
Mitochondrial Modification Techniques and Ethical Issues

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Treatment of Mitochondrial Diseases
2.1. Mitochondrial Replacement Techniques
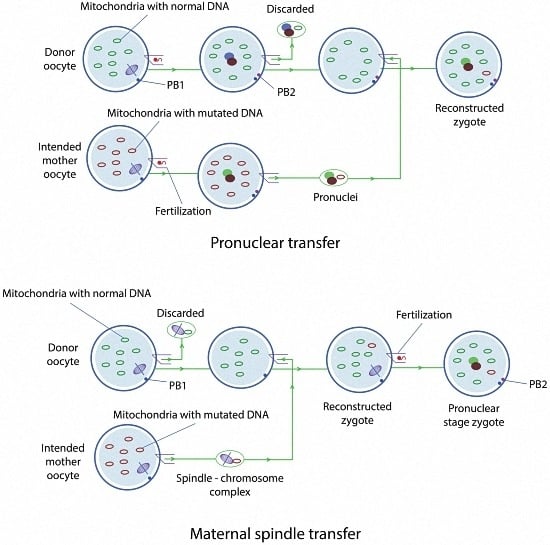
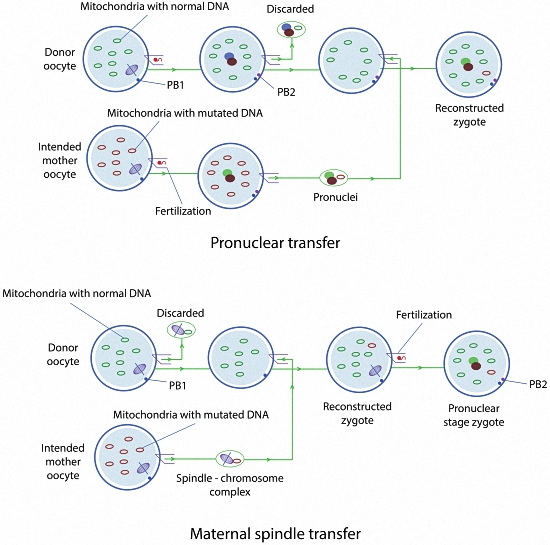
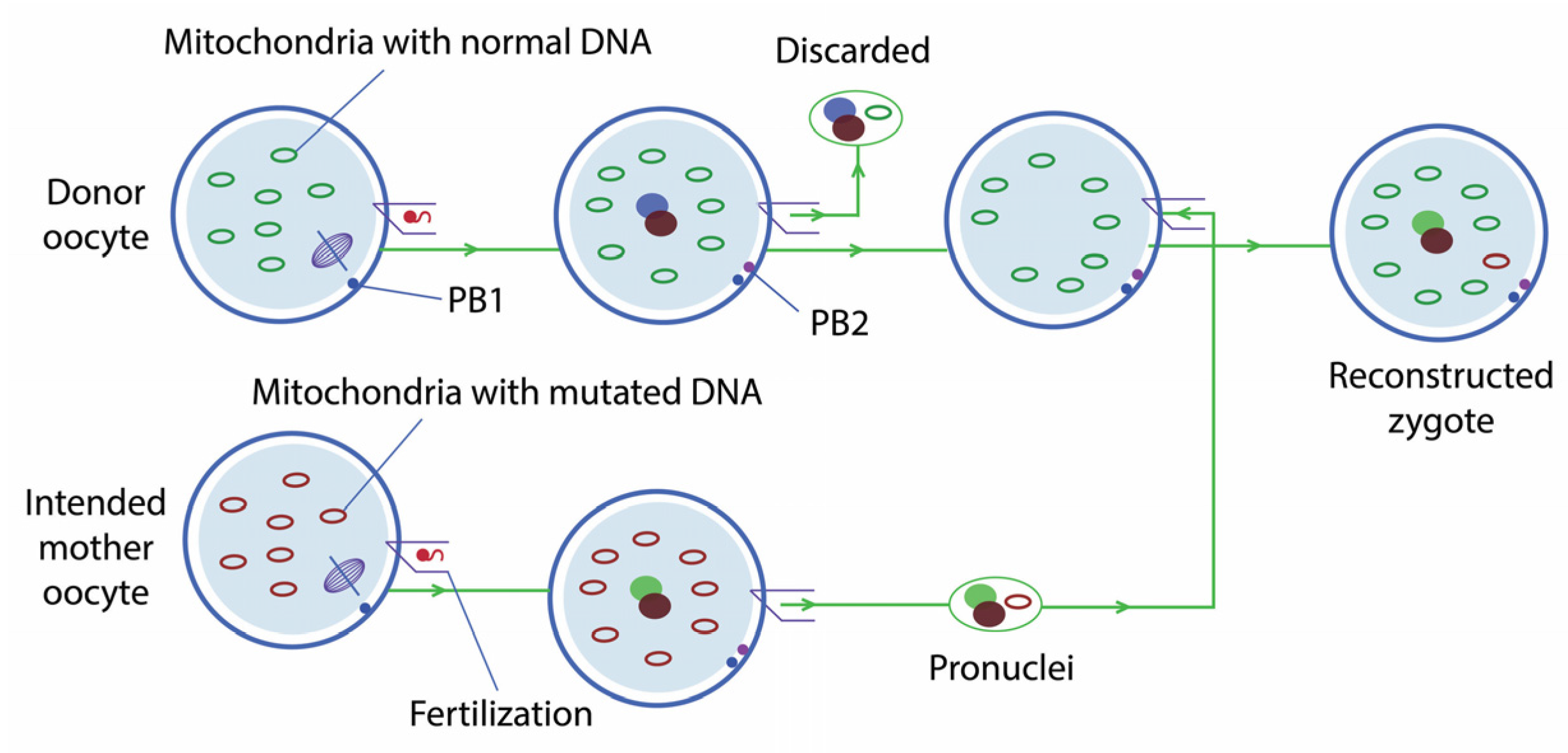
2.1.1. Pronuclear Transfer
2.1.2. Maternal Spindle Transfer
2.1.3. Polar Body Transfer
2.2. Mitochondrial Gene Editing
2.2.1. CRISPR/Cas9
2.2.2. TALENs
3. Bioethical Issues
3.1. Germline Genetic Modification
3.2. Application of Mitochondrial Transfer in Cases of Infertility
3.3. Donor-Recipient Relationship
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van der Giezen, M.; Tovar, J. Degenerate mitochondria. EMBO Rep. 2005, 6, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016. [Google Scholar] [CrossRef] [PubMed]
- Nass, S.; Nass, M.M.K. Intramitochondrial fibers with DNA characteristics II. Enzymatic and Other Hydrolytic Treatments. J. Cell Biol. 1963, 19, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010, 9, 829–840. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A.; Carelli, V.; Hirano, M. The clinical maze of mitochondrial neurology. Nat. Rev. Neurol. 2013, 9, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Elliott, H.R.; Samuels, D.C.; Eden, J.A.; Relton, C.L.; Chinnery, P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008, 83, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Howell, N.; Lightowlers, R.N.; Turnbull, D.M. Molecular pathology of MELAS and MERRF. The relationship between mutation load and clinical phenotypes. Brain 1997, 120, 1713–1721. [Google Scholar] [PubMed]
- White, S.L.; Collins, V.R.; Wolfe, R.; Cleary, M.A.; Shanske, S.; DiMauro, S.; Dahl, H.H.; Thorburn, D.R. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am. J. Hum. Genet. 1999, 65, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Nuffield Council on Bioethics. Novel Techniques for the Prevention of Mitochondrial DNA Disorders: An Ethical Review. Available online: http://nuffieldbioethics.org/wp-content/uploads/2014/06/Novel_techniques_for_the_prevention_of_mitochondrial_DNA_disorders_compressed.pdf (accessed on 23 December 2016).
