Th17 in Animal Models of Rheumatoid Arthritis

Abstract
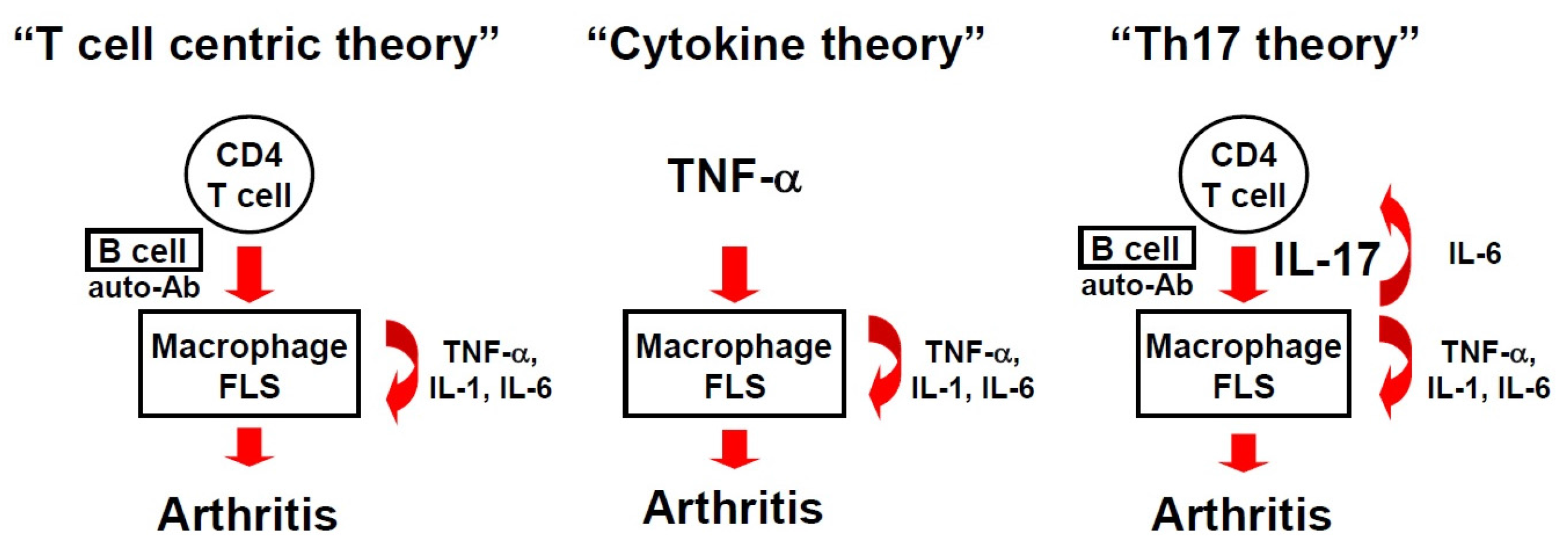
:1. A Brief History of the RA Theory
2. Animal Models of RA
2.1. Type II Collagen-Induced Arthritis
2.2. SKG Mice
2.3. K/BxN Mice
2.4. IL-1 Receptor-Antagonist Knockout Mice
2.5. gp130 F759/F759 Knock-in Mice
2.6. TNF-α Transgenic Mice
3. Role of IL-17 in the Development of Arthritis
4. Interplay between Th17 Cells and Regulatory T Cells
5. Th17 Cells in Mice and Humans
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Lundy, S.K.; Sarkar, S.; Tesmer, L.A.; Fox, D.A. Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res. Ther. 2007, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Janossy, G.; Panayi, G.; Duke, O.; Bofill, M.; Poulter, L.W.; Goldstein, G. Rheumatoid arthritis: A disease of T-lymphocyte/macrophage immunoregulation. Lancet 1981, 2, 839–842. [Google Scholar] [CrossRef]
- Klareskog, L.; Forsum, U.; Scheynius, A.; Kabelitz, D.; Wigzell, H. Evidence in support of a self-perpetuating HLA-DR-dependent delayed-type cell reaction in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 1982, 79, 3632–3636. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; Zvaifler, N.J. How important are T cells in chronic rheumatoid synovitis? Arthritis Rheum. 1990, 33, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Wendling, D.; Racadot, E.; Wijdenes, J.; Sibilia, J.; Flipo, R.M.; Cantagrel, A.; Miossec, P.; Eschard, J.P.; Macro, M.; Bertin, P.; et al. A randomized, double blind, placebo controlled multicenter trial of murine anti-CD4 monoclonal antibody therapy in rheumatoid arthritis. J. Rheumatol. 1998, 25, 1457–1461. [Google Scholar] [PubMed]
- Vermeire, K.; Heremans, H.; Vandeputte, M.; Huang, S.; Billiau, A.; Matthys, P. Accelerated collagen-induced arthritis in IFN-γ receptor-deficient mice. J. Immunol. 1997, 158, 5507–5513. [Google Scholar] [PubMed]
- Firestein, G.S.; Zvaifler, N.J. How important are T cells in chronic rheumatoid synovitis? II. T cell-independent mechanisms from beginning to end. Arthritis Rheum. 2002, 46, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol. 1996, 14, 397–440. [Google Scholar] [CrossRef] [PubMed]
- Udalova, I.A.; Mantovani, A.; Feldmann, M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [PubMed]
- Horai, R.; Saijo, S.; Tanioka, H.; Nakae, S.; Sudo, K.; Okahara, A.; Ikuse, T.; Asano, M.; Iwakura, Y. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J. Exp. Med. 2000, 191, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T.; Ishihara, K.; Kamimura, D.; Ikushima, H.; Ohtani, T.; Hirota, S.; Kobayashi, H.; Park, S.J.; Saeki, Y.; Kitamura, Y.; et al. A point mutation of Tyr-759 in interleukin 6 family cytokine receptor subunit gp130 causes autoimmune arthritis. J. Exp. Med. 2002, 196, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, M.; Durand, J.M.; Buchs, N.; Fossiez, F.; Page, G.; Frappart, L.; Miossec, P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999, 42, 963–970. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-induced arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Khachigian, L.M. Collagen antibody-induced arthritis. Nat. Protoc. 2006, 1, 2512–2516. [Google Scholar] [CrossRef] [PubMed]
