
Platelet Activating Factor Receptor and Intercellular Adhesion Molecule–1 Expression Increases in the Small Airway Epithelium and Parenchyma of Patients with Idiopathic Pulmonary Fibrosis: Implications for Microbial Pathogenesis

 ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Demographics
2.2. Immunohistochemical Staining
2.3. Small Airway and Lung Parenchyma Quantification
2.4. Statistical Analysis
3. Results
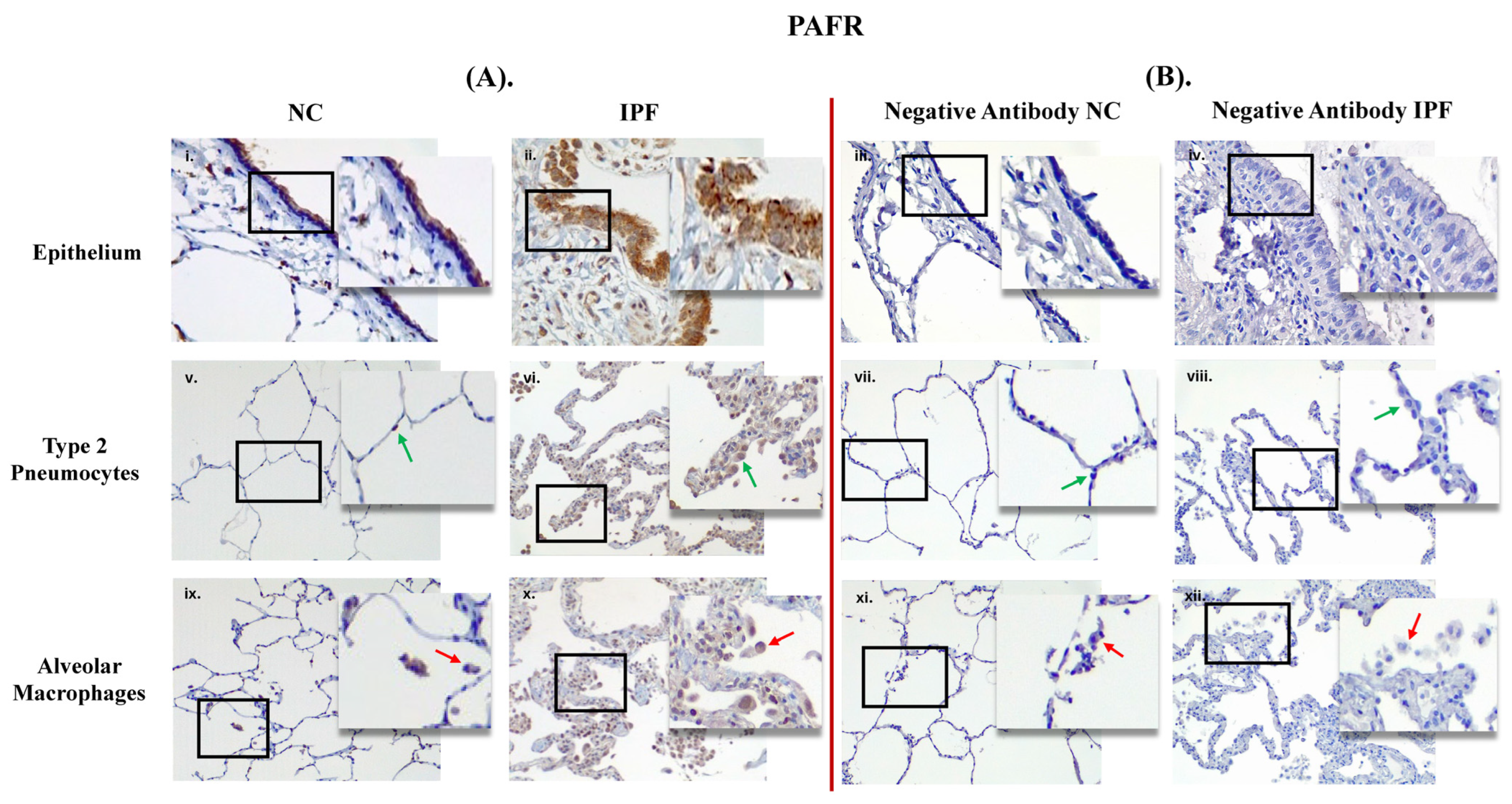
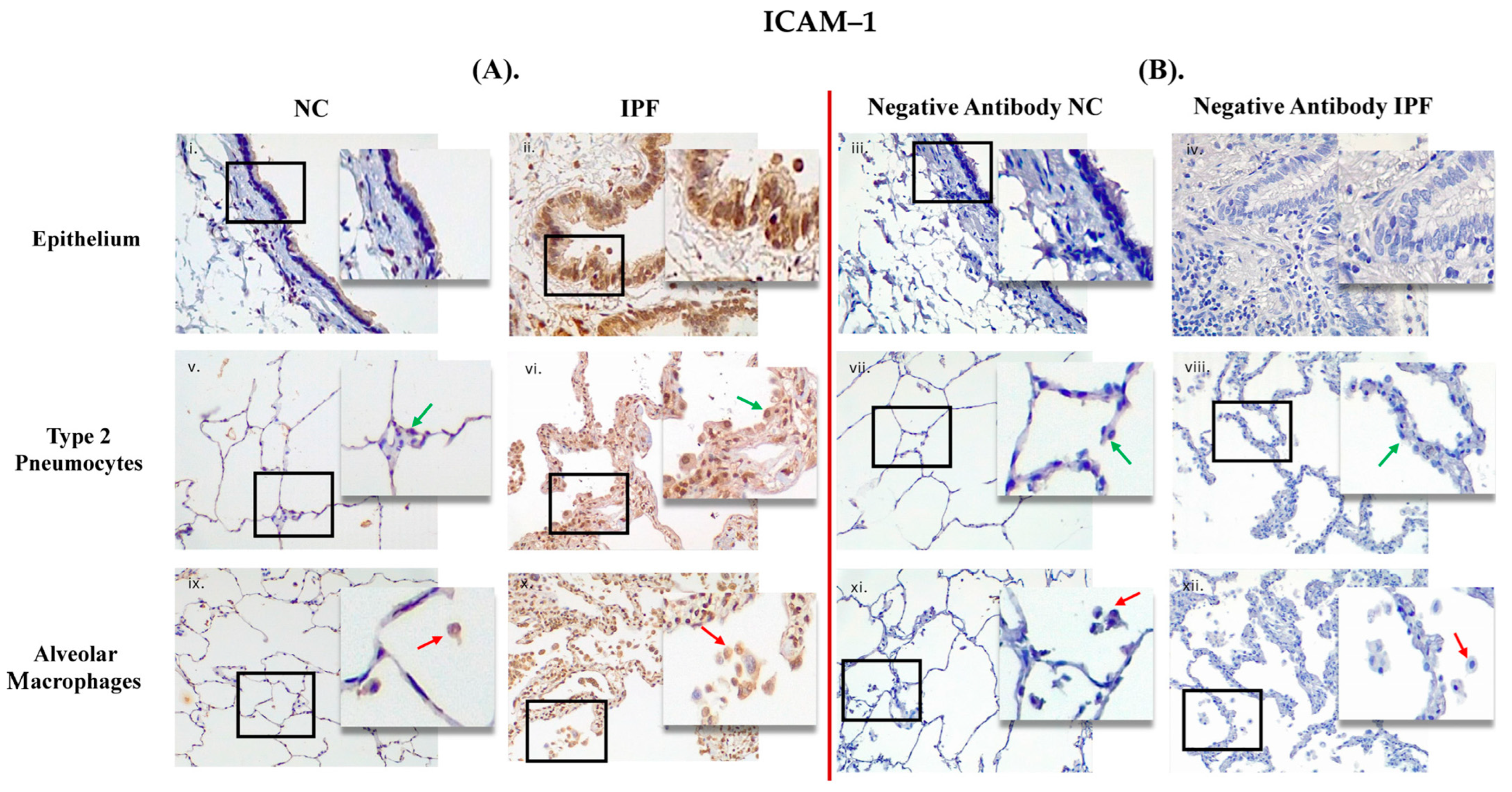
3.1. Comparison between Primary Antibody and Negative Antibody Staining on NC and IPF Lung Tissue
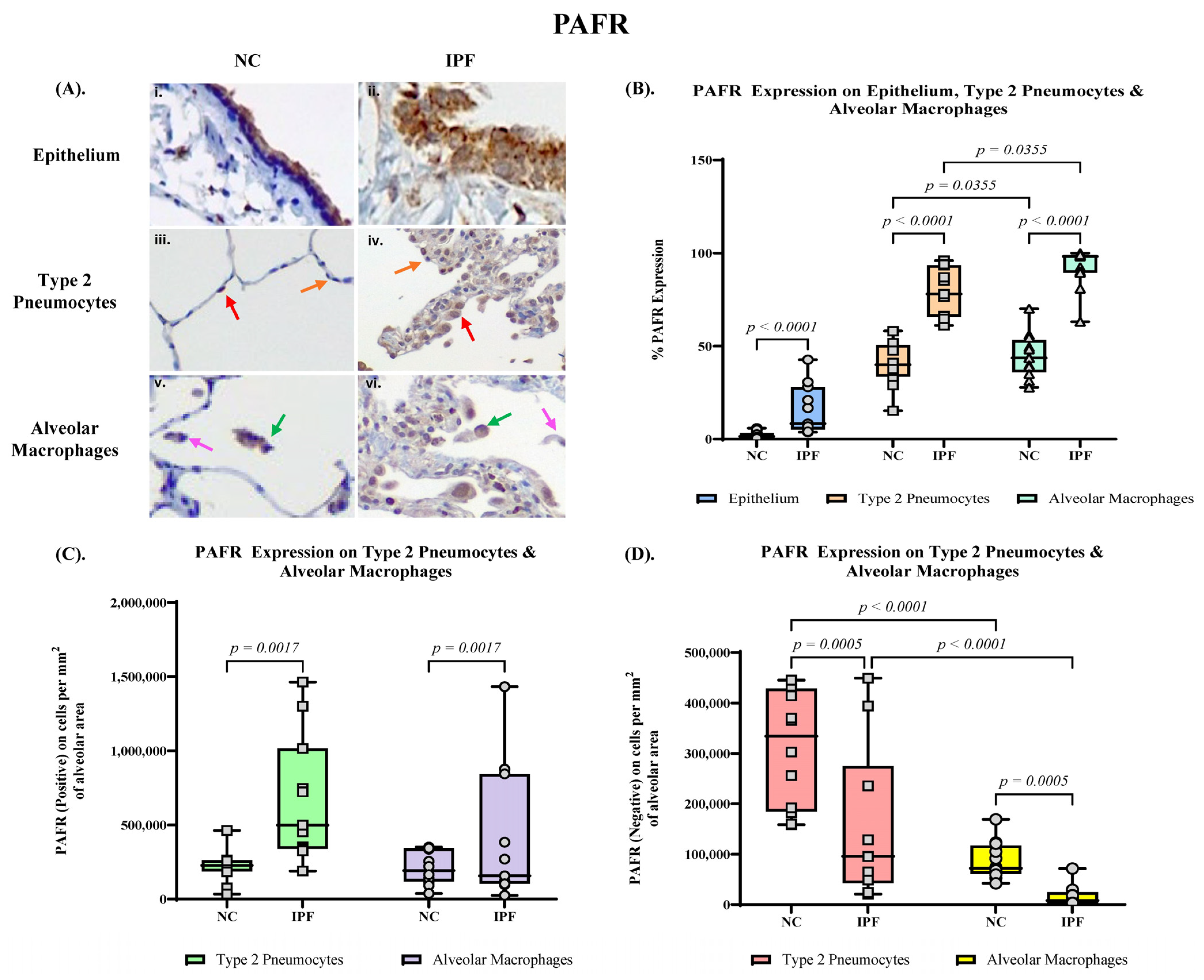
3.2. Quantification of PAFR Expression in Small Airway (SA) Epithelium, Type 2 Pneumocytes and Alveolar Macrophages
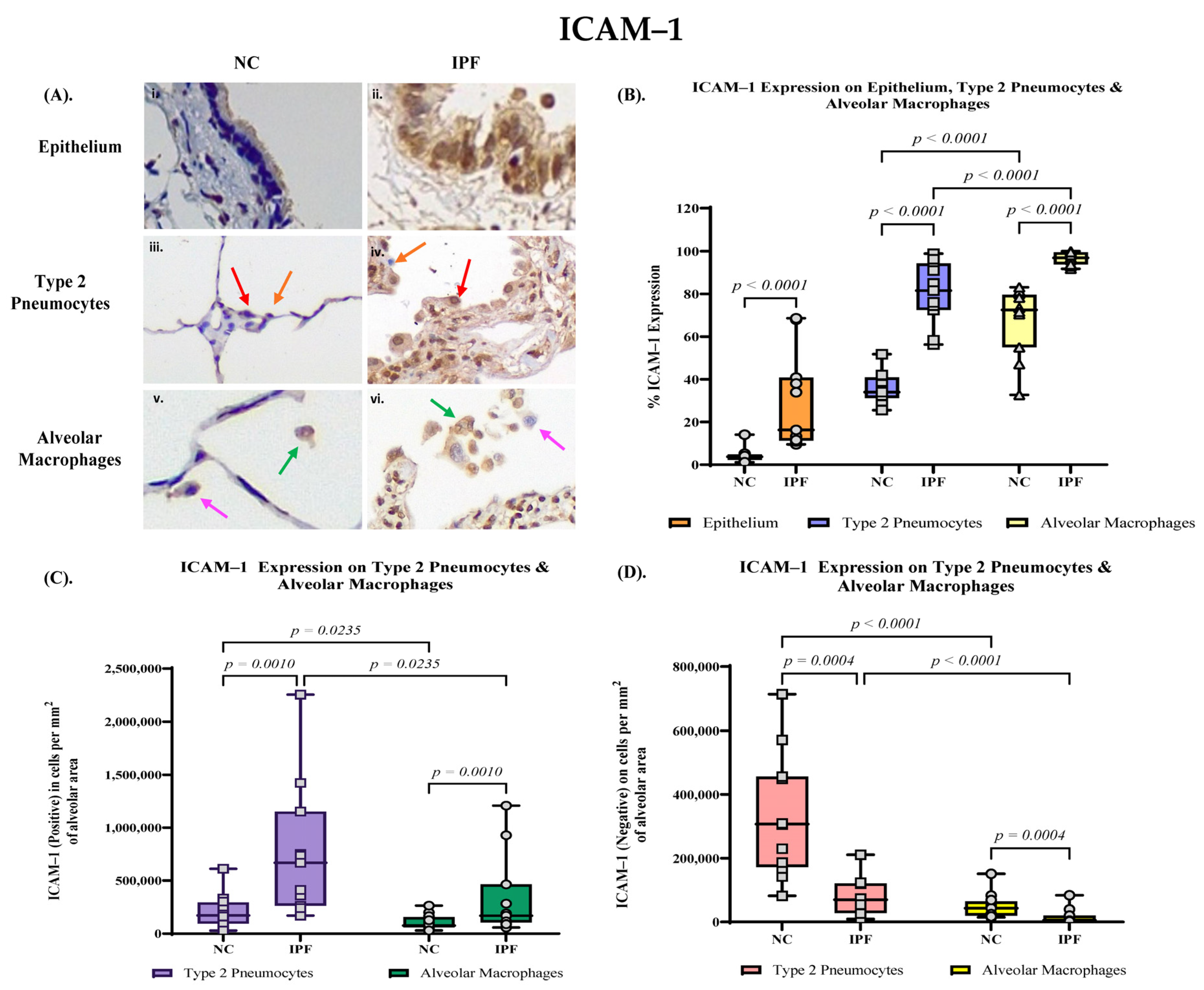
3.3. Quantification of ICAM–1 Expression in Small Airway (SA) Epithelium, Type 2 Pneumocytes and Alveolar Macrophages
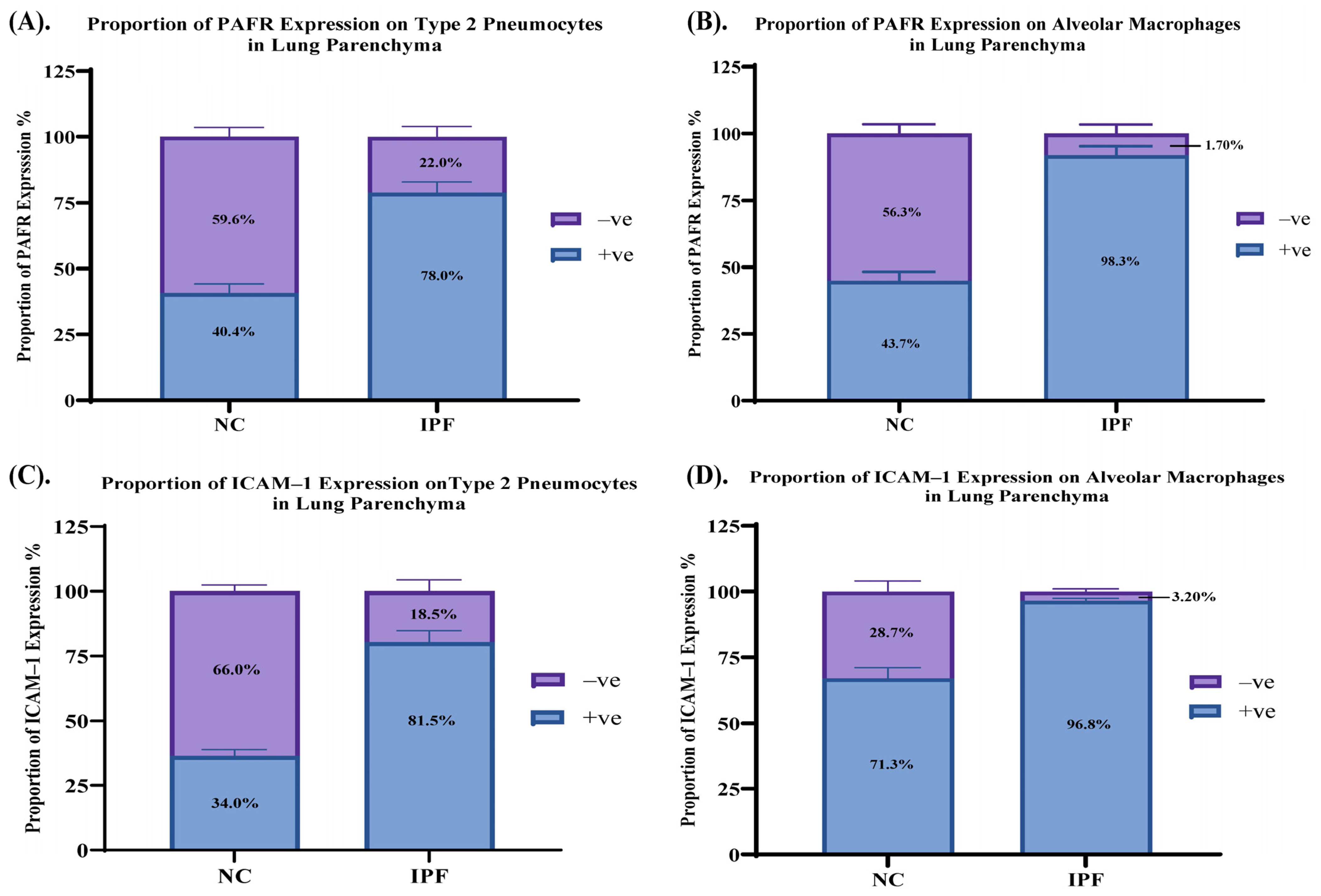
3.4. Proportion of PAFR and ICAM–1 Expression in IPF and NC in the Alveolar Area
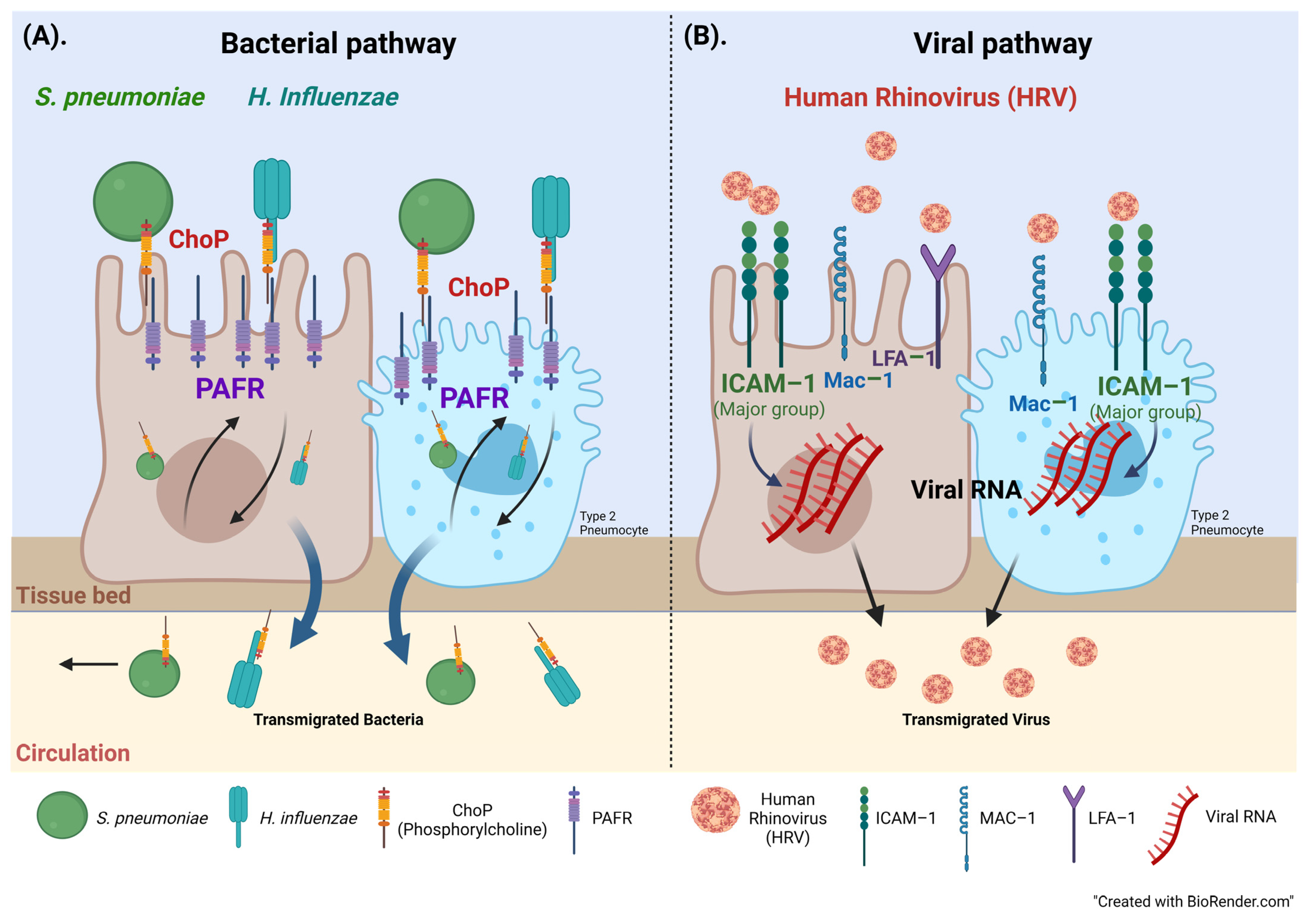
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gaikwad, A.V.; Lu, W.; Dey, S.; Bhattarai, P.; Chia, C.; Larby, J.; Haug, G.; Myers, S.; Jaffar, J.; Westall, G.; et al. Vascular remodelling in idiopathic pulmonary fibrosis patients and its detrimental effect on lung physiology: Potential role of endothelial-to-mesenchymal transition. ERJ Open Res. 2022, 8, 00571–2021. [Google Scholar] [CrossRef] [PubMed]
- Suri, G.S.; Kaur, G.; Jha, C.K.; Tiwari, M. Understanding idiopathic pulmonary fibrosis—Clinical features, molecular mechanism and therapies. Exp. Gerontol. 2021, 153, 111473. [Google Scholar] [CrossRef] [PubMed]
