NK Cell-Fc Receptors Advance Tumor Immunotherapy

{kind=link}
Abstract
:1. Introduction
2. Immune Checkpoint Inhibitors and NK Cell Fc Receptors
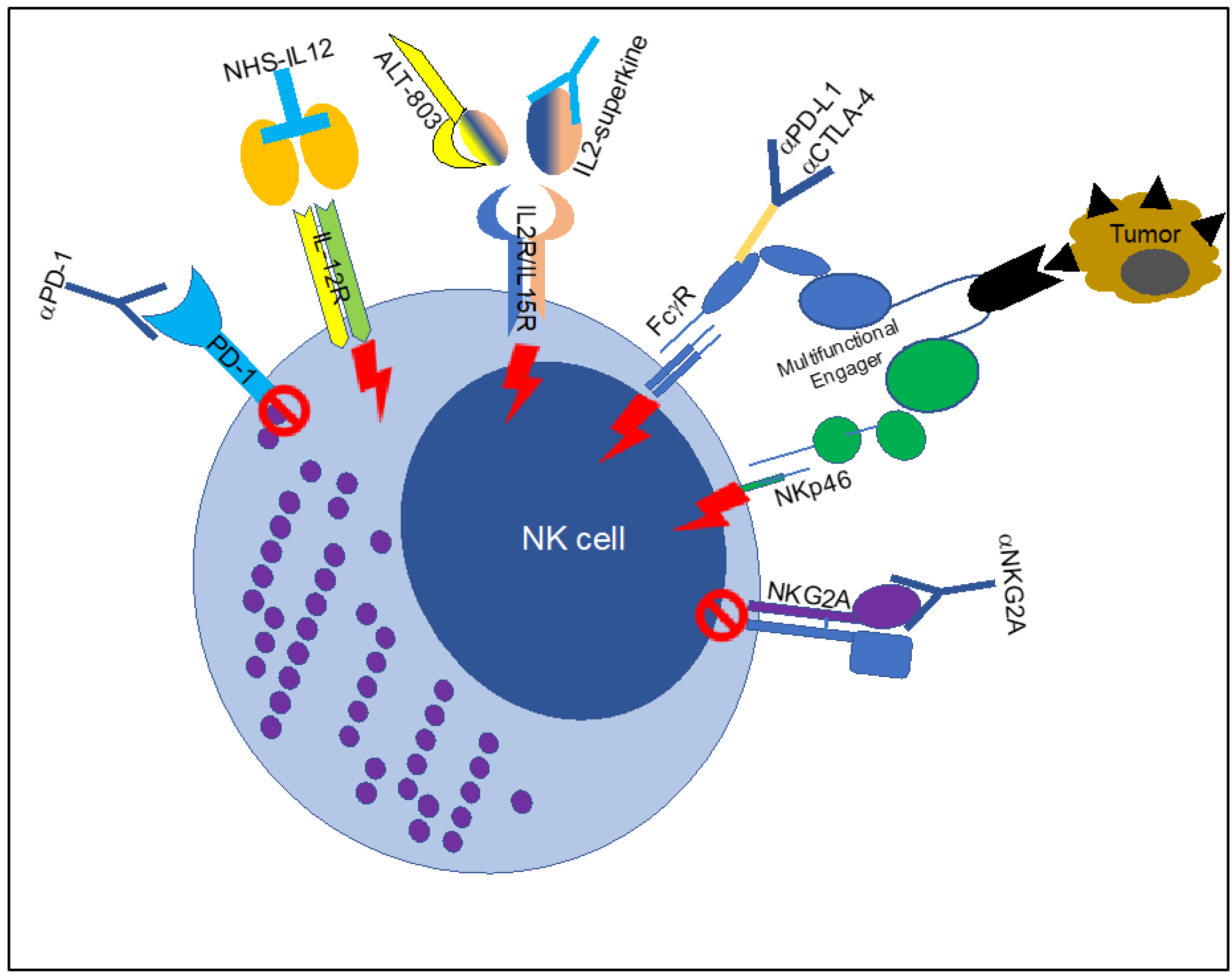
3. Enhancing Therapeutic Antibodies for Cancer Treatment by Fc Engineering
4. Activation of NK Cell Fc Receptors with Tumor-Targeting BI-Specific Antibodies and Bi-Specific or Tri-Specific Killer Engagers (Bikes, Trikes)
5. Stimulating NK Cells to Improve the Efficacy of Therapeutic Antibodies in Cancer
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Gismondi, A.; Stabile, H.; Nisti, P.; Santoni, A. Effector Functions of Natural Killer Cell Subsets in the Control of Hematological Malignancies. Front. Immunol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Vance, R.E. Self-tolerance of natural killer cells. Nat. Rev. Immunol. 2006, 6, 520–531. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar]
- Shifrin, N.; Raulet, D.H.; Ardolino, M. NK cell self tolerance, responsiveness and missing self recognition. Semin. Immunol. 2014, 26, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a Novel Triggering Surface Molecule Specifically Expressed by Activated Natural Killer Cells, Is Involved in Non-Major Histocompatibility Complex-restricted Tumor Cell Lysis. J. Exp. Med. 1998, 187, 2065–2072. [Google Scholar] [CrossRef]
- Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular Cloning of NKp46: A Novel Member of the Immunoglobulin Superfamily Involved in Triggering of Natural Cytotoxicity. J. Exp. Med. 1998, 188, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pende, D. Identification and Molecular Characterization of NKp30, a Novel Triggering Receptor Involved in Natural Cytotoxicity Mediated by Human Natural Killer Cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Azzoni, L.; Anegon, I.; Calabretta, B.; Perussia, B. Ligand binding to Fc gamma R induces c-myc-dependent apoptosis in IL-2-stimulated NK cells. J. Immunol. 1995, 154, 491–499. [Google Scholar] [PubMed]
- Trotta, R.; Kanakaraj, P.; Perussia, B. Fc gamma R-dependent mitogen-activated protein kinase activation in leukocytes: A common signal transduction event necessary for expression of TNF-alpha and early activation genes. J. Exp. Med. 1996, 184, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Galandrini, R. CD16-mediated p21ras activation is associated with Shc and p36 tyrosine phosphorylation and their binding with Grb2 in human natural killer cells. J. Exp. Med. 1996, 183, 179–186. [Google Scholar] [CrossRef]
- Galandrini, R.; Palmieri, G.; Paolini, R.; Piccoli, M.; Frati, L.; Santoni, A. Selective binding of shc-SH2 domain to tyrosine-phosphorylated zeta but not gamma-chain upon CD16 ligation on human NK cells. J. Immunol. 1997, 159, 3767–3773. [Google Scholar]
- Galandrini, R.; Micucci, F.; Tassi, I.; Cifone, M.G.; Cinque, B.; Piccoli, M.; Fati, L.; Santoni, A. Arf6: A new player in FcgammaRIIIA lymphocyte-mediated cytotoxicity. Blood 2005, 106, 577–583. [Google Scholar] [CrossRef]