- Kagari, T.; Doi, H.; Shimozato, T. The importance of IL-1 β and TNF-α, and the noninvolvement of IL-6, in the development of monoclonal antibody-induced arthritis. J. Immunol. 2002, 169, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Alonzi, T.; Fattori, E.; Lazzaro, D.; Costa, P.; Probert, L.; Kollias, G.; De Benedetti, F.; Poli, V.; Ciliberto, G. Interleukin 6 is required for the development of collagen-induced arthritis. J. Exp. Med. 1998, 187, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Hata, H.; Sakaguchi, N.; Yoshitomi, H.; Iwakura, Y.; Sekikawa, K.; Azuma, Y.; Kanai, C.; Moriizumi, E.; Nomura, T.; Nakamura, T.; et al. Distinct contribution of IL-6, TNF-α, IL-1, and IL-10 to T cell–mediated spontaneous autoimmune arthritis in mice. J. Clin. Investig. 2004, 114, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Hashimoto, M.; Yoshitomi, H.; Tanaka, S.; Nomura, T.; Yamaguchi, T.; Iwakura, Y.; Sakaguchi, N.; Sakaguchi, S. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J. Exp. Med. 2007, 204, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Benham, H.; Rehaume, L.M.; Hasnain, S.Z.; Velasco, J.; Baillet, A.C.; Ruutu, M.; Kikly, K.; Wang, R.; Tseng, H.W.; Thomas, G.P.; et al. Interleukin-23 mediates the intestinal response to microbial beta-1,3-glucan and the development of spondyloarthritis pathology in SKG mice. Arthritis Rheumatol. 2014, 66, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Pettit, A.; Ohmura, K.; Ortiz-Lopez, A.; Duchatelle, V.; Degott, C.; Gravallese, E.; Mathis, D.; Benoist, C. Critical Roles for Interleukin 1 and Tumor Necrosis Factor α in Antibody-induced Arthritis. J. Exp. Med. 2002, 196, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.P.; Wu, H.J.; Benoist, C.; Mathis, D. IL-17-producing T cells can augment autoantibody-induced arthritis. Proc. Natl. Acad. Sci. USA 2009, 106, 21789–21794. [Google Scholar] [CrossRef] [PubMed]
- Pfeifle, R.; Rothe, T.; Ipseiz, N.; Scherer, H.U.; Culemann, S.; Harre, U.; Ackermann, J.A.; Seefried, M.; Kleyer, A.; Uderhardt, S.; et al. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nat. Immunol. 2017, 18, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Koenders, M.I.; Devesa, I.; Marijnissen, R.J.; Abdollahi-Roodsaz, S.; Boots, A.M.; Walgreen, B.; di Padova, F.E.; Nicklin, M.J.; Joosten, L.A.; van den Berg, W.B. Interleukin-1 drives pathogenic Th17 cells during spontaneous arthritis in interleukin-1 receptor antagonist-deficient mice. Arthritis Rheum. 2008, 58, 3461–3470. [Google Scholar] [CrossRef] [PubMed]
- Nakae, S.; Saijo, S.; Horai, R.; Sudo, K.; Mori, S.; Iwakura, Y. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc. Natl. Acad. Sci. USA 2003, 100, 5986–5990. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.L.; Kang, J.W.; Moon, Y.M.; Nam, H.J.; Jhun, J.Y.; Heo, S.B.; Jin, H.T.; Min, S.Y.; Ju, J.H.; Park, K.S.; et al. STAT3 and NF-κB Signal Pathway Is Required for IL-23-Mediated IL-17 Production in Spontaneous Arthritis Animal Model IL-1 Receptor Antagonist-Deficient Mice. J. Immunol. 2006, 176, 5652–5661. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, M.; Ogura, H.; Ueda, N.; Tsuruoka, M.; Kitabayashi, C.; Tsuji, F.; Aono, H.; Ishihara, K.; Huseby, E.; Betz, U.A.; et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int. Immunol. 2007, 19, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Ogura, H.; Murakami, M.; Okuyama, Y.; Tsuruoka, M.; Kitabayashi, C.; Kanamoto, M.; Nishihara, M.; Iwakura, Y.; Hirano, T. Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 2008, 29, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Probert, L.; Plows, D.; Kontogeorgos, G.; Kollias, G. The type I interleukin-1 receptor acts in series with tumor necrosis factor (TNF) to induce arthritis in TNF-transgenic mice. Eur. J. Immunol. 