
Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Renal and Hepatic Fibrosis

{kind=link}
{kind=link}
Abstract
:1. Introduction
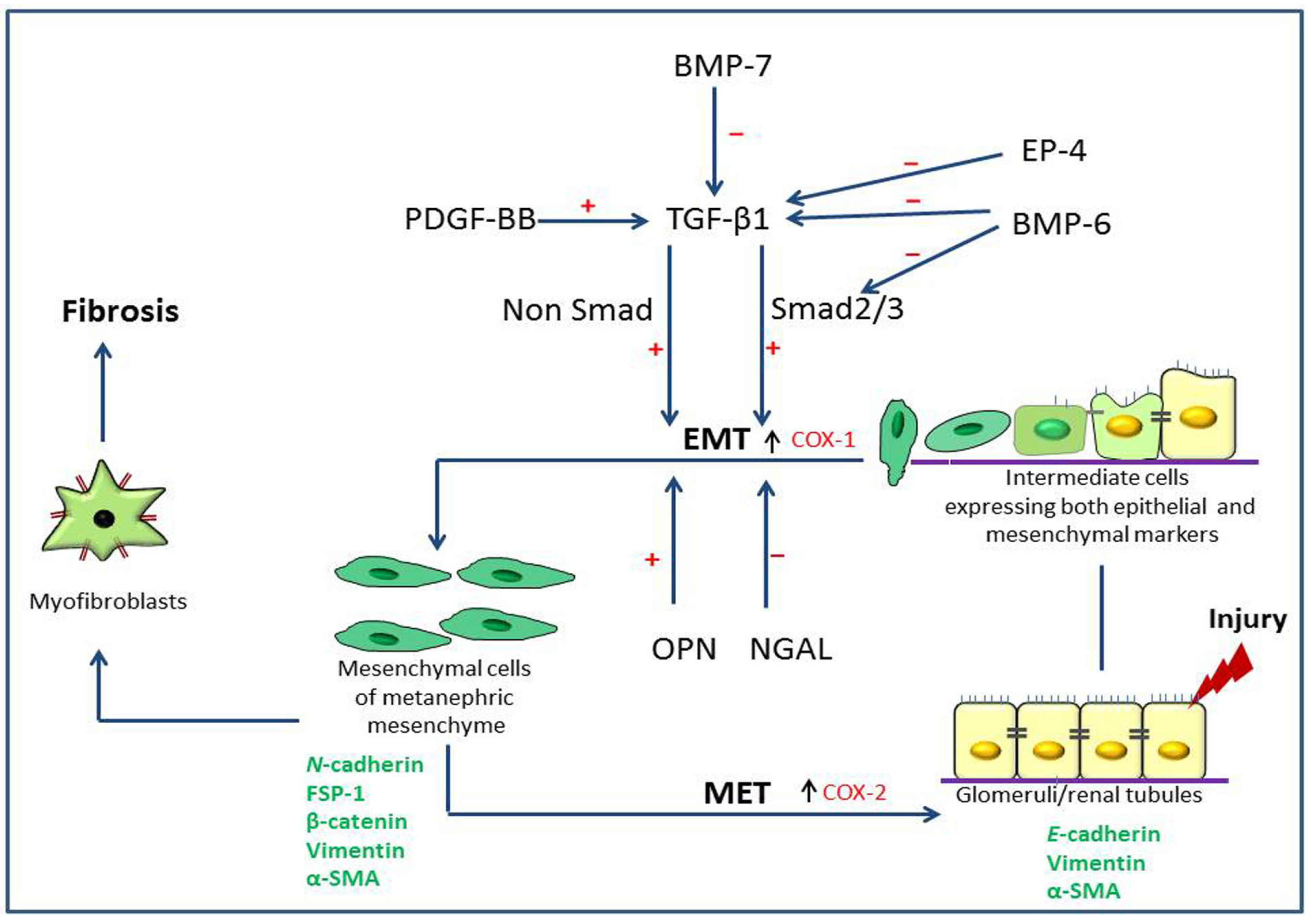
2. Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Renal Fibrosis
2.1. Renal Fibrosis and Disease Models
2.2. Expression of Type 2 EMT Markers
2.3. Growth Factors Associated with Type 2 EMT
2.4. Roles of Prostaglandins
2.5. Neutrophil Gelatinase-Associated Lipocalin (NGAL), Osteopontin (OPN) and Bone Morphogenic Protein-6 (BMP-6)

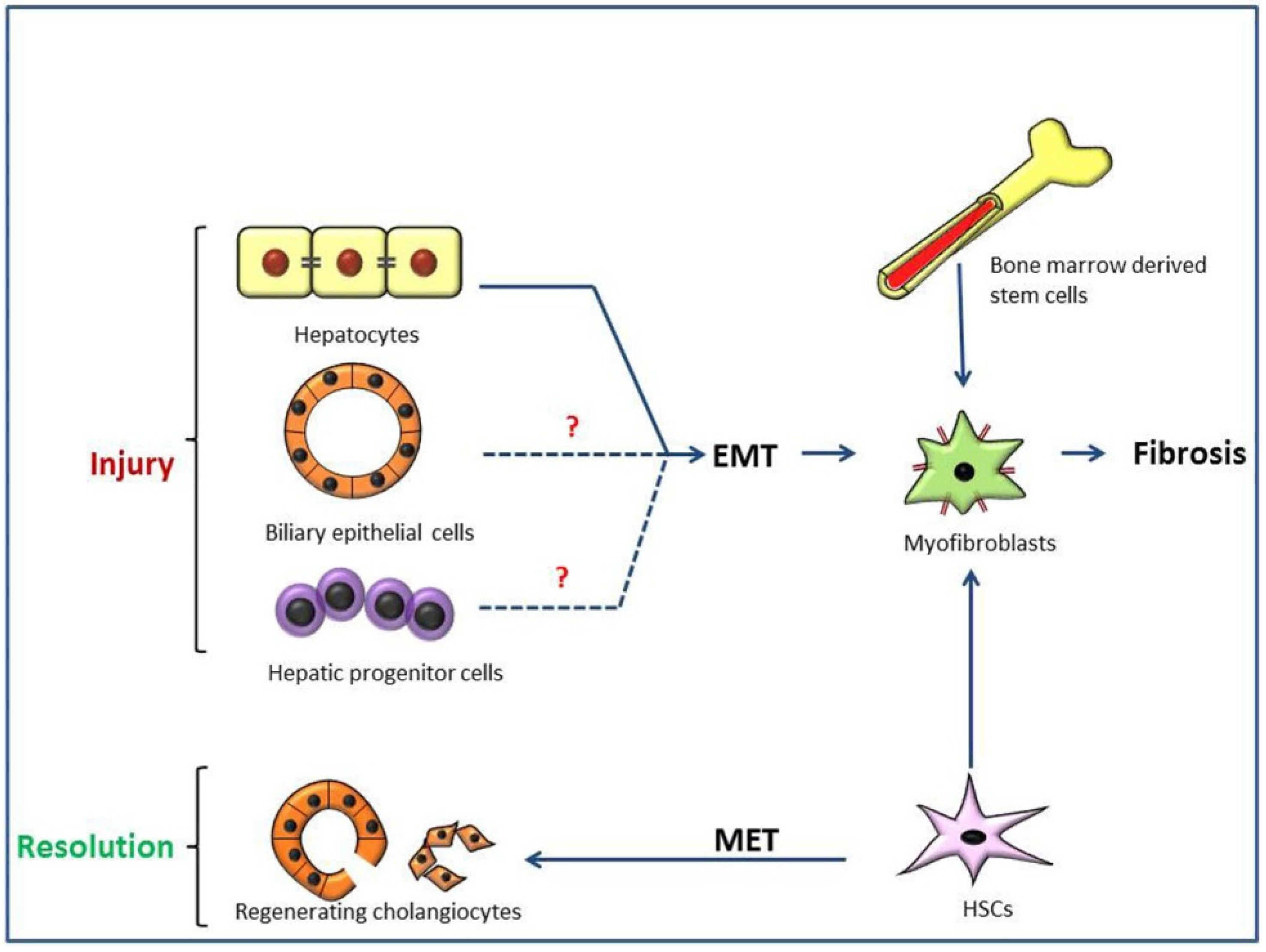
3. Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Hepatic Fibrosis
3.1. Liver Fibrosis and Disease Models
3.2. Possible Liver EMT in Thioacetamide (TAA)-Induced Rat Cirrhosis
3.3. Importance of Ductal Reaction and Possible Hepatocarcinogenesis, Instead of Type 2 EMT, in TAA-Induced Rat Cirrhosis
3.4. Regenerating Cholangiocytes Do Not Induce Type 2 EMT in α-Naphthylisothiocyanate (ANIT)-Induced Peri-Biliary Fibrosis
3.5. Type 2 EMT and Hepatic Cirrhosis

4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Diehl, A.M. Epithelial-to-mesenchymal transitions in the liver. Hepatology 2009, 50, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Kothari, A.N.; Mi, Z.; Zapf, M.; Kuo, P.C. Novel clinical therapeutics targeting the epithelial to mesenchymal transition. Clin. Transl. Med. 2014, 3, 35. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Faa, G.; Sanna, A.; Gerosa, C.; Fanni, D.; Puddu, M.; Ottonello, G.; van Eyken, P.; Fanos, V. Renal physiological regenerative medicine to prevent chronic renal failure: Should we start at birth? Clin. Chim. Acta 2015, 444, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Cook, H.T. The origin of renal fibroblasts and progression of kidney disease. Am. J. Pathol. 2010, 176, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15. [Google Scholar] [CrossRef]
- Kim, M.-K.; Maeng, Y.-I.; Sung, W.J.; Oh, H.-K.; Park, J.-B.; Yoon, G.S.; Cho, C.-H.; Park, K.-K. The differential expression of TGF-β1, ILK and wnt signaling inducing epithelial to mesenchymal transition in human renal fibrogenesis: An immunohistochemical study. Int. J. Clin. Exp. Pathol. 2013, 6, 1747–1758. [Google Scholar] [PubMed]
- Fintha, A.; Gasparics, Á.; Fang, L.; Erdei, Z.; Hamar, P.; Mózes, M.M.; Kökény, G.; Rosivall, L.; Sebe, A. Characterization and role of SCAI during renal fibrosis and epithelial-to-mesenchymal transition. Am. J. Pathol. 2013, 182, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Hendry, C.; Rumballe, B.; Moritz, K.; Little, M.H. Defining and redefining the nephron progenitor population. Pediatr. Nephrol. 2011, 26, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Cheng, H.-T.; Surendran, K. Molecular insights into segmentation along the proximal distal axis of the nephron. J. Am. Soc. Nephrol. 2007, 18, 2014–2020. [Google Scholar] [CrossRef] [PubMed]
- Horster, M.F.; Braun, G.S.; Huber, S.M. Embryonic renal epithelia: Induction, nephrogenesis, and cell differentiation. Physiol. Rev. 1999, 79, 1157–1191. [Google Scholar] [PubMed]
- Fabian, S.L.; Humphreys, B.D. What’s past is prologue: Developmental pathways and chronic allograft dysfunction. Am. J. Transpl. 2012, 12, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Swetha, G.; Chandra, V.; Phadnis, S.; Bhonde, R. Glomerular parietal epithelial cells of adult murine kidney undergo EMT to generate cells with traits of renal progenitors. J. Cell. Mol. Med. 2011, 15, 396–413. [Google Scholar] [PubMed]
- Yuasa, T.; Izawa, T.; Kuwamura, M.; Yamate, J. Thy-1 expressing mesenchymal cells in rat nephrogenesis in correlation with cells immunoreactive for α-smooth muscle actin and vimentin. J. Toxicol. Pathol. 2010, 23. [Google Scholar] [CrossRef] [PubMed]