- Heukels, P.; Moor, C.C.; von der Thüsen, J.H.; Wijsenbeek, M.S.; Kool, M. Inflammation and immunity in IPF pathogenesis and treatment. Respir. Med. 2019, 147, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.E.; Glaspole, I.; Grainge, C.; Goh, N.; Hopkins, P.M.A.; Moodley, Y.; Reynolds, P.N.; Chapman, S.; Walters, E.H.; Zappala, C.; et al. Baseline characteristics of idiopathic pulmonary fibrosis: Analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur. Respir. J. 2017, 49, 1601592. [Google Scholar] [CrossRef] [PubMed]
- Sauleda, J.; Núñez, B.; Sala, E.; Soriano, J.B. Idiopathic Pulmonary Fibrosis: Epidemiology, Natural History, Phenotypes. Med. Sci. 2018, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.; Chen, P.; Wei, Y.; Yue, H.; Chu, J.; Zhao, J.; Wang, Y.; Zhang, W.; Zhang, H.L. Viral Infection Increases the Risk of Idiopathic Pulmonary Fibrosis: A Meta-Analysis. Chest 2020, 157, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S. Progress in Understanding and Treating Idiopathic Pulmonary Fibrosis. Annu. Rev. Med. 2019, 70, 211–224. [Google Scholar] [CrossRef]
- Nakamura, Y.; Suda, T. Idiopathic Pulmonary Fibrosis: Diagnosis and Clinical Manifestations. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9 (Suppl. S1), 163–171. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, M.; Dastghaib, S.; Aftabi, S.; Aghanoori, M.R.; Alizadeh, J.; Mokarram, P.; Mehrbod, P.; Ashrafizadeh, M.; Zarrabi, A.; McAlinden, K.D.; et al. The ER Stress/UPR Axis in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Life 2020, 11, 1. [Google Scholar] [CrossRef]
- Liu, G.; Philp, A.M.; Corte, T.; Travis, M.A.; Schilter, H.; Hansbro, N.G.; Burns, C.J.; Eapen, M.S.; Sohal, S.S.; Burgess, J.K.; et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol. Ther. 2021, 225, 107839. [Google Scholar] [CrossRef]
- Meyer, K.C. Pulmonary fibrosis, part I: Epidemiology, pathogenesis, and diagnosis. Expert. Rev. Respir. Med. 2017, 11, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Chapman, K.; Ullah, S. Idiopathic Pulmonary Fibrosis. In StatPearls; StatPearls Publishing Copyright© 2024; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2024. [Google Scholar]