- Capuano, C.; Romanelli, M.; Pighi, C.; Cimino, G.; Rago, A.; Molfetta, R.; Paolini, R.; Santoni, A.; Galandrini, R. Anti-CD20 Therapy Acts via FcgammaRIIIA to Diminish Responsiveness of Human Natural Killer Cells. Cancer Res. 2015, 75, 4097–4108. [Google Scholar] [CrossRef]
- Pahl, J.H.; Koch, J.; Götz, J.-J.; Arnold, A.; Reusch, U.; Gantke, T.; Rajkovic, E.; Treder, M.S.; Cerwenka, A. CD16A Activation of NK Cells Promotes NK Cell Proliferation and Memory-Like Cytotoxicity against Cancer Cells. Cancer Immunol. Res. 2018, 6, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Hogarth, P.M.; Pietersz, G.A. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat. Rev. Drug Discov. 2012, 11, 311–331. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Metes, D.; Ernst, L.K.; Chambers, W.H.; Sulica, A.; Herberman, R.B.; Morel, A.P. Expression of functional CD32 molecules on human NK cells is determined by an allelic polymorphism of the FcgammaRIIC gene. Blood 1998, 91, 2369–2380. [Google Scholar] [PubMed]
- Sanseviero, E.; O’Brien, E.M.; Karras, J.R.; Shabaneh, T.B.; Aksoy, B.A.; Xu, W.; Zheng, C.; Yin, X.; Xu, X.; Karakousis, G.C.; et al. Anti-CTLA-4 Activates Intratumoral NK Cells and Combined with IL15/IL15Ralpha Complexes Enhances Tumor Control. Cancer Immunol. Res. 2019, 7, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Mechetina, L.V.; Najakshin, A.M.; Alabyev, B.Y.; Chikaev, N.A.; Taranin, A.V. Identification of CD16-2, a novel mouse receptor homologous to CD16/Fc gamma RIII. Immunogenetics 2002, 54, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Lux, A.; Albert, H.; Woigk, M.; Lehmann, C.; Dudziak, D.; Smith, P.; Ravetch, J.V. FcgammaRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 19396–19401. [Google Scholar] [CrossRef]
- Callahan, M.K.; Wolchok, J.D. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J. Leukoc. Boil. 2013, 94, 41–53. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Robert, C. CTLA-4 and PD-1/PD-L1 Blockade: New Immunotherapeutic Modalities with Durable Clinical Benefit in Melanoma Patients. Clin. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef]
- Vargas, F.A.; Furness, A.J.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663.e4. [Google Scholar] [CrossRef]
- Ingram, J.R.; Blomberg, O.S.; Rashidian, M.; Ali, L.; Garforth, S.; Fedorov, E.; Fedorov, A.A.; Bonanno, J.B.; Le Gall, C.; Crowley, S.; et al. Anti–CTLA-4 therapy requires an Fc domain for efficacy. Proc. Natl. Acad. Sci. USA 2018, 115, 3912–3917. [Google Scholar] [CrossRef]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, A.C.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [Green Version]