1995, 25, 1794–1797. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Durez, P.; Richards, H.B.; Supronik, J.; Dokoupilova, E.; Mazurov, V.; Aelion, J.A.; Lee, S.H.; Codding, C.E.; Kellner, H.; et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: A phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann. Rheum. Dis. 2013, 72, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Burkett, P.R.; Kuchroo, V.K. IL-17 Blockade in Psoriasis. Cell 2016, 167, 1669. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, A.; Ritchlin, C.; Rahman, P.; Puig, L.; Gottlieb, A.B.; Li, S.; Wang, Y.; Noonan, L.; Brodmerkel, C.; Song, M.; et al. Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: Results of an integrated analysis of radiographic data from the phase 3, multicentre, randomised, double-blind, placebo-controlled PSUMMIT-1 and PSUMMIT-2 trials. Ann. Rheum. Dis. 2014, 73, 1000–1006. [Google Scholar] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H. New developments in osteoimmunology. Nat. Rev. Rheumatol. 2012, 8, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, N.; Takahashi, T.; Hata, H.; Nomura, T.; Tagami, T.; Yamazaki, S.; Sakihama, T.; Matsutani, T.; Negishi, I.; Nakatsuru, S.; et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 2003, 426, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Keith, R.C.; Powers, J.L.; Redente, E.F.; Sergew, A.; Martin, R.J.; Gizinski, A.; Holers, V.M.; Sakaguchi, S.; Riches, D.W. A novel model of rheumatoid arthritis-associated interstitial lung disease in SKG mice. Exp. Lung Res. 2012, 38, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, H.; Sakaguchi, N.; Kobayashi, K.; Brown, G.D.; Tagami, T.; Sakihama, T.; Hirota, K.; Tanaka, S.; Nomura, T.; Miki, I.; et al. A role for fungal β-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J. Exp. Med. 2005, 201, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Hirota, K.; Yoshitomi, H.; Maeda, S.; Teradaira, S.; Akizuki, S.; Prieto-Martin, P.; Nomura, T.; Sakaguchi, N.; Kohl, J.; et al. Complement drives Th17 cell differentiation and triggers autoimmune arthritis. J. Exp. Med. 2010, 207, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- LeibundGut-Landmann, S.; Gross, O.; Robinson, M.J.; Osorio, F.; Slack, E.C.; Tsoni, S.V.; Schweighoffer, E.; Tybulewicz, V.; Brown, G.D.; Ruland, J.; et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 2007, 8, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Ruutu, M.; Thomas, G.; Steck, R.; Degli-Esposti, M.A.; Zinkernagel, M.S.; Alexander, K.; Velasco, J.; Strutton, G.; Tran, A.; Benham, H.; et al. Beta-glucan triggers spondylarthritis and Crohn's disease-like ileitis in SKG mice. Arthritis Rheum. 2012, 64, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Yoshitomi, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 2007, 204, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Hashimoto, M.; Hirota, K.; Ohkura, N.; Morikawa, H.; Nishikawa, H.; Tanaka, A.; Furu, M.; Ito, H.; Fujii, T.; et al. Detection of T cell responses to a ubiquitous cellular protein in autoimmune disease. Science 2014, 346, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Korganow, A.S.; Ji, H.; Mangialaio, S.; Duchatelle, V.; Pelanda, R.; Martin, T.; Degott, C.; Kikutani, H.; Rajewsky, K.; Pasquali, J.L.; et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 1999, 10, 451–461. [Google Scholar] [CrossRef]