- Moghoofei, M.; Mostafaei, S.; Kondori, N.; Armstrong, M.E.; Babaei, F. Bacterial and viral coinfection in idiopathic pulmonary fibrosis patients: The prevalence and possible role in disease progression. BMC Pulm. Med. 2022, 22, 60. [Google Scholar] [CrossRef] [PubMed]
- Mostafaei, S.; Sayad, B.; Azar, M.E.F.; Doroudian, M.; Hadifar, S.; Behrouzi, A.; Riahi, P.; Hussen, B.M.; Bayat, B.; Nahand, J.S.; et al. The role of viral and bacterial infections in the pathogenesis of IPF: A systematic review and meta-analysis. Respir. Res. 2021, 22, 53. [Google Scholar] [CrossRef]
- Sohal, S.S.; Hansbro, P.M.; Shukla, S.D.; Eapen, M.S.; Walters, E.H. Potential Mechanisms of Microbial Pathogens in Idiopathic Interstitial Lung Disease. Chest 2017, 152, 899–900. [Google Scholar] [CrossRef]
- Keyvani, H.; Moghoofei, M.; Bokharaei-Salim, F.; Mostafaei, S.; Javad Mousavi, S.A.; Monavari, S.H.; Esghaei, M. Prevalence of respiratory viruses in Iranian patients with idiopathic pulmonary fibrosis. J. Med. Microbiol. 2017, 66, 1602–1606. [Google Scholar] [CrossRef]
- Lu, W.; Eapen, M.S.; Singhera, G.K.; Markos, J.; Haug, G.; Chia, C.; Larby, J.; Brake, S.J.; Westall, G.P.; Jaffar, J.; et al. Angiotensin-Converting Enzyme 2 (ACE2), Transmembrane Peptidase Serine 2 (TMPRSS2), and Furin Expression Increases in the Lungs of Patients with Idiopathic Pulmonary Fibrosis (IPF) and Lymphangioleiomyomatosis (LAM): Implications for SARS-CoV-2 (COVID-19) Infections. J. Clin. Med. 2022, 11, 777. [Google Scholar] [CrossRef]
- Humlicek, A.L.; Pang, L.; Look, D.C. Modulation of airway inflammation and bacterial clearance by epithelial cell ICAM-1. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, L598–L607. [Google Scholar] [CrossRef]
- Novotny, L.A.; Bakaletz, L.O. Intercellular adhesion molecule 1 serves as a primary cognate receptor for the Type IV pilus of nontypeable Haemophilus influenzae. Cell. Microbiol. 2016, 18, 1043–1055. [Google Scholar] [CrossRef]