- Dahan, R.; Sega, E.; Engelhardt, J.; Selby, M.; Korman, A.J.; Ravetch, J.V. FcyRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell 2015, 28, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.; Tanaka, A.; Kibayashi, K.; Tanemura, A.; Sugiyama, D.; Wing, J.B.; Lim, E.L.; Teng, K.W.W.; Adeegbe, D.; Newell, E.W.; et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc. Natl. Acad. Sci. USA 2019, 116, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Peggs, K.S. Lost in Translation: Deciphering the Mechanism of Action of Anti-human CTLA-4. Clin. Cancer Res. 2019, 25, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, X.; Li, K.; Zhang, T. FcgammaR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front. Immunol. 2019, 10, 292. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Souza-Fonseca-Guimaraes, F.; Cursons, J.; Huntington, N.D. The Emergence of Natural Killer Cells as a Major Target in Cancer Immunotherapy. Trends Immunol. 2019, 40, 142–158. [Google Scholar] [CrossRef]
- Cerwenka, A.; Lanier, L.L. Natural killer cell memory in infection, inflammation and cancer. Nat. Rev. Immunol. 2016, 16, 112–123. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Callahan, M.K.; Kluger, H.; Postow, M.A.; Segal, N.H.; Lesokhin, A.; Atkins, M.B.; Kirkwood, J.M.; Krishnan, S.; Bhore, B.; Horak, C.; et al. Nivolumab Plus Ipilimumab in Patients with Advanced Melanoma: Updated Survival, Response, and Safety Data in a Phase I Dose-Escalation Study. J. Clin. Oncol. 2018, 36, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Rollin, L.; Larkin, J.; Ryu, H.; Lee, H.J.; Alegre-Del-Rey, E.J.; Fernández, B.D.L.N.; Briceño-Casado, P. Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 2503–2504. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Atkinson, V.; Lo, S.; Sandhu, S.; Guminski, A.D.; Brown, M.P.; Wilmott, J.S.; Edwards, J.; González, M.; Scolyer, A.R.; et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: A multicentre randomised phase 2 study. Lancet Oncol. 2018, 19, 672–681. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Quatrini, L.; Wieduwild, E.; Escaliere, B.; Filtjens, J.; Chasson, L.; Laprie, C.; Vivier, E.; Ugolini, S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018, 19, 954–962. [Google Scholar] [CrossRef]
- Munari, E.; Zamboni, G.; Lunardi, G.; Marconi, M.; Brunelli, M.; Martignoni, G.; Netto, G.J.; Quatrini, L.; Vacca, P.; Moretta, L.; et al. PD-L1 expression in non-small cell lung cancer: Evaluation of the diagnostic accuracy of a laboratory-developed test using clone E1L3N in comparison with 22C3 and SP263 assays. Hum. Pathol. 2019, 90, 54–59. [Google Scholar] [CrossRef]
- Mariotti, F.R.; Quatrini, L.; Munari, E.; Vacca, P.; Moretta, L. Innate Lymphoid Cells: Expression of PD-1 and Other Checkpoints in Normal and Pathological Conditions. Front. Immunol. 2019, 10, 910. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Pesce, S.; Muccio, L.; Carlomagno, S.; Sivori, S.; Moretta, A.; Marcenaro, E. Features of Memory-Like and PD-1(+) Human NK Cell Subsets. Front. Immunol. 2016, 7, 351. [Google Scholar] [CrossRef]
- Mariotti, F.R.; Petrini, S.; Ingegnere, T.; Tumino, N.; Besi, F.; Scordamaglia, F.; Munari, E.; Pesce, S.; Marcenaro, E.; Moretta, A.; et al. PD-1 in human NK cells: Evidence of cytoplasmic mRNA and protein expression. Oncoimmunology 2019, 8, 1557030. [Google Scholar] [CrossRef] [PubMed]
