
Liquid Biopsies for Cancer: Coming to a Patient near You


Abstract
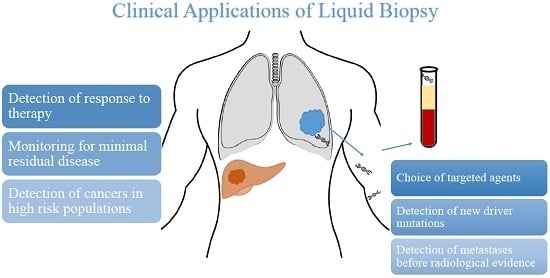
:
1. Introduction
2. Biology of ctDNA
2.1. Amount and Fragmentation of ctDNA
2.2. Methylation Profiling
3. Diagnosis, Liquid Biopsy
3.1. Circulating Tumor DNA versus Tissue Biopsy
3.2. Circulating Tumor DNA versus Circulating Tumor Cells
3.3. Circulating Tumor DNA versus Cancer Antigens
4. Personalized Cancer Treatment
4.1. Cancer Prognosis, Relapse, and Resistance
4.2. Alternative Liquid Biopsy Sources
4.3. Available Liquid Biopsy
5. Limitations
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lo, Y.D.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. The nucleic acids in blood plasma in humans. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Chiu, R.W.; Lo, Y.M. Noninvasive prenatal diagnosis empowered by high-throughput sequencing. Prenat. Diagn. 2012, 32, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, D.W.; Chudova, D.; Sehnert, A.J.; Bhatt, S.; Murray, K.; Prosen, T.L.; Garber, J.E.; Wilkins-Haug, L.; Vora, N.L.; Warsof, S.; et al. Noninvasive Prenatal Testing and Incidental Detection of Occult Maternal Malignancies. JAMA 2015, 314, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Tada, M.; Omata, M.; Kawai, S.; Saisho, H.; Ohto, M.; Saiki, R.K.; Sninsky, J.J. Detection of ras gene mutations in pancreatic juice and peripheral blood of patients with pancreatic adenocarcinoma. Cancer Res. 1993, 53, 2472–2474. [Google Scholar] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Eder, J.; Pratscher, B.; Brandt, S.; Schneller, D.; Müllegger, R.; Vogl, C.; Trautinger, F.; Brem, G.; Burgstaller, J.P. SNPase-ARMS qPCR: Ultrasensitive Mutation-Based Detection of Cell-Free Tumor DNA in Melanoma Patients. PLoS ONE 2015, 10, e0142273. [Google Scholar] [CrossRef] [PubMed]
- Lanman, R.B.; Mortimer, S.A.; Zill, O.A.; Sebisanovic, D.; Lopez, R.; Blau, S.; Collisson, E.A.; Divers, S.G.; Hoon, D.S.; Kopetz, E.S.; et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS ONE 2015, 10, e0140712. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Fleischhacker, M.; Rabien, A. Cell-free DNA in the blood as a solid tumor biomarker—A critical appraisal of the literature. Clin. Chim. Acta 2010, 411, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Jiang, P.; Chan, C.W.; Chan, K.A.; Cheng, S.H.; Wong, J.; Wong, V.W.; Wong, G.L.; Chan, S.L.; Mok, T.S.; Chan, H.L.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Robert, B.; Peyrotte, E.A.; Del Rio, M.; Ychou, M.; Molina, F.; Gongora, C.; Thierry, A.R. High fragmentation characterizes tumour-derived circulating DNA. PLoS ONE 2011, 6, e23418. [Google Scholar] [CrossRef] [PubMed]
- Madhavan, D.; Wallwiener, M.; Bents, K.; Zucknick, M.; Nees, J.; Schott, S.; Cuk, K.; Riethdorf, S.; Trumpp, A.; Pantel, K.; et al. Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res. Treat. 2014, 146, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Leszinski, G.; Lehner, J.; Gezer, U. Increased DNA integrity in colorectal cancer. In Vivo 2014, 28, 299–303. [Google Scholar] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Liggett, T.E.; Melnikov, A.; Yi, Q.; Replogle, C.; Hu, W.; Rotmensch, J.; Kamat, A.; Sood, A.K.; Levenson, V. Distinctive DNA methylation patterns of cell-free plasma DNA in women with malignant ovarian tumors. Gynecol. Oncol. 2011, 120, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.P.; Campan, M.; Hinoue, T.; Schmitz, R.F.; van der Meulen-de, A.E.; Slingerland, H.; Kok, P.J.; van Dijk, C.M.; Weisenberger, D.J.; Shen, H.; et al. Genome-scale discovery of DNA-methylation biomarkers for blood-based detection of colorectal cancer. PLoS ONE 2012, 7, e50266. [Google Scholar] [CrossRef] [PubMed]
- Mahon, K.L.; Qu, W.; Devaney, J.; Paul, C.; Castillo, L.; Wykes, R.J.; Chatfield, M.D.; Boyer, M.J.; Stockler, M.R.; Marx, G.; et al. Methylated Glutathione S-transferase 1 (mGSTP1) is a potential plasma free DNA epigenetic marker of prognosis and response to chemotherapy in castrate-resistant prostate cancer. Br. J. Cancer 2014, 1119, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Margolin, G.; Petrykowska, H.M.; Jameel, N.; Bell, D.W.; Young, A.C.; Elnitski, L. Robust Detection of DNA Hypermethylation of ZNF154 as a Pan-Cancer Locus with in Silico Modeling for Blood-Based Diagnostic Development. J. Mol. Diagn. 2016, 18, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Abramson, R. Overview of Targeted Therapies for Cancer. My Cancer Genome, 2016. Available online: https://www.mycancergenome.org/content/molecular-medicine/overview-of-targeted-therapies-for-cancer/ (accessed on 23 July 2016).