- de Paula, R.R.; Marinho, F.V.; Fahel, J.S.; Oliveira, S.C. Contribution of intercellular adhesion molecule 1 (ICAM-1) to control Mycobacterium avium infection. Microbes Infect. 2017, 19, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J. The platelet activating factor receptor: A new anti-infective target in respiratory disease? Thorax 2012, 67, 840. [Google Scholar] [CrossRef] [PubMed]
- Huffnagle, G.B.; Dickson, R.P.; Lukacs, N.W. The respiratory tract microbiome and lung inflammation: A two-way street. Mucosal Immunol. 2017, 10, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.; Batra, S.; Zemans, R.L.; Downey, G.P.; Jeyaseelan, S. Divergent functions of Toll-like receptors during bacterial lung infections. Am. J. Respir. Crit. Care Med. 2014, 190, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.A.; Nookala, V. Biochemistry of Platelet Activating Factor. In StatPearls; StatPearls Publishing Copyright© 2022; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hills, R.; Ishii, S.; Jancar, S.; McIntyre, T.; Ninio, E.; O’Neill, C.; Oliveira Rios, F.J.; Travers, J.B.; Whittaker, M. Platelet-activating factor receptor in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol. 2019, 2019, 1–9. [Google Scholar] [CrossRef]
- Soares, A.C.; Pinho, V.S.; Souza, D.G.; Shimizu, T.; Ishii, S.; Nicoli, J.R.; Teixeira, M.M. Role of the platelet-activating factor (PAF) receptor during pulmonary infection with gram negative bacteria. Br. J. Pharmacol. 2002, 137, 621–628. [Google Scholar] [CrossRef]
- Grigg, J.; Walters, H.; Sohal, S.S.; Wood-Baker, R.; Reid, D.W.; Xu, C.B.; Edvinsson, L.; Morissette, M.C.; Stämpfli, M.R.; Kirwan, M.; et al. Cigarette smoke and platelet-activating factor receptor dependent adhesion of Streptococcus pneumoniae to lower airway cells. Thorax 2012, 67, 908–913. [Google Scholar] [CrossRef]
- Iovino, F.; Brouwer, M.C.; van de Beek, D.; Molema, G.; Bijlsma, J.J.E. Signalling or binding: The role of the platelet-activating factor receptor in invasive pneumococcal disease. Cell. Microbiol. 2013, 15, 870–881. [Google Scholar] [CrossRef]