- Pesce, S.; Greppi, M.; Grossi, F.; Del Zotto, G.; Moretta, L.; Sivori, S.; Genova, C.; Marcenaro, E. PD/1-PD-Ls Checkpoint: Insight on the Potential Role of NK Cells. Front. Immunol. 2019, 10, 1242. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.-C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Blank, C.; Mackensen, A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: An update on implications for chronic infections and tumor evasion. Cancer Immunol. Immunother. 2007, 56, 739–745. [Google Scholar] [CrossRef]
- Dahan, R.; Sega, E.; Engelhardt, J.; Selby, M.; Korman, A.J.; Ravetch, J.V. FcyRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis—Erratum. Cancer Cell 2015, 28, 543. [Google Scholar] [CrossRef]
- Johnson, D.B.; Peng, C.; Sosman, J.A. Nivolumab in melanoma: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2015, 7, 97–106. [Google Scholar] [CrossRef]
- Fessas, P.; Lee, H.; Ikemizu, S.; Janowitz, T. A molecular and preclinical comparison of the PD-1–targeted T-cell checkpoint inhibitors nivolumab and pembrolizumab. Semin. Oncol. 2017, 44, 136–140. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, A.R. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliá, E.P.; Amante, A.; Pampena, M.B.; Mordoh, J.; Levy, E.M. Avelumab, an IgG1 anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front. Immunol. 2018, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Russell, J.S.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Milella, M.; Brownell, I.; et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J. Immunother. Cancer 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Liu, K.; Chai, Y.; Zhang, C.W.-H.; Gao, S.; Gao, G.F.; Qi, J. Distinct PD-L1 binding characteristics of therapeutic monoclonal antibody durvalumab. Protein Cell 2018, 9, 135–139. [Google Scholar] [CrossRef]
- Planchard, D.; Yokoi, T.; McCleod, M.J.; Fischer, J.R.; Kim, Y.-C.; Ballas, M.; Shi, K.; Soria, J.-C. A Phase III Study of Durvalumab (MEDI4736) With or Without Tremelimumab for Previously Treated Patients with Advanced NSCLC: Rationale and Protocol Design of the ARCTIC Study. Clin. Lung Cancer 2016, 17, 232–236.e1. [Google Scholar] [CrossRef]
- Tsang, K.-Y.; Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.R.; Madan, R.A.; Gulley, J.L.; Schlom, J. Antibody dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody, avelumab (MSB0010718C), on human tumor cells. J. Clin. Oncol. 2015, 33, 3038. [Google Scholar] [CrossRef]
- Hicks, K.C.; Fantini, M.; Donahue, R.N.; Schwab, A.; Knudson, K.M.; Tritsch, S.R.; Jochems, C.; Clavijo, P.E.; Allen, C.T.; Hodge, J.W.; et al. Epigenetic priming of both tumor and NK cells augments antibody-dependent cellular cytotoxicity elicited by the anti-PD-L1 antibody avelumab against multiple carcinoma cell types. OncoImmunology 2018, 7, e1466018. [Google Scholar] [CrossRef]
- Shrikant, P.; Khoruts, A.; Mescher, M.F. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity 1999, 11, 483–493. [Google Scholar] [CrossRef]