- El-Nahas, A.M. Plasticity of kidney cells: Role in kidney remodeling and scarring. Kidney Int. 2003, 64, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 2010, 21, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Runyan, C.E.; Hayashida, T.; Hubchak, S.; Curley, J.F.; Schnaper, H.W. Role of SARA (SMAD anchor for receptor activation) in maintenance of epithelial cell phenotype. J. Biol. Chem. 2009, 284, 25181–25189. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Masola, V.; Granata, S.; Bellin, G.; dalla Gassa, A.; Onisto, M.; Gambaro, G.; Lupo, A. Sulodexide alone or in combination with low doses of everolimus inhibits the hypoxia-mediated epithelial to mesenchymal transition in human renal proximal tubular cells. J. Nephrol. 2015, 28, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.K.; Zheng, G.; Hsu, T.-T.; Wang, Y.; Lee, V.W.S.; Tian, X.; Wang, Y.; Cao, Q.; Wang, Y.; Harris, D.C.H. Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am. J. Pathol. 2010, 176, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Bonner, G.; Maeshima, Y.; Colorado, P.; Müller, G.A.; Strutz, F.; Kalluri, R. Renal fibrosis: Collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am. J. Pathol. 2001, 159, 1313–1321. [Google Scholar] [CrossRef]
- Inoue, T.; Umezawa, A.; Takenaka, T.; Suzuki, H.; Okada, H. The contribution of epithelial-mesenchymal transition to renal fibrosis differs among kidney disease models. Kidney Int. 2015, 87, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, F.; Wu, J.; Shao, C.; Gao, Y. Urinary candidate biomarker discovery in a rat unilateral ureteral obstruction model. Sci. Rep. 2015, 5, 9314. [Google Scholar] [CrossRef] [PubMed]
- Ucero, A.C.; Benito-Martin, A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Sanz, A.B.; Ramos, A.M.; Berzal, S.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Unilateral ureteral obstruction: Beyond obstruction. Int. Urol. Nephrol. 2014, 46, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Zhang, J.; Xiao, Z.; Peng, X.; Qi, Y.; Du, J. Akt2 is involved in loss of epithelial cells and renal fibrosis following unilateral ureteral obstruction. PLoS ONE 2014, 9, e105451. [Google Scholar] [CrossRef] [PubMed]
- Yamate, J.; Tatsumi, M.; Nakatsuji, S.; Kuwamura, M.; Kotani, T.; Sakuma, S. Immunohistochemical observations on the kinetics of macrophages and myofibroblasts in rat renal interstitial fibrosis induced by cis-diamminedichloroplatinum. J. Comp. Pathol. 1995, 112, 27–39. [Google Scholar] [CrossRef]
- Yamate, J.; Ishida, A.; Tsujino, K.; Tatsumi, M.; Nakatsuji, S.; Kuwamura, M.; Kotani, T.; Sakuma, S. Immunohistochemical study of rat renal interstitial fibrosis induced by repeated injection of cisplatin, with special reference to the kinetics of macrophages and myofibroblasts. Toxicol. Pathol. 1996, 24, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Yamate, J.; Kuribayashi, M.; Kuwamura, M.; Kotani, T.; Ogihara, K. Differential immunoexpressions of cytoskeletons in renal epithelial and interstitial cells in rat and canine fibrotic kidneys, and in kidney-related cell lines under fibrogenic stimuli. Exp. Toxicol. Pathol. 2005, 57, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Yamate, J.; Sato, K.; Machida, Y.; Ide, M.; Sato, S.; Nakatsuji, S.; Kuwamura, M.; Kotani, T.; Sakuma, S. Cisplatin-Induced rat renal interstitial fibrasis; a possible pathogenesis based on the data. J. Toxicol. Pathol. 2000, 13, 237–247. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, C.S.; van Tubergen, E.A.; Inglehart, R.C.; D’Silva, N.J. Biomarkers of epithelial-mesenchymal transition in squamous cell carcinoma. J. Dent. Res. 2012, 92, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M. Epithelial-mesenchymal transition in chronic liver disease: Fibrogenesis or escape from death? J. Hepatol. 