- Matsumoto, I.; Staub, A.; Benoist, C.; Mathis, D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science 1999, 286, 1732–1735. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, M.; Zeder-Lutz, G.; Huang, H.; Ebel, C.; Gerber, P.; Hergueux, J.; Marchal, P.; Duchatelle, V.; Degott, C.; van Regenmortel, M.; et al. Arthritogenic Monoclonal Antibodies from K/BxN Mice. J. Exp. Med. 2002, 195, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ohmura, K.; Mahmood, U.; Lee, D.M.; Hofhuis, F.M.A.; Boackle, S.A.; Takahashi, K.; Holerts, V.M.; Walport, M.; Gerard, C.; et al. Arthritis critically dependent on innate immune system players. Immunity 2002, 16, 157–168. [Google Scholar] [CrossRef]
- Lee, D.M.; Friend, D.S.; Gurish, M.F.; Benoist, C.; Mathis, D.; Brenner, M.B. Mast cells: A cellular link between autoantibodies and inflammatory arthritis. Science 2002, 297, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.; Rajasekaran, N.; Jeisy-Walder, E.; Snapper, S.B.; Illges, H. A crucial role for macrophages in the pathology of K/BxN serum-induced arthritis. Eur. J. Immunol. 2005, 35, 3064–3073. [Google Scholar] [CrossRef] [PubMed]
- Auger, J.L.; Cowan, H.M.; Engelson, B.J.; Kashem, S.W.; Prinz, I.; Binstadt, B.A. Brief Report: Arthritis in KRN T Cell Receptor-Transgenic Mice Does Not Require Interleukin-17 or Th17 Cells. Arthritis Rheumatol. 2016, 68, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Block, K.E.; Zheng, Z.; Dent, A.L.; Kee, B.L.; Huang, H. Gut Microbiota Regulates K/BxN Autoimmune Arthritis through Follicular Helper T but Not Th17 Cells. J. Immunol. 2016, 196, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Katayama, M.; Ohmura, K.; Yukawa, N.; Terao, C.; Hashimoto, M.; Yoshifuji, H.; Kawabata, D.; Fujii, T.; Iwakura, Y.; Mimori, T. Neutrophils are essential as a source of IL-17 in the effector phase of arthritis. PLoS ONE 2013, 8, e62231. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar]
- Maeda, Y.; Takeda, K. Role of Gut Microbiota in Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y. Roles of IL-1 in the development of rheumatoid arthritis: Consideration from mouse models. Cytokine Growth Factor Rev. 2002, 13, 341–355. [Google Scholar] [CrossRef]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, T.; Ishihara, K.; Atsumi, T.; Nishida, K.; Kaneko, Y.; Miyata, T.; Itoh, S.; Narimatsu, M.; Maeda, H.; Fukada, T.; et al. Dissection of signaling cascades through gp130 in vivo: Reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity 2000, 12, 95–105. [Google Scholar] [CrossRef]
- Sawa, S.; Kamimura, D.; Jin, G.H.; Morikawa, H.; Kamon, H.; Nishihara, M.; Ishihara, K.; Murakami, M.; Hirano, T. Autoimmune arthritis associated with mutated interleukin (IL)-6 receptor gp130 is driven by STAT3/IL-7-dependent homeostatic proliferation of CD4+ T cells. J. Exp. Med. 2006, 203, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Schwarz, M. The TNF-alpha transgenic mouse model of inflammatory arthritis. Springer Semin. Immunopathol. 2003, 25, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Aidinis, V.; Plows, D.; Haralambous, S.; Armaka, M.; Papadopoulos, P.; Kanaki, M.Z.; Koczan, D.; Thiesen, H.J.; Kollias, G. Functional analysis of an arthritogenic synovial fibroblast. Arthritis Res. Ther. 2003, 5, R140–R157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Zwerina, K.; Koenders, M.; Hueber, A.; Marijnissen, R.J.; Baum, W.; Heiland, G.R.; Zaiss, M.; McLnnes, I.; Joosten, L.; van den Berg, W.; et al. Anti IL-17A therapy inhibits bone loss in TNF-α-mediated murine arthritis by modulation of the T-cell balance. Eur. J. Immunol. 