, G.; Jiang, Z.; Zheng, D. Regulation of renal fibrosis by macrophage polarization. Cell. Physiol. Biochem. 2015, 35, 1062–1069. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, H.; Hu, Y.; Ma, X.; Li, J.; Shi, Y.; Tao, M.; Wang, Y.; Zhong, Q.; Yan, D.; et al. Enhancer of zeste homolog 2 promotes renal fibrosis after acute kidney injury by inducing epithelial-mesenchymal transition and activation of M2 macrophage polarization. Cell Death Dis. 2023, 14, 253. [Google Scholar] [CrossRef]
- Deng, J.; Wu, Z.; He, Y.; Lin, L.; Tan, W.; Yang, J. Interaction Between Intrinsic Renal Cells and Immune Cells in the Progression of Acute Kidney Injury. Front. Med. 2022, 9, 954574. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, X.; Gui, Z.; Suo, C.; Tao, J.; Han, Z.; Ju, X.; Tan, R.; Gu, M.; Wang, Z. Single-nucleotide polymorphisms of matrix metalloproteinase genes are associated with graft fibrosis after kidney transplantation. Transl. Androl. Urol. 2023, 12, 375–383. [Google Scholar] [CrossRef]
- Zheng, G.; Lyons, J.G.; Tan, T.K.; Wang, Y.; Hsu, T.T.; Min, D.; Succar, L.; Rangan, G.K.; Hu, M.; Henderson, B.R.; et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am. J. Pathol. 2009, 175, 580–591. [Google Scholar] [CrossRef]
- Ke, B.; Fan, C.; Yang, L.; Fang, X. Matrix Metalloproteinases-7 and Kidney Fibrosis. Front. Physiol. 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.P.; Hansch, C. Matrix metalloproteinases (MMPs): Chemical-biological functions and (Q)SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.K.; Zheng, G.; Hsu, T.T.; Wang, Y.; Lee, V.W.; Tian, X.; Wang, Y.; Cao, Q.; Wang, Y.; Harris, D.C. Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am. J. Pathol. 2010, 176, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, Y.; Tan, R.; Xiong, M.; He, W.; Fang, L.; Wen, P.; Jiang, L.; Yang, J. Mice lacking the matrix metalloproteinase-9 gene reduce renal interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. Renal Physiol. 2010, 299, F973–F982. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, R.; Gu, X.; Wang, X.; Xi, P.; Chen, X. Exosomes from tubular epithelial cells undergoing epithelial-to-mesenchymal transition promote renal fibrosis by M1 macrophage activation. FASEB Bioadv. 2023, 5, 101–113. [Google Scholar] [CrossRef]
- Li, Q.; Liu, B.C.; Lv, L.L.; Ma, K.L.; Zhang, X.L.; Phillips, A.O. Monocytes induce proximal tubular epithelial-mesenchymal transition through NF-kappa B dependent upregulation of ICAM-1. J. Cell. Biochem. 2011, 112, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Morishita, Y.; Watanabe, M.; Nakazawa, E.; Ishibashi, K.; Kusano, E. The interaction of LFA-1 on mononuclear cells and ICAM-1 on tubular epithelial cells accelerates TGF-β1-induced renal epithelial-mesenchymal transition. PLoS ONE 2011, 6, e23267. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Zaza, G.; Gambaro, G.; Onisto, M.; Bellin, G.; Vischini, G.; Khamaysi, I.; Hassan, A.; Hamoud, S.; Nativ, O.; et al. Heparanase: A Potential New Factor Involved in the Renal Epithelial Mesenchymal Transition (EMT) Induced by Ischemia/Reperfusion (I/R) Injury. PLoS ONE 2016, 11, e0160074. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Chao, C.T.; Huang, J.W.; Hung, K.Y.; Liu, S.H.; Tarng, D.C.; Chiang, C.K. Early elimination of uremic toxin ameliorates AKI-to-CKD transition. Clin. Sci. 2021, 135, 2643–2658. [Google Scholar] [CrossRef]
- Kelly, K.J.; Williams, W.W., Jr.; Colvin, R.B.; Bonventre, J.V. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc. Natl. Acad. Sci. USA 1994, 91, 812–816. [Google Scholar] [CrossRef]
- Yang, S.; Zhong, S.; Deng, Z.; Xie, T.; Yin, G.; Wang, L.; Liu, J.; Yang, J.; Long, Z.; Jiang, X.; et al. Hyperforin regulates renal fibrosis via targeting the PI3K-AKT/ICAM1 axis. Cell. Signal. 2023, 108, 110691. [Google Scholar] [CrossRef]
- Jang, H.N.; Kim, J.H.; Jung, M.H.; Tak, T.; Jung, J.H.; Lee, S.; Jung, S.; Chang, S.H.; Kim, H.J. Human Endothelial Progenitor Cells Protect the Kidney against Ischemia-Reperfusion Injury via the NLRP3 Inflammasome in Mice. Int. J. Mol. Sci. 2022, 23, 1546. [Google Scholar] [CrossRef]
- Li, L.; Liu, H.; Zhang, Q.; Jin, H.; Tao, H.; Zhu, R.; Zhou, Z. Serum amyloid A and risks of all-cause and cardiovascular mortality in chronic kidney disease: A systematic review and dose-response meta-analysis. Ren. Fail. 2023, 45, 2250877. [Google Scholar] [CrossRef]
- Dieter, B.P.; McPherson, S.M.; Afkarian, M.; de Boer, I.H.; Mehrotra, R.; Short, R.; Barbosa-Leiker, C.; Alicic, R.Z.; Meek, R.L.; Tuttle, K.R. Serum amyloid a and risk of death and end-stage renal disease in diabetic kidney disease. J. Diabetes Complicat. 2016, 30, 1467–1472. [Google Scholar] [CrossRef]
- den Hartigh, L.J.; May, K.S.; Zhang, X.S.; Chait, A.; Blaser, M.J. Serum amyloid A and metabolic disease: Evidence for a critical role in chronic inflammatory conditions. Front. Cardiovasc. Med. 2023, 10, 1197432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhou, X.; Zou, H.; Liu, L.; Li, X.; Ruan, Y.; Xie, Y.; Shi, M.; Xiao, Y.; Wang, Y.; et al. SAA1 is transcriptionally activated by STAT3 and accelerates renal interstitial fibrosis by inducing endoplasmic reticulum stress. Exp. Cell Res. 2021, 408, 112856. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Guo, F.; Xia, Z.; Liu, J.; Mai, H.; Liang, Y.; Zhu, G.; Li, Y.; Bai, L.; Li, L.; et al. Inhibition of Fatty Acid-Binding Protein 4 Attenuated Kidney Fibrosis by Mediating Macrophage-to-Myofibroblast Transition. Front. Immunol. 2020, 11, 566535. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.E.; Teng, C.; Tunnicliffe, D.J.; Cashmore, B.A.; Strippoli, G.F. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers for adults with early (stage 1 to 3) non-diabetic chronic kidney disease. Cochrane Database Syst. Rev. 2023, 7, CD007751. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J.; Nakamura, S.; Ma, L.; Nakamura, I.; Donnert, E.; Freeman, M.; Vaughan, D.E.; Fogo, A.B. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int. 2000, 58, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Mirabito Colafella, K.M.; Bovée, D.M.; Danser, A.H.J. The renin-angiotensin-aldosterone system and its therapeutic targets. Exp. Eye Res. 2019, 186, 107680. [Google Scholar] [CrossRef] [PubMed]