2011, 55, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Roeder, S.S.; Stefanska, A.; Eng, D.G.; Kaverina, N.V.; Sunseri, M.W.; McNicholas, B.A.; Rabinovitch, P.; Engel, F.B.; Daniel, C.; Amann, K.; et al. Changes in glomerular parietal epithelial cells in mouse kidneys with advanced age. Am. J. Physiol. Renal Physiol. 2015, 309, F164–F178. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmoulière, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Masszi, A.; Fan, L.; Rosivall, L.; McCulloch, C.A.; Rotstein, O.D.; Mucsi, I.; Kapus, A. Integrity of cell-cell contacts is a critical regulator of TGF-β 1-induced epithelial-to-myofibroblast transition: Role for β-catenin. Am. J. Pathol. 2004, 165, 1955–1967. [Google Scholar] [CrossRef]
- Wu, S.; Liu, S.; Liu, Z.; Huang, J.; Pu, X.; Li, J.; Yang, D.; Deng, H.; Yang, N.; Xu, J. Classification of circulating tumor cells by epithelial-mesenchymal transition markers. PLoS ONE 2015, 10, e0123976. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kang, Y.S.; Dai, C.; Kiss, L.P.; Wen, X.; Liu, Y. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am. J. Pathol. 2008, 172, 299–308. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.W.; Gomez, E.W. Biomechanics of TGFβ-induced epithelial-mesenchymal transition: Implications for fibrosis and cancer. Clin. Transl. Med. 2014, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-β1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwano, M. EMT and TGF-β in renal fibrosis. Front. Biosci. 2010, S2, 229–238. [Google Scholar] [CrossRef]
- O’Connor, J.W.; Gomez, E.W. Cell adhesion and shape regulate TGF-β1-induced epithelial-myofibroblast transition via MRTF-A signaling. PLoS ONE 2013, 8, e83188. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Ulich, T.R.; Lacey, D.L.; Hill, D.C.; Qi, M.; Kaufman, S.A.; Van, G.Y.; Tarpley, J.E.; Yee, J.S. Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am. J. Pathol. 1996, 148, 1169–1180. [Google Scholar] [PubMed]
- Kordes, C.; Brookmann, S.; Häussinger, D.; Klonowski-Stumpe, H. Differential and synergistic effects of platelet-derived growth factor-BB and transforming growth factor-β1 on activated pancreatic stellate cells. Pancreas 2005, 31, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Su, A.; He, S.; Tian, B.; Hu, W.; Zhang, Z. MicroRNA-221 mediates the effects of PDGF-BB on migration, proliferation, and the epithelial-mesenchymal transition in pancreatic cancer cells. PLoS ONE 2013, 8, e71309. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Landerholm, T.E.; Wei, J.S.; Dong, X.R.; Wu, S.P.; Liu, X.; Nagata, K.; Inagaki, M.; Majesky, M.W. Coronary smooth muscle differentiation from proepicardial cells requires rhoA-mediated actin reorganization and p160 rho-kinase activity. Dev. Biol. 2001, 240, 404–418. [Google Scholar] [CrossRef] [PubMed]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Chang, F.-C.; Wu, C.-F.; Chou, Y.-H.; Hsu, H.-L.; Chiang, W.-C.; Shen, J.; Chen, Y.-M.; Wu, K.-D.; Tsai, T.-J.; et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011, 80, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Breyer, M.D.; Breyer, R.M. Prostaglandin E receptors and the kidney. Am. J. Physiol. Renal Physiol. 2000, 279, F12–F23. [Google Scholar] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Menter, D.G.; Dubois, R.N. Prostaglandins in cancer cell adhesion, migration, and invasion. Int. J. Cell Biol. 2012, 2012, 723419. [Google Scholar] [CrossRef] [PubMed]