- Atto, B.; Eapen, M.S.; Sharma, P.; Frey, U.; Ammit, A.J.; Markos, J.; Chia, C.; Larby, J.; Haug, G.; Weber, H.C.; et al. New therapeutic targets for the prevention of infectious acute exacerbations of COPD: Role of epithelial adhesion molecules and inflammatory pathways. Clin. Sci. 2019, 133, 1663–1703. [Google Scholar] [CrossRef]
- Bella, J.; Kolatkar, P.R.; Marlor, C.W.; Greve, J.M.; Rossmann, M.G. The structure of the two amino-terminal domains of human ICAM-1 suggests how it functions as a rhinovirus receptor and as an LFA-1 integrin ligand. Proc. Natl. Acad. Sci. USA 1998, 95, 4140–4145. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, A.K.; Rothlein, R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic. Biol. Med. 2000, 28, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, Y.A.; Gern, J.E. Rhinoviruses and Their Receptors: Implications for Allergic Disease. Curr. Allergy Asthma Rep. 2016, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Mahmood, M.Q.; Weston, S.; Latham, R.; Muller, H.K.; Sohal, S.S.; Walters, E.H. The main rhinovirus respiratory tract adhesion site (ICAM-1) is upregulated in smokers and patients with chronic airflow limitation (CAL). Respir. Res. 2017, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Moore, T.A. Viruses in Idiopathic Pulmonary Fibrosis. Etiology and Exacerbation. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S2), S186–S192. [Google Scholar] [CrossRef] [PubMed]
- Vannella, K.M.; Luckhardt, T.R.; Wilke, C.A.; van Dyk, L.F.; Toews, G.B.; Moore, B.B. Latent herpesvirus infection augments experimental pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 181, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Muller, H.K.; Latham, R.; Sohal, S.S.; Walters, E.H. Platelet-activating factor receptor (PAFr) is upregulated in small airways and alveoli of smokers and COPD patients. Respirology 2016, 21, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr266. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef]