- Heeneman, S.; Haendeler, J.; Saito, Y.; Ishida, M.; Berk, B.C. Angiotensin II induces transactivation of two different populations of the platelet-derived growth factor beta receptor. Key role for the p66 adaptor protein Shc. J. Biol. Chem. 2000, 275, 15926–15932. [Google Scholar] [CrossRef]
- Mukoyama, M.; Kuwabara, T. Role of renin-angiotensin system blockade in advanced CKD: To use or not to use? Hypertens. Res. 2022, 45, 1072–1075. [Google Scholar] [CrossRef]
- Cockfield, S.M.; Wilson, S.; Campbell, P.M.; Cantarovich, M.; Gangji, A.; Houde, I.; Jevnikar, A.M.; Keough-Ryan, T.M.; Monroy-Cuadros, F.M.; Nickerson, P.W.; et al. Comparison of the effects of standard vs low-dose prolonged-release tacrolimus with or without ACEi/ARB on the histology and function of renal allografts. Am. J. Transplant. 2019, 19, 1730–1744. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Yu, Y.; Yu, C. Antifibrotic Roles of RAAS Blockers: Update. Adv. Exp. Med. Biol. 2019, 1165, 671–691. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Zakiyanov, O.; Kalousová, M.; Zima, T.; Tesař, V. Matrix Metalloproteinases in Renal Diseases: A Critical Appraisal. Kidney Blood Press. Res. 2019, 44, 298–330. [Google Scholar] [CrossRef] [PubMed]
- Rüster, C.; Wolf, G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J. Am. Soc. Nephrol. 2011, 22, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Liu, L.; Zhang, T.; Wu, X.; Zhang, T.; Xu, Y. MKL1 mediates TGF-β-induced CTGF transcription to promote renal fibrosis. J. Cell. Physiol. 2020, 235, 4790–4803. [Google Scholar] [CrossRef] [PubMed]
- Gauer, S.; Segitz, V.; Goppelt-Struebe, M. Aldosterone induces CTGF in mesangial cells by activation of the glucocorticoid receptor. Nephrol. Dial. Transplant. 2007, 22, 3154–3159. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Mukoyama, M.; Yanagita, M.; Yokoi, H. CTGF in kidney fibrosis and glomerulonephritis. Inflamm. Regen. 2018, 38, 14. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Aten, J.; Bende, R.J.; Oemar, B.S.; Rabelink, T.J.; Weening, J.J.; Goldschmeding, R. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 1998, 53, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Rupérez, M.; Ruiz-Ortega, M.; Esteban, V.; Lorenzo, O.L.; Mezzano, S.L.; Plaza, J.J.; Egido, J. Angiotensin II increases connective tissue growth factor in the kidney. Am. J. Pathol. 2003, 163, 1937–1947. [Google Scholar] [CrossRef]
- Nakayama, T.; Azegami, T.; Hayashi, K.; Hishikawa, A.; Yoshimoto, N.; Nakamichi, R.; Sugita, E.; Itoh, H. Vaccination against connective tissue growth factor attenuates the development of renal fibrosis. Sci. Rep. 2022, 12, 10933. [Google Scholar] [CrossRef]
- Dai, H.Y.; Ma, L.N.; Cao, Y.; Chen, X.L.; Shi, H.; Fan, Y.P.; Yang, B. Protection of CTGF antibody against diabetic nephropathy in mice via reducing glomerular β-catenin expression and podocyte epithelial-mesenchymal transition. J. Cell. Biochem. 2017, 118, 3706–3712. [Google Scholar] [CrossRef]
- Wang, Q.; Usinger, W.; Nichols, B.; Gray, J.; Xu, L.; Seeley, T.W.; Brenner, M.; Guo, G.; Zhang, W.; Oliver, N.; et al. Cooperative interaction of CTGF and TGF-β in animal models of fibrotic disease. Fibrogenesis Tissue Repair 2011, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.; Ohta, N.; Ozono, S.; Kawabe, K.; Kitamura, T. Inhibition of plasminogen activator inhibitor-1 by angiotensin II receptor blockers on cyclosporine-treated renal allograft recipients. Transplant. Proc. 2005, 37, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Nie, H.; Liu, G.; Li, P.; Peng, Y.H.; Xiao, J.; Gu, W.; Li, T.S. Losartan alleviates renal fibrosis by inhibiting the biomechanical stress-induced epithelial-mesenchymal transition of renal epithelial cells. Arch. Biochem. Biophys. 2023, 748, 109770. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.M.; Fogo, A.B. The role of mineralocorticoid receptor activation in kidney inflammation and fibrosis. Kidney Int. Suppl. 2022, 12, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Matavelli, L.C.; Siragy, H.M. Reduction of aldosterone production improves renal oxidative stress and fibrosis in diabetic rats. J. Cardiovasc. Pharmacol. 2013, 61, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Chimal, J.; Jaisser, F.; Anders, H.J. The mineralocorticoid receptor in chronic kidney disease. Br. J. Pharmacol. 2022, 179, 3152–3164. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Ueda, S.; Hamada, K.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Kagawa, T.; Horino, T.; Takao, T. Aldosterone stimulates nuclear factor-kappa B activity and transcription of intercellular adhesion molecule-1 and connective tissue growth factor in rat mesangial cells via serum- and glucocorticoid-inducible protein kinase-1. Clin. Exp. Nephrol. 2012, 16, 81–88. [Google Scholar] [CrossRef]