- Okegawa, T.; Jonas, P.E.; DeSchryver, K.; Kawasaki, A.; Needleman, P. Metabolic and cellular alterations underlying the exaggerated renal prostaglandin and thromboxane synthesis in ureter obstruction in rabbits. Inflammatory response involving fibroblasts and mononuclear cells. J. Clin. Investig. 1983, 71, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Whinnery, M.A.; Shaw, J.O.; Beck, N. Thromboxane B2 and prostaglandin E2 in the rat kidney with unilateral ureteral obstruction. Am. J. Physiol. 1982, 242, F220–F225. [Google Scholar] [PubMed]
- Nakagawa, N.; Yuhki, K.; Kawabe, J.; Fujino, T.; Takahata, O.; Kabara, M.; Abe, K.; Kojima, F.; Kashiwagi, H.; Hasebe, N.; et al. The intrinsic prostaglandin E2-EP4 system of the renal tubular epithelium limits the development of tubulointerstitial fibrosis in mice. Kidney Int. 2012, 82, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Loutzenhiser, K.; Loutzenhiser, R. Biphasic actions of prostaglandin E2 on the renal afferent arteriole: Role of EP3 and EP4 receptors. Circ. Res. 2000, 86, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Narayanan, N.K.; Lasano, S.; Narayanan, B. Modulation of PGE2-induced EP4 expression on snail signaling and the impact on epithelial-mesenchymal transition: Significance of EP4 antagonism. Anticancer Res. 2011, 31, 4347–4357. [Google Scholar] [PubMed]
- Park, J.Y.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin. Immunol. 2006, 119, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Caughey, G.E.; Cleland, L.G.; Penglis, P.S.; Gamble, J.R.; James, M.J. Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: Selective up-regulation of prostacyclin synthesis by COX-2. J. Immunol. 2001, 167, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Dinchuk, J.E.; Car, B.D.; Focht, R.J.; Johnston, J.J.; Jaffee, B.D.; Covington, M.B.; Contel, N.R.; Eng, V.M.; Collins, R.J.; Czerniak, P.M. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature 1995, 378, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Kömhoff, M.; Wang, J.L.; Cheng, H.F.; Langenbach, R.; McKanna, J.A.; Harris, R.C.; Breyer, M.D. Cyclooxygenase-2-selective inhibitors impair glomerulogenesis and renal cortical development. Kidney Int. 2000, 57, 414–422. [Google Scholar] [CrossRef]
- Stacey, D.W. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr. Opin. Cell Biol. 2003, 15, 158–163. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Baldin, V.; Lukas, J.; Marcote, M.J.; Pagano, M.; Draetta, G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993, 7, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Izawa, T.; Juniantito, V.; Kuwamura, M.; Sugiura, K.; Takeuchi, T.; Yamate, J. Involvement of endogenous prostaglandin E2 in tubular epithelial regeneration through inhibition of apoptosis and epithelial-mesenchymal transition in cisplatin-induced rat renal lesions. Histol. Histopathol. 2010, 25, 995–1007. [Google Scholar] [PubMed]
- Yamamoto, E.; Izawa, T.; Kuwamura, M.; Yamate, J. Immunohistochemical expressions of main PGE2 biosynthesis-related enzymes and PGE2 receptor in rat nephrogenesis. J. Toxicol. Pathol. 2011, 24, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Donato, V.; Coppolino, G.; Campo, S.; Buemi, A.; Lacquaniti, A.; Buemi, M. Neutrophil gelatinase-associated lipocalin (NGAL) as a marker of kidney damage. Am. J. Kidney Dis. 2008, 52, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, V.S.; Ferguson, M.A.; Bonventre, J.V. Biomarkers of acute kidney injury. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Cernaro, V.; Lacquaniti, A.; Donato, V.; Fazio, M.R.; Buemi, A.; Buemi, M. Fibrosis, regeneration and cancer: What is the link? Nephrol. Dial. Transpl. 2012, 27, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Nickolas, T.L. Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase–associated lipocalin for diagnosing acute kidney injury. Ann. Intern. Med. 2008, 148, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Mori, K.; Ma, Q.; Kelly, C.; Barasch, J.; Devarajan, P. Neutrophil gelatinase-associated lipocalin: A novel early urinary biomarker for cisplatin nephrotoxicity. Am. J. Nephrol. 2004, 24, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Wu, L.L.; Huang, X.R.; Yang, N.; Gilbert, R.E.; Cooper, M.E.; Johnson, R.J.; Lai, K.N.; Lan, H.Y. Osteopontin expression in progressive renal injury in remnant kidney: Role of angiotensin II. Kidney Int. 2000, 58, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Sakatsume, M.; Nishi, S.; Narita, I.; Arakawa, M.; Gejyo, F. Expression, roles, receptors, and regulation of osteopontin in the kidney. Kidney Int. 2001, 60, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Shevde, L.A.; Samant, R.S. Role of osteopontin in the pathophysiology of cancer. Matrix Biol. 2014, 37, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, E.; Tonomura, Y.; Kondo, C.; Masuno, K.; Fujisawa, K.; Tsuchiya, N.; Matsushima, S.; Torii, M.; Takasu, N.; Izawa, T.; et al. Involvement of neutrophil gelatinase-associated lipocalin and osteopontin in renal tubular regeneration and interstitial fibrosis after cisplatin-induced renal failure. Exp. Toxicol. Pathol. 2014, 66, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Yano, R.; Golbar, H.M.; Izawa, T.; Sawamoto, O.; Kuwamura, M.; Yamate, J. Participation of bone morphogenetic protein (BMP)-6 and osteopontin in cisplatin (CDDP)-induced rat renal fibrosis. Exp. Toxicol. Pathol. 2015, 67, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Chen, X.; Deng, Z.; Tan, R.; Liu, C.; Lu, P.; Zhang, W.; Gu, M. Osteopontin mediating cyclosporine A induced epithelial-to-mesenchymal transition on rat renal tubular epithelial cells. Cell Biol. Int. 2014, 38, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, L.; Moses, M.A. Overexpression of neutrophil gelatinase-associated lipocalin (NGAL) induces epithelial-mesenchymal transition in MCF-7 breast cancer cells. Cancer Res. 2005, 65, 1328. [Google Scholar]
- Gheorgheosu, D.; Jung, M.; Ören, B.; Schmid, T.; Dehelean, C.; Muntean, D.; Brüne, B. Betulinic acid suppresses NGAL-induced epithelial-to-mesenchymal transition in melanoma. Biol. Chem. 2013, 394, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Toyono, T.; Murakami, T.; Akamine, A. Transforming growth factor-β superfamily members expressed in rat incisor pulp. Arch. Oral Biol. 1998, 43, 745–751. [Google Scholar] [CrossRef]
- Long, J.; Badal, S.S.; Wang, Y.; Chang, B.H.J.; Rodriguez, A.; Danesh, F.R. MicroRNA-22 is a master regulator of bone morphogenetic protein-7/6 homeostasis in the kidney. J. Biol. Chem. 2013, 288, 36202–36214. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Koka, V.; Lan, H.Y. Transforming growth factor-β and Smad signalling in kidney diseases. Nephrology 2005, 10, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Kim, S.I.; Choi, M.E. Therapeutic targets for treating fibrotic kidney diseases. Transl. Res. 2015, 165, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M.; Chung, A.C.K.; Lan, H.Y. Role of the TGF-β/BMP-7/Smad pathways in renal diseases. Clin. Sci. 2013, 124, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Manson, S.R.; Niederhoff, R.A.; Hruska, K.A.; Austin, P.F. The BMP-7-Smad1/5/8 pathway promotes kidney repair after obstruction induced renal injury. J. Urol. 2011, 185, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.D.; Phillips, A.; Fraser, D. Bone morphogenetic protein-7 inhibits proximal tubular epithelial cell Smad3 signaling via increased SnoN expression. Am. J. Pathol. 2010, 176, 1139–11347. [Google Scholar] [CrossRef] [PubMed]