- Mosser, J.L.; Tomasz, A. Choline-containing teichoic acid as a structural component of pneumococcal cell wall and its role in sensitivity to lysis by an autolytic enzyme. J. Biol. Chem. 1970, 245, 287–298. [Google Scholar] [CrossRef]
- Serino, L.; Virji, M. Genetic and functional analysis of the phosphorylcholine moiety of commensal Neisseria lipopolysaccharide. Mol. Microbiol. 2002, 43, 437–448. [Google Scholar] [CrossRef]
- Weiser, J.N.; Shchepetov, M.; Chong, S.T. Decoration of lipopolysaccharide with phosphorylcholine: A phase-variable characteristic of Haemophilus influenzae. Infect. Immun. 1997, 65, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Smani, Y.; Docobo-Pérez, F.; López-Rojas, R.; Domínguez-Herrera, J.; Ibáñez-Martínez, J.; Pachón, J. Platelet-activating factor receptor initiates contact of Acinetobacter baumannii expressing phosphorylcholine with host cells. J. Biol. Chem. 2012, 287, 26901–26910. [Google Scholar] [CrossRef] [PubMed]
- Barbier, M.; Oliver, A.; Rao, J.; Hanna, S.L.; Goldberg, J.B.; Albertí, S. Novel phosphorylcholine-containing protein of Pseudomonas aeruginosa chronic infection isolates interacts with airway epithelial cells. J. Infect. Dis. 2008, 197, 465–473. [Google Scholar] [CrossRef]
- Swords, W.E.; Buscher, B.A.; Ver Steeg Ii, K.; Preston, A.; Nichols, W.A.; Weiser, J.N.; Gibson, B.W.; Apicella, M.A. Non-typeable Haemophilus influenzae adhere to and invade human bronchial epithelial cells via an interaction of lipooligosaccharide with the PAF receptor. Mol. Microbiol. 2000, 37, 13–27. [Google Scholar] [CrossRef]
- Blaas, D.; Fuchs, R. Mechanism of human rhinovirus infections. Mol. Cell. Pediatr. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Thibaut, H.J.; Lacroix, C.; De Palma, A.M.; Franco, D.; Decramer, M.; Neyts, J. Toward antiviral therapy/prophylaxis for rhinovirus-induced exacerbations of chronic obstructive pulmonary disease: Challenges, opportunities, and strategies. Rev. Med. Virol. 2016, 26, 21–33. [Google Scholar] [CrossRef]