- Wilson, I.J.; Carling, P.J.; Alston, C.L.; Floros, V.I.; Pyle, A.; Hudson, G.; Sallevelt, S.C.; Lamperti, C.; Carelli, V.; Bindoff, L.A.; et al. Mitochondrial DNA sequence characteristics modulate the size of the genetic bottleneck. Hum. Mol. Genet. 2016, 25, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Howell, N.; Lightowlers, R.N.; Turnbull, D.M. Genetic counseling and prenatal diagnosis for mtDNA disease. Am. J. Hum. Genet. 1998, 63, 1908–1911. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, D.R.; Dahl, H.H. Mitochondrial disorders: Genetics, counseling, prenatal diagnosis and reproductive options. Am. J. Med. Genet. 2001, 106, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.T.; Herbert, M.; Lamb, V.K.; Chinnery, P.F.; Taylor, R.W.; Lightowlers, R.N.; Craven, L.; Cree, L.; Gardner, J.L.; Turnbull, D.M. Transmission of mitochondrial DNA disorders: Possibilities for the future. Lancet 2006, 368, 87–89. [Google Scholar] [CrossRef]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012, 18, CD004426. [Google Scholar]
- HFEA Approves Licence Application to Use Gene Editing in Research. Available online: http://www.hfea.gov.uk/10187.html (accessed on 23 December 2016).
- HFEA Reconvenes Independent Expert Panel and Launches Call for Evidence. Available online: http://www.hfea.gov.uk/10363.html (accessed on 23 December 2016).
- Scientific Review of the Safety and Efficacy of Methods to Avoid Mitochondrial Disease through Assisted Conception: 2016 Update. Available online: http://www.hfea.gov.uk/docs/Fourth_scientific_review_mitochondria_2016.PDF (accessed on 23 December 2016).
- HFEA Permits Cautious Use of Mitochondrial Donation in Treatment, Following Advice from Scientific Experts. Available online: http://www.hfea.gov.uk/10563.html (accessed on 23 December 2016).
- Yamada, M.; Emmanuele, V.; Sanchez-Quintero, M.J.; Sun, B.; Lallos, G.; Paull, D.; Zimmer, M.; Pagett, S.; Prosser, R.W.; Sauer, M.V.; et al. Genetic Drift Can Compromise Mitochondrial Replacement by Nuclear Transfer in Human Oocytes. Cell Stem Cell 2016, 18, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Hyslop, L.A.; Blakeley, P.; Craven, L.; Richardson, J.; Fogarty, N.M.; Fragouli, E.; Lamb, M.; Wamaitha, S.E.; Prathalingam, N.; Zhang, Q.; et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 2016, 534, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; MartinezRedondo, P.; Ma, H.; Lee, Y.; et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 2016, 540, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Samuels, D.C.; Wonnapinij, P.; Chinnery, P.F. Preventing the transmission of pathogenic mitochondrial DNA mutations: Can we achieve long-term benefits from germ-line gene transfer? Hum. Reprod. 2013, 28, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Amato, P.; Sparman, M.; Woodward, J.; Sanchis, D.M.; Ma, H.; Gutierrez, N.M.; Tippner-Hedges, R.; Kang, E.; Lee, H.S.; et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature 2013, 493, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Tuppen, H.A.; Greggains, G.D.; Harbottle, S.J.; Murphy, J.L.; Cree, L.M.; Murdoch, A.P.; Chinnery, P.F.; Taylor, R.W.; Lightowlers, R.N.; et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 2010, 465, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sha, H.; Ji, D.; Zhang, H.L.; Chen, D.; Cao, Y.; Zhu, J. Polar body genome transfer for preventing the transmission of inherited mitocondrial diseases. Cell 2014, 157, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- National Academies of Sciences, Engineering, and Medicine. Mitochondrial Replacement Techniques: Ethical, Social, and Policy Considerations; National Academies Press: Washington, DC, USA, 2016. [Google Scholar]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Bacman, S.R.; Peralta, S.; Falk, M.J.; Chomyn, A.; Chan, D.C.; Williams, S.L.; Moraes, C.T. MitoTALEN: A General Approach to Reduce Mutant mtDNA Loads and Restore Oxidative Phosphorylation Function in Mitochondrial Diseases. Mol. Ther. 2015, 23, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Minczuk, M.; Papworth, M.A.; Kolasinska, P.; Murphy, M.P.; Klug, A. Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc. Natl. Acad. Sci. USA 2006, 103, 19689–19694. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.; Wolfe, R.; Hendrickx, A.T.; de Coo, I.F.; de Die, C.E.; Geraedts, J.P.; Chinnery, P.F.; Smeets, H.J. PGD and heteroplasmic mitochondrial DNA point mutations: A systematic review estimating the chance of healthy offspring. Hum. Reprod. Update 2012, 18, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhuang, G.; Zeng, Y.; Grifo, J.; Acosta, C.; Shu, Y.; Liu, H. Pregnancy derived from human zygote pronuclear transfer in a patient who had arrested embryos after IVF. Reprod. Biomed. Online 2016, 33, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Luo, S.; Chavez-Badiola, A.; Liu, Z.; Yang, M.; Munne, S.; Konstantinidis, M.; Wells, D.; Huang, T. First live birth using human oocytes reconstituted by spindle nuclear transfer for mitochondrial DNA mutation causing Leigh syndrome. Fertil. Steril. 2016, 106, e375–e376. [Google Scholar] [CrossRef]
- Review of the Safety and Efficacy of Polar Body Transfer to Avoid Mitochondrial Disease. Available online: http://www.hfea.gov.uk/docs/2014–10–07_-_Polar_Body_Transfer_Review_-_Final.PDF (accessed on 23 December 2016).