- Li, R.X.; Yiu, W.H.; Tang, S.C.W. Role of bone morphogenetic protein-7 in renal fibrosis. Front. Physiol. 2015, 6, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dendooven, A.; van Oostrom, O.; van der Giezen, D.M.; Leeuwis, J.W.; Snijckers, C.; Joles, J.A.; Robertson, E.J.; Verhaar, M.C.; Nguyen, T.Q.; Goldschmeding, R. Loss of endogenous bone morphogenetic protein-6 aggravates renal fibrosis. Am. J. Pathol. 2011, 178, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.-D.; Yang, S.; Zhang, J.; Zhu, T.-H. BMP6 reverses TGF-β1-induced changes in HK-2 cells: Implications for the treatment of renal fibrosis. Acta Pharmacol. Sin. 2009, 30, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Ainslie, G.R.; Kutanzi, K.; Pogribny, I.P.; Muskhelishvili, L.; Izawa, T.; Yamate, J.; Kosyk, O.; Shymonyak, S.; Bradford, B.U.; et al. Molecular mechanisms of fibrosis-associated promotion of liver carcinogenesis. Toxicol. Sci. 2013, 132, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Wijesundera, K.K.; Izawa, T.; Tennakoon, A.H.; Murakami, H.; Golbar, H.M.; Katou-Ichikawa, C.; Tanaka, M.; Kuwamura, M.; Yamate, J. M1- and M2-macrophage polarization in rat liver cirrhosis induced by thioacetamide (TAA), focusing on Iba1 and galectin-3. Exp. Mol. Pathol. 2014, 96, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Goodman, Z.D.; Ishak, K.G. Histopathology of hepatitis C virus infection. Semin. Liver Dis. 1995, 15, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration: Alternative epithelial pathways. Int. J. Biochem. Cell Biol. 2011, 43, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.A. Reversibility of liver fibrosis. Gastroenterol. Hepatol. 2013, 9, 737–739. [Google Scholar]
- Stickel, F. Alcoholic cirrhosis and hepatocellular carcinoma. Adv. Exp. Med. Biol. 2015, 815, 113–130. [Google Scholar] [PubMed]
- Iwaisako, K.; Taura, K.; Koyama, Y.; Takemoto, K.; Asagiri, M. Strategies to detect hepatic myofibroblasts in liver cirrhosis of different etiologies. Curr. Pathobiol. Rep. 2014, 2, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Luedde, T.; Sauerbruch, T.; Scholten, D.; Streetz, K.; Tacke, F.; Tolba, R.; Trautwein, C.; Trebicka, J.; Weiskirchen, R. Experimental liver fibrosis research: Update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Tennakoon, A.H.; Izawa, T.; Wijesundera, K.K.; Katou-Ichikawa, C.; Tanaka, M.; Golbar, H.M.; Kuwamura, M.; Yamate, J. Analysis of glial fibrillary acidic protein (GFAP)-expressing ductular cells in a rat liver cirrhosis model induced by repeated injections of thioacetamide (TAA). Exp. Mol. Pathol. 2015, 98, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Golbar, H.M.; Izawa, T.; Wijesundera, K.K.; Tennakoon, A.H.; Katou-Ichikawa, C.; Tanaka, M.; Kuwamura, M.; Yamate, J. Expression of nestin in remodelling of α-naphthylisothiocyanate-induced acute bile duct injury in rats. J. Comp. Pathol. 2014, 151, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Masumi, S.; Moriyama, M.; Kannan, Y.; Ohta, M.; Sugano, T.; Yamate, J. Population of hepatic macrophages and response of perfused liver to platelet-activating factor during production of thioacetamide-induced cirrhosis in rats. Hepatology 1996, 24, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Yovchev, M.I.; Xue, Y.; Shafritz, D.A.; Locker, J.; Oertel, M. Repopulation of the fibrotic/cirrhotic rat liver by transplanted hepatic stem/progenitor cells and mature hepatocytes. Hepatology 2014, 59, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Desmet, V.J.; Krstulović, B.; van Damme, B. Histochemical study of rat liver in α-naphthyl isothiocyanate (ANIT) induced cholestasis. Am. J. Pathol. 1968, 52, 401–421. [Google Scholar] [PubMed]