- Abedini, A.; Sánchez-Navaro, A.; Wu, J.; Klötzer, K.A.; Ma, Z.; Poudel, B.; Doke, T.; Balzer, M.S.; Frederick, J.; Cernecka, H.; et al. Single-cell transcriptomics and chromatin accessibility profiling elucidate the kidney-protective mechanism of mineralocorticoid receptor antagonists. J. Clin. Investig. 2024, 134, e157165. [Google Scholar] [CrossRef]
- Huang, L.L.; Nikolic-Paterson, D.J.; Ma, F.Y.; Tesch, G.H. Aldosterone induces kidney fibroblast proliferation via activation of growth factor receptors and PI3K/MAPK signalling. Nephron Exp. Nephrol. 2012, 120, e115–e122. [Google Scholar] [CrossRef]
- Palacios-Ramirez, R.; Lima-Posada, I.; Bonnard, B.; Genty, M.; Fernandez-Celis, A.; Hartleib-Geschwindner, J.; Foufelle, F.; Lopez-Andres, N.; Bamberg, K.; Jaisser, F. Mineralocorticoid Receptor Antagonism Prevents the Synergistic Effect of Metabolic Challenge and Chronic Kidney Disease on Renal Fibrosis and Inflammation in Mice. Front. Physiol. 2022, 13, 859812. [Google Scholar] [CrossRef]
- Bhuiyan, A.S.; Rafiq, K.; Kobara, H.; Masaki, T.; Nakano, D.; Nishiyama, A. Effect of a novel nonsteroidal selective mineralocorticoid receptor antagonist, esaxerenone (CS-3150), on blood pressure and renal injury in high salt-treated type 2 diabetic mice. Hypertens. Res. 2019, 42, 892–902. [Google Scholar] [CrossRef]
- Barrera-Chimal, J.; Lima-Posada, I.; Bakris, G.L.; Jaisser, F. Mineralocorticoid receptor antagonists in diabetic kidney disease—Mechanistic and therapeutic effects. Nat. Rev. Nephrol. 2022, 18, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Bakris, G.L.; Kolkhof, P. Novel non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Br. J. Pharmacol. 2022, 179, 3220–3234. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Bhatt, D.L.; Cosentino, F.; Marx, N.; Rotstein, O.; Pitt, B.; Pandey, A.; Butler, J.; Verma, S. Non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Eur. Heart J. 2022, 43, 2931–2945. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U. Cardiovascular and Renal Benefit of Novel Non-steroidal Mineralocorticoid Antagonists in Patients with Diabetes. Curr. Cardiol. Rep. 2023, 25, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.; Iatridi, F.; Ferro, C.; Alexandrou, M.E.; Fernandez-Fernandez, B.; Kanbay, M.; Mallamaci, F.; Nistor, I.; Rossignol, P.; Wanner, C.; et al. Mineralocorticoid receptor antagonist use in chronic kidney disease with type 2 diabetes: A clinical practice document by the European Renal Best Practice (ERBP) board of the European Renal Association (ERA). Clin. Kidney J. 2023, 16, 1885–1907. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Nowack, C.; Kolkhof, P.; Ferreira, A.C.; Schloemer, P.; Filippatos, G.; et al. Design and baseline characteristics of the Finerenone in Reducing Kidney Failure and Disease Progression in Diabetic Kidney Disease trial. Am. J. Nephrol. 2019, 50, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P.; Anker, S.D.; Filippatos, G.; Pitt, B.; Ruilope, L.M.; Birkenfeld, A.L.; McGill, J.B.; Rosas, S.E.; Joseph, A.; Gebel, M.; et al. Finerenone in Patients With Chronic Kidney Disease and Type 2 Diabetes by Sodium-Glucose Cotransporter 2 Inhibitor Treatment: The FIDELITY Analysis. Diabetes Care 2022, 45, 2991–2998. [Google Scholar] [CrossRef]
- Chen, X.; Li, X.; Zhang, K.; Lian, K.; Zhang, W.; Song, Y.; Kan, C.; Zhang, J.; Han, F.; Sun, X.; et al. The role of a novel mineralocorticoid receptor antagonist, finerenone, in chronic kidney disease: Mechanisms and clinical advances. Clin. Exp. Nephrol. 2024, 28, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, K.; Millier, A.; Smela, B.; Olewinska, E.; Schmedt, N.; Mernagh, P.; Kovesdy, C.P. Real-world evidence for steroidal mineralocorticoid receptor antagonists in patients with chronic kidney disease. J. Nephrol. 2023, 36, 1135–1167. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.Y.M.; Strippoli, G.F.M. Aldosterone Antagonists in Addition to Renin Angiotensin System Antagonists for Preventing the Progression of CKD: Editorial Summary of a Cochrane Review. Am. J. Kidney Dis. 2021, 77, 810–812. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.; Pelle, M.C.; Zaffina, I.; Tassone, B.; Pujia, R.; Ricchio, M.; Serra, R.; Sciacqua, A.; Michael, A.; Andreucci, M.; et al. Sodium-Glucose Co-transporter-2 Inhibitors and Nephroprotection in Diabetic Patients: More Than a Challenge. Front. Med. 2021, 8, 654557. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. CREDENCE Trial Investigators Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Li, S.; Kang, B.; Zhou, J. The current role of sodium-glucose cotransporter 2 inhibitors in type 2 diabetes mellitus management. Cardiovasc. Diabetol. 2022, 21, 83. [Google Scholar] [CrossRef]
- Maffei, P.; Bettini, S.; Busetto, L.; Dassie, F. SGLT2 Inhibitors in the Management of Type 1 Diabetes (T1D): An Update on Current Evidence and Recommendations. Diabetes Metab. Syndr. Obes. 2023, 16, 3579–3598. [Google Scholar] [CrossRef]
- Fonseca-Correa, J.I.; Correa-Rotter, R. Sodium-Glucose Cotransporter 2 Inhibitors Mechanisms of Action: A Review. Front. Med. 2021, 8, 777861. [Google Scholar] [CrossRef]
- Thomson, S.C.; Vallon, V. Effects of SGLT2 inhibitor and dietary NaCl on glomerular hemodynamics assessed by micropuncture in diabetic rats. Am. J. Physiol. Renal Physiol. 2021, 320, F761–F771. [Google Scholar] [CrossRef]