- Brunet, J.-F.; Denizot, F.; Luciani, M.-F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.-G.; Golstein, P. A new member of the immunoglobulin superfamily—CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Kerdiles, Y.M.; Stone, E.L.; Beisner, D.R.; Beisner, D.L.; McGargill, M.A.; Ch’En, I.L.; Stockmann, C.; Katayama, C.D.; Hedrick, S.M. Foxo transcription factors control regulatory T cell development and function. Immunity 2010, 33, 890–904. [Google Scholar] [CrossRef]
- Samstein, R.M.; Arvey, A.; Josefowicz, S.Z.; Peng, X.; Reynolds, A.; Sandstrom, R.; Neph, S.; Sabo, P.; Kim, J.M.; Liao, W.; et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 2012, 151, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.D.; Plitas, G.; Rudra, D.; Lee, S.Y.; Rudensky, A.Y. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J. Exp. Med. 2013, 210, 2435–2466. [Google Scholar] [CrossRef] [PubMed]
- Caudana, P.; Núñez, N.G.; De La Rochere, P.; Pinto, A.; Denizeau, J.; Alonso, R.; Niborski, L.L.; Lantz, O.; Sedlik, C.; Piaggio, E. IL2/Anti-IL2 Complex Combined with CTLA-4, But Not PD-1, Blockade Rescues Antitumor NK Cell Function by Regulatory T-cell Modulation. Cancer Immunol. Res. 2019, 7, 443–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, F.A.; Furness, A.J.; Solomon, I.; Joshi, K.; Mekkaoui, L.; Lesko, M.H.; Rota, E.M.; Dahan, R.; Georgiou, A.; Sledzinska, A.; et al. Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 2017, 46, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Coe, D.; Begom, S.; Addey, C.; White, M.; Dyson, J.; Chai, J.-G. Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1367–1377. [Google Scholar] [CrossRef]
- Mahne, A.E.; Mauze, S.; Joyce-Shaikh, B.; Xia, J.; Bowman, E.P.; Beebe, A.M.; Cua, D.J.; Jain, R. Dual Roles for Regulatory T-cell Depletion and Costimulatory Signaling in Agonistic GITR Targeting for Tumor Immunotherapy. Cancer Res. 2017, 77, 1108–1118. [Google Scholar] [CrossRef]
- Wang, X.; Mathieu, M.; Brezski, R.J. IgG Fc engineering to modulate antibody effector functions. Protein Cell 2018, 9, 63–73. [Google Scholar] [CrossRef]
- Ferrara, C.; Grau, S.; Jäger, C.; Sondermann, P.; Brünker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef]
- Li, W.; Zhu, Z.; Chen, W.; Feng, Y.; Dimitrov, D.S. Crystallizable Fragment Glycoengineering for Therapeutic Antibodies Development. Front. Immunol. 2017, 8, 1554. [Google Scholar] [CrossRef] [Green Version]
- Buettner, M.J.; Shah, S.R.; Saeui, C.T.; Ariss, R.; Yarema, K.J. Improving Immunotherapy Through Glycodesign. Front. Immunol. 2018, 9, 2485. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; De La Serna, J.; Dilhuydy, M.-S.; Illmer, T.; et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Ratner, M. Genentech’s glyco-engineered antibody to succeed Rituxan. Nat. Biotechnol. 