- Lesage, G.; Glaser, S.; Ueno, Y.; Alvaro, D.; Baiocchi, L.; Kanno, N.; Phinizy, J.L.; Francis, H.; Alpini, G. Regression of cholangiocyte proliferation after cessation of ANIT feeding is coupled with increased apoptosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G182–G190. [Google Scholar] [PubMed]
- Tjandra, K.; Sharkey, K.A.; Swain, M.G. Progressive development of a Th1-type hepatic cytokine profile in rats with experimental cholangitis. Hepatology 2000, 31, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.-H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. 2014, 111, E3297–E3305. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, X.; Brenner, D.; Kisseleva, T. Novel perspectives on the origins of the hepatic myofibroblasts. Cell Health Cytoskelet. 2015, 7, 111–119. [Google Scholar] [CrossRef]
- Brenner, D.A.; Kisseleva, T.; Scholten, D.; Paik, Y.H.; Iwaisako, K.; Inokuchi, S.; Schnabl, B.; Seki, E.; de Minicis, S.; Oesterreicher, C.; et al. Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair 2012, 5, S17. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Kaimori, A.; Potter, J.; Kaimori, J.-Y.; Wang, C.; Mezey, E.; Koteish, A. Transforming growth factor-β1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 2007, 282, 22089–22101. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.-L.; Dai, C.; Michalopoulos, G.K.; Liu, Y. Hepatocyte growth factor attenuates liver fibrosis induced by bile duct ligation. Am. J. Pathol. 2006, 168, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Porrello, A.; Jung, Y.; Yang, L.; Popov, Y.; Choi, S.S.; Witek, R.P.; Alpini, G.; Venter, J.; Vandongen, H.M.; et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J. Clin. Investig. 2008, 118, 3331–3342. [Google Scholar] [CrossRef] [PubMed]
- Yovchev, M.I.; Grozdanov, P.N.; Zhou, H.; Racherla, H.; Guha, C.; Dabeva, M.D. Identification of adult hepatic progenitor cells capable of repopulating injured rat liver. Hepatology 2008, 47, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Díaz, R.; Kim, J.W.; Hui, J.-J.; Li, Z.; Swain, G.P.; Fong, K.S.K.; Csiszar, K.; Russo, P.A.; Rand, E.B.; Furth, E.E.; et al. Evidence for the epithelial to mesenchymal transition in biliary atresia fibrosis. Hum. Pathol. 2008, 39, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Desmet, V.; Roskams, T.; van Eyken, P. Ductular reaction in the livers. Pathol. Res. Pract. 1995, 191, 513–524. [Google Scholar] [CrossRef]
- Zhou, H.; Rogler, L.E.; Teperman, L.; Morgan, G.; Rogler, C.E. Identification of hepatocytic and bile ductular cell lineages and candidate stem cells in bipolar ductular reactions in cirrhotic human liver. Hepatology 2007, 45, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Clouston, A.D.; Powell, E.E.; Walsh, M.J.; Richardson, M.M.; Demetris, A.J.; Jonsson, J.R. Fibrosis correlates with a ductular reaction in hepatitis C: Roles of impaired replication, progenitor cells and steatosis. Hepatology 2005, 41, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, K.D.; Zaret, K.S. Distinct populations of endoderm cells converge to generate the embryonic liver bud and ventral foregut tissues. Dev. Biol. 2005, 280, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Strazzabosco, M.; Fabris, L. Development of the bile ducts: Essentials for the clinical hepatologist. J. Hepatol. 2012, 56, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, R.; Suñer, R.E.; van Hul, N.; Kopp, J.L.; Beaudry, J.-B.; Cordi, S.; Antoniou, A.; Raynaud, P.; Lepreux, S.; Jacquemin, P.; et al. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology 2011, 141, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, E.; Carpino, G.; Cardinale, V.; Franchitto, A.; Onori, P.; Alvaro, D. New insights into liver stem cells. Dig. Liver Dis. 2009, 41, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.I.; Xiang, Y.-Y.; Lobe, C.; Filmus, J. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. 2005, 65, 6245–6254. [Google Scholar] [CrossRef] [PubMed]