- Mojica, F.J.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. CRISPR, the disruptor. Nat. News 2015, 522, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D.; Berg, P.; Botchan, M.; Carroll, D.; Charo, R.A.; Church, G.; Corn, J.E.; Daley, G.Q.; Doudna, J.A.; Fenner, M.; et al. A prudent path forward for genomic engineering and germline gene modification. Science 2015, 348, 36–38. [Google Scholar] [CrossRef] [PubMed]
- Lanphier, E.; Urnov, F.; Haecker, S.E.; Werner, M.; Smolenski, J. Don’t edit the human germ line. Nature 2015, 519, 410–411. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Xu, Y.; Zhang, X.; Ding, C.; Huang, R.; Zhang, Z.; Lv, J.; Xie, X.; Chen, Y.; Li, Y.; et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 2015, 6, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; He, W.; Huang, Y.; Yu, Q.; Chen, Y.; Gao, X.; Sun, X.; Fan, Y. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. J. Assist. Reprod. Genet. 2016, 33, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.I.; Kim, S.; Lee, Y.S.; Shin, J.H.; Lee, Y. Efficient Mitochondrial Genome Editing by CRISPR/Cas9. Biomed. Res. Int. 2015, 2015, 305716:1–305716:10. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucl. Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Ocampo, A.; Suzuki, K.; Luo, J.; Bacman, S.R.; Williams, S.L.; Sugawara, A.; Okamura, D.; Tsunekawa, Y.; Wu, J.; et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 2015, 23, 161, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Baylis, F. The ethics of creating children with three genetic parents. Reprod. Biomed. Online 2013, 26, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Darnovsky, M. A slippery slope to human germline modification. Nature 2013, 499, 127. [Google Scholar] [CrossRef] [PubMed]
- Newson, A.J.; Wrigley, A. Is Mitochondrial Donation Germ-Line Gene Therapy? Classifications and Ethical Implications. Bioethics 2017, 31, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Jones, D. The Other Woman: Evaluating the Language of “Three Parent” Embryos. Clin. Eth. 2015, 10, 97–106. [Google Scholar] [CrossRef]
- Nisker, J. The Latest Thorn by Any Other Name: Germ-Line Nuclear Transfer in the Name of “Mitochondrial Replacement”. J. Obstet. Gynaecol. Can. 2015, 37, 829–831. [Google Scholar] [CrossRef]
- Baylis, F. Human Nuclear Genome Transfer (So-Called Mitochondrial Replacement): Clearing the Underbrush. Bioethics 2017, 31, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, K.A. Maternal effect genes: Findings and effects on mouse embryo development. Clin. Exp. Reprod. Med. 2014, 41, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, D.; Group Leader of Mitchondrial Research, Murdoch Childrens Research Institute. Personal communication, 2017.