- Patel, K.M.; Tsui, D.W. The translational potential of circulating tumour DNA in oncology. Clin. Biochem. 2015, 48, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Yap, T.A.; Pope, L.; Cassidy, A.M.; Dukes, J.P.; Riisnaes, R.; Massard, C.; Cassier, P.A.; Miranda, S.; Clark, J.; et al. Multi-purpose utility of circulating plasma DNA testing in patients with advanced cancers. PLoS ONE 2012, 7, e47020. [Google Scholar] [CrossRef] [PubMed]
- Couraud, S.; Vaca-Paniagua, F.; Villar, S.; Oliver, J.; Schuster, T.; Blanché, H.; Girard, N.; Trédaniel, J.; Guilleminault, L.; Gervais, R.; et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: A proof-of-concept study from BioCAST/IFCT-1002. Clin Cancer Res. 2014, 20, 4613–4624. [Google Scholar] [CrossRef] [PubMed]
- Lebofsky, R.; Decraene, C.; Bernard, V.; Kamal, M.; Blin, A.; Leroy, Q.; Frio, T.R.; Pierron, G.; Callens, C.; Bieche, I.; et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol. Oncol. 2015, 9, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Peppercorn, J. Toward improved understanding of the ethical and clinical issues surrounding mandatory research biopsies. J. Clin. Oncol. 2013, 31, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auer, M.; Ulz, P.; Geigl, J.B.; Speicher, M.R. Circulating tumor cells and DNA as liquid biopsies. Genome Med. 2013, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Freidin, M.B.; Freydina, D.V.; Leung, M.; Fernandez, A.M.; Nicholson, A.G.; Lim, E. Circulating Tumor DNA Outperforms Circulating Tumor Cells for KRAS Mutation Detection in Thoracic Malignancies. Clin. Chem. 2015, 61, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.A.; Stebbing, J. Circulating free DNA in the management of breast cancer. Ann. Transl. Med. 2014, 2, 3. [Google Scholar] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer [EURTAC]: A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Yong, E. Cancer biomarkers: Written in blood. Nature 2014, 511, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.L.; Pallisgaard, N.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Circulating free DNA as biomarker and source for mutation detection in metastatic colorectal cancer. PLoS ONE 2015, 10, e0108247. [Google Scholar] [CrossRef] [PubMed]
- Martignetti, J.A.; Camacho-Vanegas, O.; Priedigkeit, N.; Camacho, C.; Pereira, E.; Lin, L.; Garnar-Wortzel, L.; Miller, D.; Losic, B.; Shah, H.; et al. Personalized ovarian cancer disease surveillance and detection of candidate therapeutic drug target in circulating tumor DNA. Neoplasia 2014, 16, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Menon, U.; Griffin, M.; Gentry-Maharaj, A. Ovarian cancer screening—Current status, future directions. Gynecol. Oncol. 2014, 132, 490–495. [Google Scholar] [CrossRef] [PubMed]
- De Cuba, E.M.; Snaebjornsson, P.; Heideman, D.A.; van Grieken, N.C.; Bosch, L.J.; Fijneman, R.J.; Belt, E.; Bril, H.; Stockmann, H.B.; Hooijberg, E.; et al. Prognostic value of BRAF and KRAS mutation status in stage II and III microsatellite instable colon cancers. Int. J. Cancer 2016, 138, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Criscitiello, C.; Locatelli, M.; Milano, M.; Curigliano, G. Liquid biopsies for solid tumors: Understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol. Ther. 2016, 157, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.H.; Dahlgren, M.; Schulz, R.; Grabau, D.; van Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, T.K.; Sequist, L.V.; Heymach, J.V.; Riely, G.J.; Jänne, P.A.; Koch, W.H.; Sullivan, J.P.; Fox, D.B.; Maher, R.; Muzikansky, A.; et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin. Cancer Res. 2016, 22, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Hovelson, D.H.; Tomlins, S.A. The Role of Next-Generation Sequencing in Castration-Resistant Prostate Cancer Treatment. Cancer J. 2016, 22, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. Prostate cancer: Genetics of mCRPC tracked in ctDNA. Nat. Rev. Urol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain tumor mutations detected in cerebral spinal fluid. Clin. Chem. 2015, 61, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Goessl, C.; Muller, M.; Straub, B.; Miller, K. DNA alterations in body fluids as molecular tumor markers for urological malignancies. Eur. Urol. 2002, 41, 668–676. [Google Scholar] [CrossRef]
- Husain, H.; Kosco, K.; Guerrero, S.; Lu, T.T.; Vibat, C.R.T.; Erlander, M.G.; Melnikova, V. Detection of EGFR T790M mutation in urinary circulating tumor DNA from metastatic non-small cell lung cancer patients. Ann. Oncol. 2015, 26, i10. [Google Scholar] [CrossRef]
- Su, Y.H.; Wang, M.; Brenner, D.E.; Norton, P.A.; Block, T.M. Detection of mutated K-ras DNA in urine, plasma, and serum of patients with colorectal carcinoma or adenomatous polyps. Ann. N. Y. Acad. Sci. 2008, 1137, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Springer, S.; Mulvey, C.L.; Silliman, N.; Schaefer, J.; Sausen, M.; James, N.; Rettig, E.M.; Guo, T.; Pickering, C.R.; et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci. Transl. Med. 2015, 7, 293ra104. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.; Giardina, T.; Carrello, A.; Spagnolo, D.V.; Amanuel, B. Detection of EGFR mutational profile by direct dideoxy sequencing in cytology and non-cytology biopsy samples. Pathology 2014, 46, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Benlloch, S.; Martí-Ciriquián, J.L.; Galbis-Caravajal, J.M.; Martín, C.; Sánchez-Payá, J.; Rodríguez-Paniagua, J.M.; Romero, S.; Massutí, B. Cell-free DNA concentration in pleural fluid and serum: Quantitative approach and potential prognostic factor in patients with cancer and pleural effusions. Clin. Lung Cancer 2006, 8, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Garner, D. Clinical application of DNA ploidy to cervical cancer screening: A review. World J. Clin. Oncol. 2014, 55, 931–965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qi, J.; Wu, Y.Q.; Zhang, P.; Jiang, J.; Wang, Q.X.; Zhu, Y.Q. Accuracy of early detection of colorectal tumours by stool methylation markers: A meta-analysis. World J. Gastroenterol. 2014, 20, 14040–14050. [Google Scholar] [CrossRef] [PubMed]
- Uchida, J.; Kato, K.; Kukita, Y.; Kumagai, T.; Nishino, K.; Daga, H.; Nagatomo, I.; Inoue, T.; Kimura, M.; Oba, S.; et al. Diagnostic accuracy of noninvasive genotyping of EGFR in lung cancer patients by deep sequencing of plasma cell-free DNA. Clin. Chem. 2015, 61, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- PathwayGenomics. Liquid Biopsy for the Detection and Monitoring of Cancer: Analysis of 96 Hotspot Mutations via Plasma Derived Circulating Tumor DNA. White Paper. Available online: https://d2q8958pmybfsy.cloudfront.net/wp-content/uploads/2014/07/T-1032.001-CancerIntercept_WhitePaper.pdf?280a6d2015 (accessed on 4 August 2016).