2012, 42, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Miossec, P. Interleukin-17 in rheumatoid arthritis: If T cells were to contribute to inflammation and destruction through synergy. Arthritis Rheum. 2003, 48, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Kolls, J.K.; Linden, A. Interleukin-17 family members and inflammation. Immunity 2004, 21, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-α, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar] [PubMed]
- Cho, J.H.; Gregersen, P.K. Genomics and the multifactorial nature of human autoimmune disease. N. Engl. J. Med. 2011, 365, 1612–1623. [Google Scholar] [PubMed]
- Sakaguchi, S. Regulatory T cells: Key controllers of immunologic self-tolerance. Cell 2000, 101, 455–458. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Pasare, C.; Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 2003, 299, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Valencia, X.; Stephens, G.; Goldbach-Mansky, R.; Wilson, M.; Shevach, E.M.; Lipsky, P.E. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood 2006, 108, 253–261. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.J.; Thomas, H.E.; Pai, S.; Santamaria, P.; Iwakura, Y.; Steptoe, R.J.; Kay, T.W.H.; Thomas, R. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J. Immunol. 2006, 176, 7278–7287. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Hue, S.; Buonocore, S.; Arancibia-Carcamo, C.V.; Ahern, P.P.; Iwakura, Y.; Maloy, K.J.; Powrie, F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 2008, 28, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, E.J.; Prakken, B.J.; van Wijk, F. T cells out of control—Impaired immune regulation in the inflamed joint. Nat. Rev. Rheumatol. 2013, 9, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Noack, M.; Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E.; Koenders, M.I.; van den Berg, W.B. The role of T-cell interleukin-17 in conducting destructive arthritis: Lessons from animal models. Arthritis Res. Ther. 2005, 7, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta-Rodriguez, E.V.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 2007, 8, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; De Palma, R.; Santarlasci, V.; Maggi, L.; Capone, M.; Frosali, F.; Rodolico, G.; Querci, V.; Abbate, G.; Angeli, R.; et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J. Exp. Med. 2008, 205, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Usui, T.; Kobayashi, S.; Iguchi-Hashimoto, M.; Ito, H.; Yoshitomi, H.; Nakamura, T.; Shimizu, M.; Kawabata, D.; Yukawa, N.; et al. Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis Rheum. 2009, 60, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Noordenbos, T.; Yeremenko, N.; Gofita, I.; van de Sande, M.; Tak, P.P.; Canete, J.D.; Baeten, D. Interleukin-17-positive mast cells contribute to synovial inflammation in spondylarthritis. Arthritis Rheum. 2012, 64, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, B.W.; Lassere, M.N.; Edmonds, J.P.; Juhasz, K.M.; Bird, P.A.; Lee, C.S.; Shnier, R.; Portek, I.J. Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: A two-year prospective study (the DAMAGE study cohort). Arthritis Rheum. 