- Guo, R.; Wang, P.; Zheng, X.; Cui, W.; Shang, J.; Zhao, Z. SGLT2 inhibitors suppress epithelial-mesenchymal transition in podocytes under diabetic conditions via downregulating the IGF1R/PI3K pathway. Front. Pharmacol. 2022, 13, 897167. [Google Scholar] [CrossRef]
- Cai, A.; Shen, J.; Yang, X.; Shao, X.; Gu, L.; Mou, S.; Che, X. Dapagliflozin alleviates renal inflammation and protects against diabetic kidney diseases, both dependent and independent of blood glucose levels. Front. Immunol. 2023, 14, 1205834. [Google Scholar] [CrossRef] [PubMed]
- Pirklbauer, M.; Sallaberger, S.; Staudinger, P.; Corazza, U.; Leierer, J.; Mayer, G.; Schramek, H. Empagliflozin Inhibits IL-1β-Mediated Inflammatory Response in Human Proximal Tubular Cells. Int. J. Mol. Sci. 2021, 22, 5089. [Google Scholar] [CrossRef]
- Chen, X.; Hocher, C.F.; Shen, L.; Krämer, B.K.; Hocher, B. Reno- and cardioprotective molecular mechanisms of SGLT2 inhibitors beyond glycemic control: From bedside to bench. Am. J. Physiol. Cell Physiol. 2023, 325, C661–C681. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Steiger, S.; Anders, H.J. Sodium glucose transporter-2 inhibition has no renoprotective effects on non-diabetic chronic kidney disease. Physiol. Rep. 2017, 5, e13228. [Google Scholar] [CrossRef] [PubMed]
- Cassis, P.; Locatelli, M.; Cerullo, D.; Corna, D.; Buelli, S.; Zanchi, C.; Villa, S.; Morigi, M.; Remuzzi, G.; Benigni, A.; et al. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight 2018, 3, e98720. [Google Scholar] [CrossRef] [PubMed]
- Rajasekeran, H.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.; Lovshin, J.A.; Lytvyn, Y.; Bjornstad, P.; Lai, V.; Tse, J.; Cham, L.; et al. Dapagliflozin in focal segmental glomerulosclerosis: A combined human-rodent pilot study. Am. J. Physiol. Renal Physiol. 2018, 314, F412–F422. [Google Scholar] [CrossRef]
- Greeviroj, P.; Puapatanakul, P.; Phannajit, J.; Takkavatakarn, K.; Kittanamongkolchai, W.; Boonchaya-Anant, P.; Katavetin, P.; Praditpornsilpa, K.; Eiam-Ong, S.; Susantitaphong, P. Effect of canagliflozin in non-diabetic obese patients with albuminuria: A randomized, double-blind, placebo-controlled trial. Clin. Nephrol. 2023, 100, 224–230. [Google Scholar] [CrossRef]
- Doi, Y.; Hamano, T.; Yamaguchi, S.; Sakaguchi, Y.; Kaimori, J.Y.; Isaka, Y. Mediators between canagliflozin and renoprotection vary depending on patient characteristics: Insights from the CREDENCE trial. Diabetes Obes. Metab. 2023, 25, 2944–2953. [Google Scholar] [CrossRef]
- Yi, T.W.; Smyth, B.; Di Tanna, G.L.; Arnott, C.; Cardoza, K.; Kang, A.; Pollock, C.; Agarwal, R.; Bakris, G.; Charytan, D.M.; et al. Kidney and Cardiovascular Effects of Canagliflozin According to Age and Sex: A Post Hoc Analysis of the CREDENCE Randomized Clinical Trial. Am. J. Kidney Dis. 2023, 82, 84–96.e1. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, C.C.J.; Gansevoort, R.T. Sodium-glucose cotransporter 2 inhibitors: Extending the indication to non-diabetic kidney disease? Nephrol. Dial. Transplant. 2020, 35, i33–i42. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y. Targeting TGF-β signaling in kidney fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [PubMed]
- Sartiani, L.; Bartolucci, G.; Pallecchi, M.; Spinelli, V.; Cerbai, E. Pharmacological basis of the antifibrotic effects of pirfenidone: Mechanistic insights from cardiac in-vitro and in-vivo models. Front. Cardiovasc. Med. 2022, 9, 751499. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Naito, Y.; Weng, H.; Ma, X.; Endo, K.; Kito, N.; Yanagawa, N.; Yu, Y.; Li, J.; Iwai, N. Renoprotective mechanisms of pirfenidone in hypertension-induced renal injury: Through anti-fibrotic and anti-oxidative stress pathways. Biomed. Res. 2013, 34, 309–319. [Google Scholar] [CrossRef]
- Chen, J.F.; Ni, H.F.; Pan, M.M.; Liu, H.; Xu, M.; Zhang, M.H.; Liu, B.C. Pirfenidone inhibits macrophage infiltration in 5/6 nephrectomized rats. Am. J. Physiol. Renal Physiol. 2013, 304, F676–F685. [Google Scholar] [CrossRef]
- Bai, X.; Nie, P.; Lou, Y.; Zhu, Y.; Jiang, S.; Li, B.; Luo, P. Pirfenidone is a renal protective drug: Mechanisms, signaling pathways, and preclinical evidence. Eur. J. Pharmacol. 2021, 911, 174503. [Google Scholar] [CrossRef]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef]
- RamachandraRao, S.P.; Zhu, Y.; Ravasi, T.; McGowan, T.A.; Toh, I.; Dunn, S.R.; Okada, S.; Shaw, M.A.; Sharma, K. Pirfenidone is renoprotective in diabetic kidney disease. J. Am. Soc. Nephrol. 2009, 20, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, X.; Wang, B.; Nie, Y.; Wen, J.; Wang, Q.; Gu, C. Pirfenidone suppresses the MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology 2017, 22, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ren, J.; Hu, Q.; Deng, Y.; Chen, G.; Guo, K.; Li, R.; Li, Y.; Wu, L.; Wang, G.; et al. Oral pirfenidone protects against fibrosis by inhibiting fibroblast proliferation and TGF-β signaling in a murine colitis model. Biochem. Pharmacol. 2016, 117, 57–67. [Google Scholar] [CrossRef]
- Zhang, L.; Li, W.; Liu, X.; Guo, J.; Wu, X.; Wang, J. Niclosamide inhibits TGF-β1-induced fibrosis of human Tenon’s fibroblasts by regulating the MAPK-ERK1/2 pathway. Exp. Eye Res. 2023, 235, 109628. [Google Scholar] [CrossRef]
- Samarakoon, R.; Overstreet, J.M.; Higgins, S.P.; Higgins, P.J. TGF-β1 → SMAD/p53/USF2 → PAI-1 transcriptional axis in ureteral obstruction-induced renal fibrosis. Cell Tissue Res. 2012, 347, 117–128. [Google Scholar] [CrossRef]