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.; Hofman, V.; Long, E.; Bordone, O.; Selva, E.; Washetine, K.; Marquette, C.H.; Hofman, P. Current challenges for detection of circulating tumor cells and cell-free circulating nucleic acids, and their characterization in non-small cell lung carcinoma patients. What is the best blood substrate for personalized medicine? Ann. Transl. Med. 2014, 2, 107. [Google Scholar] [PubMed]
- Reck, M.; Hagiwara, K.; Han, B.; Tjulandin, S.; Grohe, C.; Yokoi, T.; Morabito, A.; McCormack, R.; Ratcliffe, M.; Normanno, N. 35O_PR Investigating the utility of circulating-free tumor DNA(ctDNA) in plasma for the detection of epidermal growth factor receptor(EGFR) mutation status in European and Japanese patients (PTS) with advanced non-small cell lung cancer(ANSCLC): Assess study. Ann. Oncol. 2015, 26, i58–i59. [Google Scholar]
- Hou, W.; Bonkovsky, H.L. Non-coding RNAs in hepatitis C-induced hepatocellular carcinoma: Dysregulation and implications for early detection, diagnosis and therapy. World J. Gastroenterol. 2013, 19, 7836–7845. [Google Scholar] [CrossRef] [PubMed]
- Kishikawa, T.; Otsuka, M.; Ohno, M.; Yoshikawa, T.; Takata, A.; Koike, K. Circulating RNAs as new biomarkers for detecting pancreatic cancer. World J. Gastroenterol. 2015, 21, 8527–8540. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.M.; Fischer, M.; Blees, J.; Zech, M.; Siegwolf, R.T.; Saurer, M. A novel methylation derivatization method for δ18O analysis of individual carbohydrates by gas chromatography/pyrolysis-isotope ratio mass spectrometry. Rapid Commun. Mass Spectrom. 2016, 30, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, Y.; Kosaka, N.; Konishi, Y.; Ohta, H.; Okamoto, H.; Sonoda, H.; Nonaka, R.; Yamamoto, H.; Ishii, H.; Mori, M.; et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun. 2014, 5, 3591. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Type of Cancer with ctDNA Detection | Results | References |
|---|---|---|
| Breast cancer | ctDNA-based detection preceded clinical detection of metastasis in 86% of patients | [47] |
| Breast cancer | 55 non-metastatic breast cancer patients on neo-adjuvant chemotherapy; in the immediate post-operative period, 19% of available patients had detectible ctDNA, representing minimal residual disease (MRD), and 86% of these women went on to relapse during the study period | [40,48] |
| Colorectal cancer | metastatic colorectal cancer demonstrated 100% diagnostic sensitivity and specificity for mutant BRAF detection and 92% sensitivity/98% specificity for seven tested KRAS point mutations | [41] |
| Lung cancer | With tumor tissue DNA used as a reference, ctDNA demonstrated a specificity of 86% for PI3KCA exon 9, 88% for EGFR exon 19, and 100% for other measured amplicons, with an 87% (62%–96%) overall average specificity. Certain PIK3CA and EGFR hot-spot mutations were detected in ctDNA but not in the tissue DNA | [49] |
| Prostate cancer | Tumor DNA samples from the blood of 97 patients with castration-resistant prostate cancer at different times during the course of treatment with abiraterone revealed androgen receptor amplifications were present from the beginning and correlated with abiraterone resistance | [50,51] |
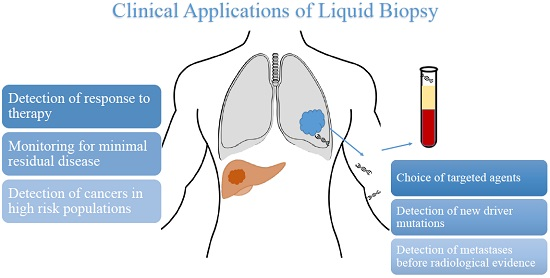
| Potential Uses of Liquid Biopsy |
|---|
| Detection of cancers in high-risk population |
| Monitoring for minimal residual disease |
| Detection of metastases before radiological evidence |
| Detection of response to therapy |
| Choice of targeted agent |
| Detection of new driver mutations |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnamurthy, N.; Spencer, E.; Torkamani, A.; Nicholson, L. Liquid Biopsies for Cancer: Coming to a Patient near You. J. Clin. Med. 2017, 6, 3. https://doi.org/10.3390/jcm6010003
Krishnamurthy N, Spencer E, Torkamani A, Nicholson L. Liquid Biopsies for Cancer: Coming to a Patient near You. Journal of Clinical Medicine. 2017; 6(1):3. https://doi.org/10.3390/jcm6010003
Chicago/Turabian StyleKrishnamurthy, Nithya, Emily Spencer, Ali Torkamani, and Laura Nicholson. 2017. "Liquid Biopsies for Cancer: Coming to a Patient near You" Journal of Clinical Medicine 6, no. 1: 3. https://doi.org/10.3390/jcm6010003
APA StyleKrishnamurthy, N., Spencer, E., Torkamani, A., & Nicholson, L. (2017). Liquid Biopsies for Cancer: Coming to a Patient near You. Journal of Clinical Medicine, 6(1), 3. https://doi.org/10.3390/jcm6010003