- Bella, J.; Rossmann, M.G. ICAM-1 receptors and cold viruses. Pharm. Acta Helv. 2000, 74, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Brake, S.J.; Eapen, M.S.; McAlinden, K.D.; Markos, J.; Haug, G.; Larby, J.; Chia, C.; Hardikar, A.; Singhera, G.K.; Hackett, T.L.; et al. SARS-CoV-2 (COVID-19) Adhesion Site Protein Upregulation in Small Airways, Type 2 Pneumocytes, and Alveolar Macrophages of Smokers and COPD—Possible Implications for Interstitial Fibrosis. Int. J. Chron. Obstruct. Pulmon. Dis. 2022, 17, 101–115. [Google Scholar] [CrossRef]
- Santos, G.; Lai, X.; Eberhardt, M.; Vera, J. Bacterial Adherence and Dwelling Probability: Two Drivers of Early Alveolar Infection by Streptococcus pneumoniae Identified in Multi-Level Mathematical Modeling. Front. Cell. Infect. Microbiol. 2018, 8, 159. [Google Scholar] [CrossRef]
- Rijneveld, A.W.; Weijer, S.; Florquin, S.; Speelman, P.; Shimizu, T.; Ishii, S.; van der Poll, T. Improved host defense against pneumococcal pneumonia in platelet-activating factor receptor-deficient mice. J. Infect. Dis. 2004, 189, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Radin, J.N.; Orihuela, C.J.; Murti, G.; Guglielmo, C.; Murray, P.J.; Tuomanen, E.I. beta-Arrestin 1 participates in platelet-activating factor receptor-mediated endocytosis of Streptococcus pneumoniae. Infect. Immun. 2005, 73, 7827–7835. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Control (NC) | IPF | |
| Factors | Values * | |
| Total Number (n) | 12 | 11 |
| Age (Years) | 39 ± 16.5 | 63 ± 4.85 |
| Gender (Female/Male) | 6/6 | 5/6 |
| Smoking status (n): Current smoker/Ex-smoker/Never | Non-smoker | 0/5/6 |
| Smoking Packs Per Year | - | 16.55 ± 22.56 |
| Respiratory Function Parameters | ||
| FEV1 (L) * | NA | 1.67 ± 0.42 |
| FVC (L) † | NA | 1.89 ± 0.45 |
| DLCO ‡ (mL/min/mmHg) | NA | 5.98 ± 3.12 |