- Jiménez-Uribe, A.P.; Gómez-Sierra, T.; Aparicio-Trejo, O.E.; Orozco-Ibarra, M.; Pedraza-Chaverri, J. Backstage players of fibrosis: NOX4, mTOR, HDAC, and S1P; companions of TGF-β. Cell. Signal. 2021, 87, 110123. [Google Scholar] [CrossRef]
- Tao, Y.; Tang, C.; Wei, J.; Shan, Y.; Fang, X.; Li, Y. Nr4a1 promotes renal interstitial fibrosis by regulating the p38 MAPK phosphorylation. Mol. Med. 2023, 29, 63. [Google Scholar] [CrossRef]
- Wang, B.; Yao, K.; Wise, A.F.; Lau, R.; Shen, H.H.; Tesch, G.H.; Ricardo, S.D. miR-378 reduces mesangial hypertrophy and kidney tubular fibrosis via MAPK signalling. Clin. Sci. 2017, 131, 411–423. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukagawa, M.; Kuroda, T.; Hata, S.; Iwasaki, Y.; Nemoto, M.; Shirai, K.; Yamauchi, S.; Margolin, S.B.; Shimizu, F.; et al. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int. Suppl. 1997, 63, S239–S243. [Google Scholar] [PubMed]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Pirfenidone treatment decreases transforming growth factor-beta1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am. J. Transplant. 2002, 2, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Effect of pirfenidone on apoptosis-regulatory genes in chronic cyclosporine nephrotoxicity. Transplantation 2005, 79, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.Z.; He, J.M.; Zhang, H.X.; Yu, Z.H.; Zhang, Z.W.; Zhou, H. Renoprotective effects of pirfenidone on chronic renal allograft dysfunction by reducing renal interstitial fibrosis in a rat model. Life Sci. 2019, 233, 116666. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J.B. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.E.; Kopp, J.B. Pirfenidone: An anti-fibrotic therapy for progressive kidney disease. Expert Opin. Investig. Drugs 2010, 19, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Saritas, T.; Kramann, R. Kidney Allograft Fibrosis: Diagnostic and Therapeutic Strategies. Transplantation 2021, 105, e114–e130. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, L.; Velez Diaz, L.A.; Varma, S.; Chowdhary, A.; Bapat, P.; Pan, H.; Kukreja, G.; Palabindela, P.; Selvam, S.A.; Kalra, K. Lifestyle Modifications and Nutritional and Therapeutic Interventions in Delaying the Progression of Chronic Kidney Disease: A Review. Cureus 2023, 15, e34572. [Google Scholar] [CrossRef] [PubMed]
- Daehn, I.S.; Duffield, J.S. The glomerular filtration barrier: A structural target for novel kidney therapies. Nat. Rev. Drug Discov. 2021, 20, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Bharti, N.; Agrawal, V.; Kamthan, S.; Prasad, N.; Agarwal, V. Blood TGF-β1 and miRNA-21-5p levels predict renal fibrosis and outcome in IgA nephropathy. Int. Urol. Nephrol. 2023, 55, 1557–1564. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Lamas, S.; Ortiz, A. Antifibrotic Agents for the Management of CKD: A Review. Am. J. Kidney Dis. 2022, 80, 251–263. [Google Scholar] [CrossRef]
- Armstrong, A. Sanofi walks away from kidney disease therapy, leaving $25M in milestones out of reach for Regulus. Fierce Biotech. 2022. Available online: https://www.fiercebiotech.com/biotech/sanofi-walks-away-kidney-disease-therapy-leaving-25m-milestones-out-reach-regulus (accessed on 28 January 2024).
- Zarjou, A.; Yang, S.; Abraham, E.; Agarwal, A.; Liu, G. Identification of a microRNA signature in renal fibrosis: Role of miR-21. Am. J. Physiol. Renal Physiol. 2011, 301, F793–F801. [Google Scholar] [CrossRef] [PubMed]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef]
- Guo, J.; Song, W.; Boulanger, J.; Xu, E.Y.; Wang, F.; Zhang, Y.; He, Q.; Wang, S.; Yang, L.; Pryce, C.; et al. Dysregulated Expression of microRNA-21 and Disease-Related Genes in Human Patients and in a Mouse Model of Alport Syndrome. Hum. Gene Ther. 2019, 30, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Rubel, D.; Boulanger, J.; Craciun, F.; Xu, E.Y.; Zhang, Y.; Phillips, L.; Callahan, M.; Weber, W.; Song, W.; Ngai, N.; et al. Anti-microRNA-21 Therapy on Top of ACE Inhibition Delays Renal Failure in Alport Syndrome Mouse Models. Cells 2022, 11, 594. [Google Scholar] [CrossRef] [PubMed]
- Gluba-Sagr, A.; Franczyk, B.; Rysz-Górzyńska, M.; Ławiński, J.; Rysz, J. The Role of miRNA in Renal Fibrosis Leading to Chronic Kidney Disease. Biomedicines 2023, 11, 2358. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, X.; Dong, Y.; Liu, X.; Xu, L.; Liu, Y.; Wu, Y.; Wang, C.; Liu, H. PDGFβ receptor-targeted delivery of truncated transforming growth factor β receptor type II for improving the in vitro and in vivo anti-renal fibrosis activity via strong inactivation of TGF-β1/Smad signaling pathway. Naunyn Schmiedebergs Arch. Pharmacol. 2024, 397, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.L.; Liu, T.T.; Lan, H.Y. TGF-Beta as a Master Regulator of Diabetic Nephropathy. Int. J. Mol. Sci. 2021, 22, 7881. [Google Scholar] [CrossRef]
- Mohammad, H.M.F.; Galal Gouda, S.; Eladl, M.A.; Elkazaz, A.Y.; Elbayoumi, K.S.; Farag, N.E.; Elshormilisy, A.; Al-Ammash, B.B.; Hegazy, A.; Abdelkhalig, S.M.; et al. Metformin suppresses LRG1 and TGFβ1/ALK1-induced angiogenesis and protects against ultrastructural changes in rat diabetic nephropathy. Biomed. Pharmacother. 2023, 158, 114128. [Google Scholar] [CrossRef]
- Pan, Z.; Yang, K.; Wang, H.; Xiao, Y.; Zhang, M.; Yu, X.; Xu, T.; Bai, T.; Zhu, H. MFAP4 deficiency alleviates renal fibrosis through inhibition of NF-κB and TGF-β/Smad signaling pathways. FASEB J. 2020, 34, 14250–14263. [Google Scholar] [CrossRef]