2006, 54, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Nakashima, Y.; Okazaki, K.; Mawatari, T.; Fukushi, J.I.; Kaibara, N.; Hori, A.; Iwamoto, Y.; Yoshikai, Y. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 1299–1304. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Agarwal, S.K.; Ilivanova, E.; Xu, X.L.; Miao, Y.; Zhuang, Y.; Nnane, I.; Radziszewski, W.; Greenspan, A.; Beutler, A.; et al. A randomised phase II study evaluating the efficacy and safety of subcutaneously administered ustekinumab and guselkumab in patients with active rheumatoid arthritis despite treatment with methotrexate. Ann. Rheum. Dis. 2017, 76, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, I.E.; Suzuki, E.; Chao, C.C.; Gorman, D.; Adda, S.; Maverakis, E.; Zarbalis, K.; Geissler, R.; Asio, A.; Blumenschein, W.M.; et al. IL-17A gene transfer induces bone loss and epidermal hyperplasia associated with psoriatic arthritis. Ann. Rheum. Dis. 2015, 74, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Alzabin, S.; Abraham, S.M.; Taher, T.E.; Palfreeman, A.; Hull, D.; McNamee, K.; Jawad, A.; Pathan, E.; Kinderlerer, A.; Taylor, P.C.; et al. Incomplete response of inflammatory arthritis to TNFalpha blockade is associated with the Th17 pathway. Ann. Rheum. Dis. 2012, 71, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Sutton, C.E.; Cua, D.; Mills, K.H. Therapeutic potential of targeting IL-17. Nat. Immunol. 2012, 13, 1022–1025. [Google Scholar]
- Kotake, S.; Yago, T.; Kobshigawa, T.; Nanke, Y. The Plasticity of Th17 Cells in the Pathogenesis of Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 67. [Google Scholar] [CrossRef] [PubMed]
- Lyakh, L.; Trinchieri, G.; Provezza, L.; Carra, G.; Gerosa, F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol. Rev. 2008, 226, 112–131. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Models | Effectors | IL-17 | IFN-γ | TNF-α | IL-1 | IL-6 | IL-12 p35 | IL-12/IL-23 p40 | IL-23 p19 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| CIA | CD4 T, autoAbs | ↓↓↓ | ↑ | ↓↓ | ↓↓↓ | ↓↓↓ | ↑ | ↓↓↓ | ↓↓↓ | [7,19,23] |
| CAIA | autoAbs | ND | ND | ↓↓ | ↓↓↓ | → | ND | ND | ND | [22] |
| SKG | CD4 T | ↓↓↓ | ↑ | ↓↓ | ↓↓ | ↓↓↓ | ND | ND | ↓↓↓ | [25,26,27] |
| K/BxN | CD4 T, autoAbs | ↓ | → | ↓↓ | ↓↓↓ | ND | → | ND | ↓ | [28,29,30,31] |
| K/BxN serum transfer | autoAbs | → | ND | ↓↓ | ↓↓↓ | → | ND | ND | ND | [28,29] |
| IL-1Ra KO | CD4 T, synoviocytes | ↓↓↓ | ND | ↓↓↓ | ↓↓↓ | ND | ND | ND | ↓↓↓ | [32,33,34] |
| Gp130 F759 | CD4 T, synoviocytes | ↓↓↓ | ND | ND | ND | ↓↓↓ | ND | ND | ND | [35,36] |
| TNF-α Tg | synoviocytes | (↓) | ND | ↓↓↓ | ↓↓↓ | → | ND | ND | ND | [23,37] |
| RA | CD4 T, autoAbs, synoviocytes | ↓ | → | ↓↓↓ | ↓ | ↓↓↓ | ND | → | ND | [9,38] |
| PsA | CD4 T, synoviocytes | ↓↓↓ | ND | ↓↓↓ | ↓↓ | ND | ND | ↓↓↓ | ND | [39,40] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashimoto, M. Th17 in Animal Models of Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 73. https://doi.org/10.3390/jcm6070073
Hashimoto M. Th17 in Animal Models of Rheumatoid Arthritis. Journal of Clinical Medicine. 2017; 6(7):73. https://doi.org/10.3390/jcm6070073
Chicago/Turabian StyleHashimoto, Motomu. 2017. "Th17 in Animal Models of Rheumatoid Arthritis" Journal of Clinical Medicine 6, no. 7: 73. https://doi.org/10.3390/jcm6070073
APA StyleHashimoto, M. (2017). Th17 in Animal Models of Rheumatoid Arthritis. Journal of Clinical Medicine, 6(7), 73. https://doi.org/10.3390/jcm6070073