| DLCO (%) | NA | 25.33 ± 12.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahzad, A.M.; Lu, W.; Dey, S.; Bhattarai, P.; Gaikwad, A.V.; Jaffar, J.; Westall, G.; Sutherland, D.; Singhera, G.K.; Hackett, T.-L.; et al. Platelet Activating Factor Receptor and Intercellular Adhesion Molecule–1 Expression Increases in the Small Airway Epithelium and Parenchyma of Patients with Idiopathic Pulmonary Fibrosis: Implications for Microbial Pathogenesis. J. Clin. Med. 2024, 13, 2126. https://doi.org/10.3390/jcm13072126
Shahzad AM, Lu W, Dey S, Bhattarai P, Gaikwad AV, Jaffar J, Westall G, Sutherland D, Singhera GK, Hackett T-L, et al. Platelet Activating Factor Receptor and Intercellular Adhesion Molecule–1 Expression Increases in the Small Airway Epithelium and Parenchyma of Patients with Idiopathic Pulmonary Fibrosis: Implications for Microbial Pathogenesis. Journal of Clinical Medicine. 2024; 13(7):2126. https://doi.org/10.3390/jcm13072126
Chicago/Turabian StyleShahzad, Affan Mahmood, Wenying Lu, Surajit Dey, Prem Bhattarai, Archana Vijay Gaikwad, Jade Jaffar, Glen Westall, Darren Sutherland, Gurpreet Kaur Singhera, Tillie-Louise Hackett, and et al. 2024. "Platelet Activating Factor Receptor and Intercellular Adhesion Molecule–1 Expression Increases in the Small Airway Epithelium and Parenchyma of Patients with Idiopathic Pulmonary Fibrosis: Implications for Microbial Pathogenesis" Journal of Clinical Medicine 13, no. 7: 2126. https://doi.org/10.3390/jcm13072126
APA StyleShahzad, A. M., Lu, W., Dey, S., Bhattarai, P., Gaikwad, A. V., Jaffar, J., Westall, G., Sutherland, D., Singhera, G. K., Hackett, T. -L., Eapen, M. S., & Sohal, S. S. (2024). Platelet Activating Factor Receptor and Intercellular Adhesion Molecule–1 Expression Increases in the Small Airway Epithelium and Parenchyma of Patients with Idiopathic Pulmonary Fibrosis: Implications for Microbial Pathogenesis. Journal of Clinical Medicine, 13(7), 2126. https://doi.org/10.3390/jcm13072126