- Arvaniti, E.; Moulos, P.; Vakrakou, A.; Chatziantoniou, C.; Chadjichristos, C.; Kavvadas, P.; Charonis, A.; Politis, P.K. Whole-transcriptome analysis of UUO mouse model of renal fibrosis reveals new molecular players in kidney diseases. Sci. Rep. 2016, 6, 26235. [Google Scholar] [CrossRef]
- Isakova, T.; Yanucil, C.; Faul, C. A Klotho-Derived Peptide as a Possible Novel Drug to Prevent Kidney Fibrosis. Am. J. Kidney Dis. 2022, 80, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.; Tan, H.; Hong, X.; Yuan, Q.; Hou, F.F.; Zhou, L.; Liu, Y. Klotho-derived peptide 1 inhibits cellular senescence in the fibrotic kidney by restoring Klotho expression via posttranscriptional regulation. Theranostics 2024, 14, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Suo, X.G.; Wang, F.; Xu, C.H.; He, X.Y.; Wang, J.N.; Zhang, Y.; Ni, W.J.; Lu, H.; Ji, M.L.; He, Y.; et al. Targeted inhibition of TGF-β type I receptor by AZ12601011 protects against kidney fibrosis. Eur. J. Pharmacol. 2022, 929, 175116. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Cao, M.; Ouyang, H.; Chen, Z.; Hu, G.; Li, Q. Recent advances in the development of HIPK2 inhibitors as anti-renal fibrosis agents. Future Med. Chem. 2023, 15, 453–465. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, J.; Tang, A.; Xu, L.; Huang, Y. A novel peptide ligand-coated nano-siRNA-lipoplex technology for kidney targeted gene therapy. Am. J. Transl. Res. 2022, 14, 7362–7377. [Google Scholar] [PubMed]
- Chung, Y.H.; Huang, G.K.; Kang, C.H.; Cheng, Y.T.; Kao, Y.H.; Chien, Y.S. MicroRNA-26a-5p Restoration Ameliorates Unilateral Ureteral Obstruction-Induced Renal Fibrosis in Mice Through Modulating TGF-β Signaling. Lab. Investig. J. Tech. Methods Pathol. 2023, 103, 100131. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ji, J.; Zhao, T.; Wang, E.; Zhang, A. Exosome-encapsulated miR-26a attenuates aldosterone-induced tubulointerstitial fibrosis by inhibiting the CTGF/SMAD3 signaling pathway. Int. J. Mol. Med. 2023, 51, 11. [Google Scholar] [CrossRef]
- Grampp, S.; Goppelt-Struebe, M. Receptor-independent modulation of TGF-β-induced pro-fibrotic pathways by relaxin-2 in human primary tubular epithelial cells. Cell Tissue Res. 2018, 374, 619–627. [Google Scholar] [CrossRef]
- Sassoli, C.; Nistri, S.; Chellini, F.; Bani, D. Human Recombinant Relaxin (Serelaxin) as Anti-fibrotic Agent: Pharmacology, Limitations and Actual Perspectives. Curr. Mol. Med. 2022, 22, 196–208. [Google Scholar] [CrossRef]
- Sasser, J.M. The emerging role of relaxin as a novel therapeutic pathway in the treatment of chronic kidney disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R559–R565. [Google Scholar] [CrossRef]
- Ding, C.; Wang, B.; Lai, X.F.; Guo, Y.; Tesch, G.; Ding, X.; Zheng, J.; Tian, P.; Ricardo, S.; Shen, H.H.; et al. Cellular delivery of relaxin-2 mRNA as a potential treatment for kidney fibrosis. Mater. Today Bio 2023, 21, 100716. [Google Scholar] [CrossRef]
- Ren, J.; Crowley, S.D. Twist1: A Double-Edged Sword in Kidney Diseases. Kidney Dis. 2020, 6, 247–257. [Google Scholar] [CrossRef]
- Ning, X.; Zhang, K.; Wu, Q.; Liu, M.; Sun, S. Emerging role of Twist1 in fibrotic diseases. J. Cell. Mol. Med. 2018, 22, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Bai, Y.; Feng, Y.; Zhang, Q.; Diao, Z.; Liu, W. Renalase Prevents Renal Fibrosis by Inhibiting Endoplasmic Reticulum Stress and Down-Regulating GSK-3β/Snail Signaling. Int. J. Med. Sci. 2023, 20, 669–681. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef]
- Qi, R.; Wang, J.; Jiang, Y.; Qiu, Y.; Xu, M.; Rong, R.; Zhu, T. Snai1-induced partial epithelial-mesenchymal transition orchestrates p53-p21-mediated G2/M arrest in the progression of renal fibrosis via NF-κB-mediated inflammation. Cell Death Dis. 2021, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Chen, G.; Yang, Q.; Liu, Y.; Zhou, T. Potential Therapeutic Effect and Mechanisms of Mesenchymal Stem Cells-Extracellular Vesicles in Renal Fibrosis. Front. Cell Dev. Biol. 2022, 10, 824752. [Google Scholar] [CrossRef]
- Alfaifi, M.; Eom, Y.W.; Newsome, P.N.; Baik, S.K. Mesenchymal stromal cell therapy for liver diseases. J. Hepatol. 2018, 68, 1272–1285. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.M.; Shih, T.E.; Lu, K.Y.; Tsai, S.F.; Harn, H.J.; Ho, L.I. Mesenchymal Stem Cell Therapy of Pulmonary Fibrosis: Improvement with Target Combination. Cell Transplant. 2018, 27, 1581–1587. [Google Scholar] [CrossRef]
- Kulus, M.; Sibiak, R.; Stefańska, K.; Zdun, M.; Wieczorkiewicz, M.; Piotrowska-Kempisty, H.; Jaśkowski, J.M.; Bukowska, D.; Ratajczak, K.; Zabel, M.; et al. Mesenchymal Stem/Stromal Cells Derived from Human and Animal Perinatal Tissues-Origins, Characteristics, Signaling Pathways, and Clinical Trials. Cells 2021, 10, 3278. [Google Scholar] [CrossRef]
- Hass, R.; Kasper, C.; Böhm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef]
- da Silva Meirelles, L.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006, 119, 2204–2213. [Google Scholar] [CrossRef]
- Sudo, K.; Kanno, M.; Miharada, K.; Ogawa, S.; Hiroyama, T.; Saijo, K.; Nakamura, Y. Mesenchymal progenitors able to differentiate into osteogenic, chondrogenic, and/or adipogenic cells in vitro are present in most primary fibroblast-like cell populations. Stem Cells 2007, 25, 1610–1617. [Google Scholar] [CrossRef]
- Matsui, F.; Babitz, S.K.; Rhee, A.; Hile, K.L.; Zhang, H.; Meldrum, K.K. Mesenchymal stem cells protect against obstruction-induced renal fibrosis by decreasing STAT3 activation and STAT3-dependent MMP-9 production. Am. J. Physiol. Renal Physiol. 2017, 312, F25–F32. [Google Scholar] [CrossRef]