- Bredenoord, A.L.; Pennings, G.; de Wert, G. Ooplasmic and nuclear transfer to prevent mitochondrial DNA disorders: Conceptual and normative issues. Hum. Reprod. Update 2008, 14, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, K.; Dowling, D.K.; Morrow, E.H. Mitochondrial replacement, evolution and the clinic. Science 2013, 341, 1345–1346. [Google Scholar] [CrossRef] [PubMed]
- Roubertoux, P.L.; Sluyter, F.; Carlier, M.; Marcet, B.; Maarouf-Veray, F.; Chérif, C.; Marican, C.; Arrechi, P.; Godin, F.; Jamon, M.; et al. Mitochondrial DNA modifies cognition in interaction with the nuclear genome and age in mice. Nat. Genet. 2003, 35, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Lease, L.R.; Winnier, D.A.; Williams, J.T.; Dyer, T.D.; Almasy, L.; Mahaney, M.C. Mitochondrial genetic effects on latent class variables associated with susceptibility to alcoholism. BMC Genet. 2005, 6, S158. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Acín-Pérez, R.; Fernández-Silva, P.; Movilla, N.; Pérez-Martos, A.; Rodriguez de Cordoba, S.; Gallardo, M.E.; Enríquez, J.A. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 2006, 38, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Totsuka, Y.; Atomi, Y.; Kaneda, H.; Lindahl, K.F.; Imai, H.; Yonekawa, H. Decreased physical performance of congenic mice with mismatch between the nuclear and the mitochondrial genome. Genes Genet. Syst. 1998, 73, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, P.; Morrow, E.H.; Dowling, D.K. Experimental evidence supports a sexspecific selective sieve in mitochondrial genome evolution. Science 2011, 332, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Camus, M.F.; Clancy, D.J.; Dowling, D.K. Dowling, Mitochondria, maternal inheritance, and male aging. Curr. Biol. 2012, 22, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Clancy, D.J. Variation in mitochondrial genotype has substantial lifespan effects which may be modulated by nuclear background. Aging Cell 2008, 7, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.K.; Maklakov, A.A.; Friberg, U.; Hailer, F. Applying the genetic theories of ageing to the cytoplasm: Cytoplasmic genetic covariation for fitness and lifespan. J. Evol. Biol. 2009, 22, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Maklakov, A.A.; Friberg, U.; Dowling, D.K.; Arnqvist, G. Within-population variation in cytoplasmic genes affects female life span and aging in Drosophila melanogaster. Evolution 2006, 60, 2081–2086. [Google Scholar] [CrossRef] [PubMed]
- Clancy, D.J.; Hime, G.R.; Shirras, A.D. Cytoplasmic male sterility in Drosophila melanogaster associated with a mitochondrial CYTB variant. Heredity 2011, 107, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Yee, W.K.; Sutton, K.L.; Dowling, D.K. In vivo male fertility is affected by naturally occurring mitochondrial haplotypes. Curr. Biol. 2013, 23, R55–R56. [Google Scholar] [CrossRef] [PubMed]
- Montooth, K.L.; Meiklejohn, C.D.; Abt, D.N.; Rand, D.M. Mitochondrial-nuclear epistasis affects fitness within species but does not contribute to fixed incompatibilities between species of Drosophila. Evolution 2010, 64, 3364–3379. [Google Scholar] [CrossRef] [PubMed]
- Sackton, T.B.; Haney, R.A.; Rand, D.M. Cytonuclear coadaptation in Drosophila: Disruption of cytochrome c oxidase activity in backcross genotypes. Evolution 2003, 57, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.K.; Abiega, K.C.; Arnqvist, G. Temperature-specific outcomes of cytoplasmic-nuclear interactions on egg-to-adult development time in seed beetles. Evolution 2007, 61, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Arnqvist, G.; Dowling, D.K.; Eady, P.; Gay, L.; Tregenza, T.; Tuda, M.; Hosken, D.J. Genetic architecture of metabolic rate: Environment specific epistasis between mitochondrial and nuclear genes in an insect. Evolution 2010, 64, 3354–3363. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.K.; Nowostawski, A.L.; Arnqvist, G. Effects of cytoplasmic genes on sperm viability and sperm morphology in a seed beetle: Implications for sperm competition theory? J. Evol. Biol. 2007, 20, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.K.; Meerupati, T.; Arnqvist, G. Cytonuclear interactions and the economics of mating in seed beetles. Am. Nat. 2010, 176, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Burton, R.S.; Ellison, C.K.; Harrison, J.S. The sorry state of F2 hybrids: Consequences of rapid mitochondrial DNA evolution in allopatric populations. Am. Nat. 2006, 168, S14–S24. [Google Scholar] [CrossRef] [PubMed]