- Yuzugullu, H.; Benhaj, K.; Ozturk, N.; Senturk, S.; Celik, E.; Toylu, A.; Tasdemir, N.; Yilmaz, M.; Erdal, E.; Akcali, K.C.; et al. Canonical Wnt signaling is antagonized by noncanonical Wnt5a in hepatocellular carcinoma cells. Mol. Cancer 2009, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yan, H.-X.; Chen, L.; Liu, Q.; He, Y.-Q.; Yu, L.-X.; Zhang, S.-H.; Huang, D.-D.; Tang, L.; Kong, X.-N.; et al. Wnt/β-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008, 68, 4287–4295. [Google Scholar] [CrossRef] [PubMed]
- Sell, S.; Leffert, H.L. Liver cancer stem cells. J. Clin. Oncol. 2008, 26, 2800–2805. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, X.; Xu, Y.; Li, X.; Ren, S.; Zhou, Y.; Duan, Y.; Zern, M.; Zhang, H.; Chen, G.; et al. Hepatic progenitor cells contribute to the progression of 2-acetylaminofluorene/carbon tetrachloride-induced cirrhosis via the non-canonical Wnt pathway. PLoS ONE 2015, 10, e0130310. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, T.; Cong, M.; Wu, X.; Bai, Y.; Yin, C.; An, W.; Wang, B.; Jia, J.; You, H. Expression of extracellular matrix genes in cultured hepatic oval cells: An origin of hepatic stellate cells through transforming growth factor β? Liver Int. 2009, 29, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Sicklick, J.K.; Choi, S.S.; Bustamante, M.; McCall, S.J.; Pérez, E.H.; Huang, J.; Li, Y.-X.; Rojkind, M.; Diehl, A.M. Evidence for epithelial-mesenchymal transitions in adult liver cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G575–G583. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Zeisberg, M.; Kalluri, R. Transcriptional regulation of epithelial-mesenchymal transition. J. Clin. Investig. 2007, 117, 304–306. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.S.; Diaz, R.; Hui, J.-J.; Yanger, K.; Zong, Y.; Alpini, G.; Stanger, B.Z.; Wells, R.G. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology 2011, 53, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Kordes, C.; Sawitza, I.; Müller-Marbach, A.; Ale-Agha, N.; Keitel, V.; Klonowski-Stumpe, H.; Häussinger, D. CD133+ hepatic stellate cells are progenitor cells. Biochem. Biophys. Res. Commun. 2007, 352, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.; Vandongen, H.M.; Huang, J.; Alpini, G.D.; Diehl, A.M. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells 2008, 26, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tennakoon, A.H.; Izawa, T.; Kuwamura, M.; Yamate, J. Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Renal and Hepatic Fibrosis. J. Clin. Med. 2016, 5, 4. https://doi.org/10.3390/jcm5010004
Tennakoon AH, Izawa T, Kuwamura M, Yamate J. Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Renal and Hepatic Fibrosis. Journal of Clinical Medicine. 2016; 5(1):4. https://doi.org/10.3390/jcm5010004
Chicago/Turabian StyleTennakoon, Anusha H., Takeshi Izawa, Mitsuru Kuwamura, and Jyoji Yamate. 2016. "Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Renal and Hepatic Fibrosis" Journal of Clinical Medicine 5, no. 1: 4. https://doi.org/10.3390/jcm5010004
APA StyleTennakoon, A. H., Izawa, T., Kuwamura, M., & Yamate, J. (2016). Pathogenesis of Type 2 Epithelial to Mesenchymal Transition (EMT) in Renal and Hepatic Fibrosis. Journal of Clinical Medicine, 5(1), 4. https://doi.org/10.3390/jcm5010004