- Zhuang, Q.; Ma, R.; Yin, Y.; Lan, T.; Yu, M.; Ming, Y. Mesenchymal Stem Cells in Renal Fibrosis: The Flame of Cytotherapy. Stem Cells Int. 2019, 2019, 8387350. [Google Scholar] [CrossRef]
- Kameishi, S.; Dunn, C.M.; Oka, M.; Kim, K.; Cho, Y.K.; Song, S.U.; Grainger, D.W.; Okano, T. Rapid and effective preparation of clonal bone marrow-derived mesenchymal stem/stromal cell sheets to reduce renal fibrosis. Sci. Rep. 2023, 13, 4421. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Liu, G.; Sun, A.; Wang, J.; Cheng, J.; Wang, W.; Liu, X.; Nie, H.; Guan, G. Mesenchymal stem cells ameliorate diabetic glomerular fibrosis in vivo and in vitro by inhibiting TGF-β signalling via secretion of bone morphogenetic protein 7. Diab. Vasc. Dis. Res. 2014, 11, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shan, S.K.; Guo, B.; Li, F.; Zheng, M.H.; Lei, L.M.; Xu, Q.S.; Ullah, M.; Xu, F.; Lin, X.; et al. The Multi-Therapeutic Role of MSCs in Diabetic Nephropathy. Front. Endocrinol. 2021, 12, 671566. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, J.; Liao, G.; Zhang, J.; Chen, Y.; Li, L.; Li, L.; Liu, F.; Chen, B.; Guo, G.; et al. Early intervention with mesenchymal stem cells prevents nephropathy in diabetic rats by ameliorating the inflammatory microenvironment. Int. J. Mol. Med. 2018, 41, 2629–2639. [Google Scholar] [CrossRef]
- Wang, J.; Lin, Y.; Chen, X.; Liu, Y.; Zhou, T. Mesenchymal stem cells: A new therapeutic tool for chronic kidney disease. Front. Cell Dev. Biol. 2022, 10, 910592. [Google Scholar] [CrossRef]
- Ishiuchi, N.; Nakashima, A.; Doi, S.; Kanai, R.; Maeda, S.; Takahashi, S.; Nagao, M.; Masaki, T. Serum-free medium and hypoxic preconditioning synergistically enhance the therapeutic effects of mesenchymal stem cells on experimental renal fibrosis. Stem Cell Res. Ther. 2021, 12, 472. [Google Scholar] [CrossRef]
- Musiał-Wysocka, A.; Kot, M.; Majka, M. The Pros and Cons of Mesenchymal Stem Cell-Based Therapies. Cell Transplant. 2019, 28, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Kheradmand, A.; Hashemitabar, M.; Kheradmand, P.; Valizadeh, F.; Kavosh, A. Protective Effect of Wharton’s Jelly-derived Mesenchymal Stem Cells on Renal Fibrosis in Rats with Unilateral Ureteral Obstruction. Eur. Urol. Open Sci. 2020, 20, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Gopalarethinam, J.; Nair, A.P.; Iyer, M.; Vellingiri, B.; Subramaniam, M.D. Advantages of mesenchymal stem cell over the other stem cells. Acta Histochem 2023, 125, 152041. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Pan, Q.; Chen, T.; Liao, S.; Li, S.; Li, A.; Chen, S.; Chen, J.; Xiao, Z.; Su, H.; et al. hUC-MSC transplantation therapy effects on lupus-prone MRL/lpr mice at early disease stages. Stem Cell Res. Ther. 2023, 14, 211. [Google Scholar] [CrossRef] [PubMed]
- Borys-Wójcik, S.; Brązert, M.; Jankowski, M.; Ożegowska, K.; Chermuła, B.; Piotrowska-Kempisty, H.; Bukowska, D.; Antosik, P.; Pawelczyk, L.; Nowicki, M.; et al. Human Wharton’s jelly mesenchymal stem cells: Properties, isolation and clinical applications. J. Biol. Regul. Homeost. Agents 2019, 33, 119–123. [Google Scholar] [PubMed]
- Maires, M.P.C.; Pereira, K.R.; Silva, E.K.V.B.; Souza, V.H.R.; Teles, F.; Barbosa, P.F.; Garnica, M.R.; Ornellas, F.M.; Noronha, I.L.; Fanelli, C. Synergic Renoprotective Effects of Combined ASC Therapy with RAAS Blockade in Experimental Advanced CKD. Stem Cells Int. 2022, 2022, 5111782. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhuang, S. Src family kinases in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2017, 313, F721–F728. [Google Scholar] [CrossRef] [PubMed]
- Caner, A.; Asik, E.; Ozpolat, B. SRC signaling in cancer and tumor microenvironment. Adv. Exp. Med. Biol. 2021, 1270, 57–71. [Google Scholar] [CrossRef]
- Hu, M.; Che, P.; Han, X.; Cai, G.Q.; Liu, G.; Antony, V.; Luckhardt, T.; Siegal, G.P.; Zhou, Y.; Liu, R.M.; et al. Therapeutic targeting of SRC kinase in myofibroblast differentiation and pulmonary fibrosis. J. Biol. Regul. Homeost. Agents 2014, 351, 87–95. [Google Scholar] [CrossRef]
- Pham, H.; Birtolo, C.; Chheda, C.; Yang, W.; Rodriguez, M.D.; Liu, S.T.; Gugliotta, G.; Lewis, M.S.; Cirulli, V.; Pandol, S.J.; et al. Essential Role of Lyn in Fibrosis. Front. Physiol. 2016, 7, 387. [Google Scholar] [CrossRef]
- Mendoza, F.A.; Piera-Velazquez, S.; Jimenez, S.A. Tyrosine kinases in the pathogenesis of tissue fibrosis in systemic sclerosis and potential therapeutic role of their inhibition. Transl. Res. 2021, 231, 139–158. [Google Scholar] [CrossRef]
- Li, N.; Lin, G.; Zhang, H.; Sun, J.; Gui, M.; Liu, Y.; Li, W.; Liu, J.; Tang, J. Src Family Kinases: A Potential Therapeutic Target for Acute Kidney Injury. Biomolecules 2022, 12, 984. [Google Scholar] [CrossRef]
- Gao, R.; Ma, Z.; Ma, M.; Yu, J.; Chen, J.; Li, Z.; Shetty, S.; Fu, J. Deletion of Src family kinase Lyn aggravates endotoxin-induced lung inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1376–L1381. [Google Scholar] [CrossRef]
- Tsantikos, E.; Gottschalk, T.A.; Maxwell, M.J.; Hibbs, M.L. Role of the Lyn Tyrosine Kinase in the Development of Autoimmune Disease. Int. J. Clin. Rheumatol. 2014, 9, 519–535. [Google Scholar] [CrossRef]
- Liu, F.; Wang, L.; Qi, H.; Wang, J.; Wang, Y.; Jiang, W.; Xu, L.; Liu, N.; Zhuang, S. Nintedanib, a triple tyrosine kinase inhibitor, attenuates renal fibrosis in chronic kidney disease. Clin. Sci. 2017, 131, 2125–2143. [Google Scholar] [CrossRef]