- Hamzelou, J. Exclusive: World’s first baby born with new “3 parent” technique. New Sci. 2016. Available online: https://www.newscientist.com/article/2107219-exclusive-worlds-first-baby-born-with-new-3-parent-technique/ (accessed on 3 February 2017). [Google Scholar]
- Schatten, H.; Sun, Q.Y.; Prather, R. The impact of mitochondrial function/dysfunction on IVF and new treatment possibilities for infertility. Reprod. Biol. Endocrinol. 2014, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Scott, R.; Alikani, M.; Schimmel, T.; Munné, S.; Levron, J.; Wu, L.; Brenner, C.; Warner, C.; Willadsen, S. Ooplasmic transfer in mature human oocytes. Mol. Hum. Reprod. 1998, 4, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Yabuuchi, A.; Beyhan, Z.; Kagawa, N.; Mori, C.; Ezoe, K.; Kato, K.; Aono, F.; Takehara, Y.; Kato, O. Prevention of mitochondrial disease inheritance by assisted reproductive technologies: Prospects and challenges. Biochim. Biophys. Acta 2012, 1820, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Scott, R.; Schimmel, T.; Levron, J.; Willadsen, S. Birth of infant after transfer of anucleate donor oocyte cytoplasm into recipient eggs. Lancet 1997, 350, 186–187. [Google Scholar] [CrossRef]
- Barritt, J.A.; Brenner, C.A.; Willadsen, S.; Cohen, J. Spontaneous and artificial changes in human ooplamsic mitochondria. Hum. Reprod. 2000, 15, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Barritt, J.A.; Brenner, C.A.; Malter, H.E.; Cohen, J. Rebuttal: Interooplasmic transfers in humans. Reprod. Biomed. Online 2001, 3, 47–88. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Reproductive medicine. Eggs’ power plants energize new IVF debate. Science 2015, 348, 14–15. [Google Scholar] [CrossRef] [PubMed]
- The Incredible, Surprising, Controversial, New Way to Make a Baby. Available online: http://www.ovascience.com/files/TIME_Magazine_May_2015.pdf (accessed on 23 December 2016).
- Coghlan, A. Exclusive: ‘3-parent’ baby method already used for infertility. New Sci. 2016. Available online: https://www.newscientist.com/article/2108549-exclusive-3-parent-baby-method-already-used-for-infertility/ (accessed on 23 December 2016). [Google Scholar]
- Cook, M. Ethics ignored in ‘3-person embryo’ technique. BioEdge 2016. Available online: http://www.bioedge.org/bioethics/ethics-ignored-in-3-person-embryo-technique/12045 (accessed on 23 December 2016). [Google Scholar]
- Ishii, T. Reproductive medicine involving genome editing: Clinical uncertainties and embryological needs. Reprod. Biomed. Online 2017, 34, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C. The girl with three biological parents. BBC News Mag. 2014. Available online: http://www.bbc.com/news/magazine-28986843?OCID=fbasia&ocid=socialflow_facebook (accessed on 4 February 2017). [Google Scholar]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Tatay, L.; Hernández-Andreu, J.M.; Aznar, J. Mitochondrial Modification Techniques and Ethical Issues. J. Clin. Med. 2017, 6, 25. https://doi.org/10.3390/jcm6030025
Gómez-Tatay L, Hernández-Andreu JM, Aznar J. Mitochondrial Modification Techniques and Ethical Issues. Journal of Clinical Medicine. 2017; 6(3):25. https://doi.org/10.3390/jcm6030025
Chicago/Turabian StyleGómez-Tatay, Lucía, José M. Hernández-Andreu, and Justo Aznar. 2017. "Mitochondrial Modification Techniques and Ethical Issues" Journal of Clinical Medicine 6, no. 3: 25. https://doi.org/10.3390/jcm6030025
APA StyleGómez-Tatay, L., Hernández-Andreu, J. M., & Aznar, J. (2017). Mitochondrial Modification Techniques and Ethical Issues. Journal of Clinical Medicine, 6(3), 25. https://doi.org/10.3390/jcm6030025