- Feng, L.; Li, W.; Chao, Y.; Huan, Q.; Lu, F.; Yi, W.; Jun, W.; Binbin, C.; Na, L.; Shougang, Z. Synergistic Inhibition of Renal Fibrosis by Nintedanib and Gefitinib in a Murine Model of Obstructive Nephropathy. Kidney Dis. 2021, 7, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Jamadar, A.; Suma, S.M.; Mathew, S.; Fields, T.A.; Wallace, D.P.; Calvet, J.P.; Rao, R. The tyrosine-kinase inhibitor Nintedanib ameliorates autosomal-dominant polycystic kidney disease. Cell Death Dis. 2021, 12, 947. [Google Scholar] [CrossRef] [PubMed]
- Dorotea, D.; Jiang, S.; Pak, E.S.; Son, J.B.; Choi, H.G.; Ahn, S.M.; Ha, H. Pan-Src kinase inhibitor treatment attenuates diabetic kidney injury via inhibition of Fyn kinase-mediated endoplasmic reticulum stress. Exp. Mol. Med. 2022, 54, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, D.; Asanuma, M.; Miyazaki, I.; Tachibana, H.; Wada, J.; Sogawa, N.; Sugaya, T.; Kitamura, S.; Maeshima, Y.; Shikata, K.; et al. High glucose increases metallothionein expression in renal proximal tubular epithelial cells. Exp. Diabetes Res. 2011, 2011, 534872. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.J.; Wu, Y.J.; Chen, L.J.; Ko, B.S.; Chang, T.C.; Wu, Y.J.; Liang, S.M.; Jan, Y.J.; Liou, J.Y. Reduced Expression of Metallothionein-I/II in Renal Proximal Tubules Is Associated with Advanced Chronic Kidney Disease. Toxins 2021, 13, 568. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Ma, L.; Zhou, X.; Ponnusamy, M.; Tang, J.; Zhuang, M.A.; Tolbert, E.; Bayliss, G.; Bai, J.; Zhuang, S. Src inhibition blocks renal interstitial fibroblast activation and ameliorates renal fibrosis. Kidney Int. 2016, 89, 68–81. [Google Scholar] [CrossRef]
- Chen, M.; Menon, M.C.; Wang, W.; Fu, J.; Yi, Z.; Sun, Z.; Liu, J.; Li, Z.; Mou, L.; Banu, K.; et al. HCK induces macrophage activation to promote renal inflammation and fibrosis via suppression of autophagy. Nat. Commun. 2023, 14, 4297. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wang, Q.; Liu, Y.; Wei, J.; Chen, X. Renal adverse reactions of tyrosine kinase inhibitors in the treatment of tumours: A Bayesian network meta-analysis. Front. Pharmacol. 2022, 13, 1023660. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Uehara, A.; Suzuki, T.; Sekine, R.; Yazawa, M.; Ichikawa, D.; Koike, J.; Shibagaki, Y. Nintedanib-induced glomerular microangiopathy: A case report. CEN Case Rep. 2020, 9, 295–300. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Current Status | Treatment | Description | References |
|---|---|---|---|
| In clinical use | Renin-angiotensin blockers | Inhibit the RAAS, slowing CKD progression and kidney fibrosis; also reduces the activity of TGF-β, PAI1, and PDGF, all molecules involved in promoting glomerular damage. | [168,169,170,171,172,183] |
| In clinical use | Mineralocorticoid receptor (MR) antagonists | Inhibit the activation of aldosterone at the MR, reducing inflammation and proteinuria; block the transcription of various inflammatory mediators that contribute to kidney fibrosis such as PAI1, TGF-β, NF-kB, and IL-6. | [194,195,196,197,198,199,200,201,202,203,204] |
| In clinical use | Sodium-glucose cotransporter-2 (SGLT-2) inhibitors | Inhibit glucose reuptake by the kidney via SGLT-2 transporters located in the proximal tubules; promote lower macrophage-mediated inflammation and cytokines such as IL-6, TNF-α, IFNγ, NF-κβ, TLR-4, and TGF-β, thus reducing glomerular fibrosis. They also lower glomerular pressure and improve renal hemodynamics. Protective against ESRD even in patients without type 2 diabetes. | [205,206,207,208,209,210,211,219,220,221,222] |
| Experimental | Pirfenidone | Antifibrotic orally administered drug; works mainly via inhibition of TGF-β and MAPK signalling. Has anti-inflammatory and antioxidant activity. | [223,224,225,230,231,232,233,234,235,236] |
| Experimental | MicroRNA silencing | Antisense oligonucleotides that silence specific fibrosis-related microRNAs are being explored. Lademirsen, an anti-microRNA-21 drug, failed in human studies, even though microRNA21 is deregulated in kidney fibrosis. Antisense oligonucleotides that silence other microRNAs may be a viable treatment. | [245,246,247,248,249,250,251,252,253] |
| Experimental | Mesenchymal stem cells (MSC) | Multi-potent adult stem cells that demonstrate anti-fibrotic and anti-inflammatory abilities and can induce repair and regeneration. | [272,279] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiss, A.B.; Jacob, B.; Zubair, A.; Srivastava, A.; Johnson, M.; De Leon, J. Fibrosis in Chronic Kidney Disease: Pathophysiology and Therapeutic Targets. J. Clin. Med. 2024, 13, 1881. https://doi.org/10.3390/jcm13071881
Reiss AB, Jacob B, Zubair A, Srivastava A, Johnson M, De Leon J. Fibrosis in Chronic Kidney Disease: Pathophysiology and Therapeutic Targets. Journal of Clinical Medicine. 2024; 13(7):1881. https://doi.org/10.3390/jcm13071881
Chicago/Turabian StyleReiss, Allison B., Berlin Jacob, Aarij Zubair, Ankita Srivastava, Maryann Johnson, and Joshua De Leon. 2024. "Fibrosis in Chronic Kidney Disease: Pathophysiology and Therapeutic Targets" Journal of Clinical Medicine 13, no. 7: 1881. https://doi.org/10.3390/jcm13071881
APA StyleReiss, A. B., Jacob, B., Zubair, A., Srivastava, A., Johnson, M., & De Leon, J. (2024). Fibrosis in Chronic Kidney Disease: Pathophysiology and Therapeutic Targets. Journal of Clinical Medicine, 13(7), 1881. https://doi.org/10.3390/jcm13071881