2014, 32, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Ollila, A.T.; Sahin, I.; Olszewski, A.J. Mogamulizumab: A new tool for management of cutaneous T-cell lymphoma. OncoTargets Ther. 2019, 12, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Ishida, T.; Hatake, K.; Taniwaki, M.; Ando, K.; Tobinai, K.; Fujimoto, K.; Yamamoto, K.; Miyamoto, T.; Uike, N.; et al. Multicenter Phase II Study of Mogamulizumab (KW-0761), a Defucosylated Anti-CC Chemokine Receptor 4 Antibody, in Patients with Relapsed Peripheral T-Cell Lymphoma and Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2014, 32, 1157–1163. [Google Scholar] [CrossRef]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A phase 1 study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Goletz, C.; Lischke, T.; Harnack, U.; Schiele, P.; Danielczyk, A.; Rühmann, J.; Goletz, S. Glyco-Engineered Anti-Human Programmed Death-Ligand 1 Antibody Mediates Stronger CD8 T Cell Activation Than Its Normal Glycosylated and Non-Glycosylated Counterparts. Front. Immunol. 2018, 9, 1614. [Google Scholar] [CrossRef] [Green Version]
- Davis, Z.B.; Vallera, D.A.; Miller, J.S.; Felices, M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin. Immunol. 2017, 31, 64–75. [Google Scholar] [CrossRef]
- Moore, G.L.; Bautista, C.; Pong, E.; Nguyen, D.-H.T.; Jacinto, J.; Eivazi, A.; Muchhal, U.S.; Karki, S.; Chu, S.Y.; Lazar, G.A. A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens. mAbs 2011, 3, 546–557. [Google Scholar] [CrossRef]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 2015, 125, 4024–4031. [Google Scholar] [CrossRef]
- Märklin, M.; Hagelstein, I.; Koerner, S.P.; Rothfelder, K.; Pfluegler, M.S.; Schumacher, A.; Grosse-Hovest, L.; Jung, G.; Salih, H.R. Bispecific NKG2D-CD3 and NKG2D-CD16 fusion proteins for induction of NK and T cell reactivity against acute myeloid leukemia. J. Immunother. Cancer 2019, 7, 143. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef]
- Perales-Puchalt, A.; Duperret, E.K.; Yang, X.; Hernandez, P.; Wojtak, K.; Zhu, X.; Jung, S.-H.; Tello-Ruiz, E.; Wise, M.C.; Montaner, L.J.; et al. DNA-encoded bispecific T cell engagers and antibodies present long-term antitumor activity. JCI Insight 2019, 4, e126086. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, C.J.; Knochelmann, H.M.; Smith, A.S.; Wyatt, M.M.; Rivera, G.O.R.; Arhontoulis, D.C.; Bartee, E.; Li, Z.; Rubinstein, M.P.; Paulos, C.M. Fueling Cancer Immunotherapy With Common Gamma Chain Cytokines. Front. Immunol. 2019, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. OncoImmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, A.M.; Bates, D.L.; Ring, A.M.; Krieg, C.; Lin, J.T.; Su, L.; Moraga, I.L.; Raeber, M.E.; Bowman, G.R.; Novick, P.; et al. Exploiting a natural conformational switch to engineer an interleukin-2 ’superkine’. Nature 2012, 484, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Ardolino, M.; Azimi, C.S.; Iannello, A.; Trevino, T.N.; Horan, L.; Zhang, L.; Deng, W.; Ring, A.M.; Fischer, S.; Garcia, K.C.; et al. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J. Clin. Investig. 2014, 124, 4781–4794. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Steel, J.C.; Zhang, M.; Morris, J.C.; Waldmann, T.A. Simultaneous blockade of multiple immune system inhibitory checkpoints enhances antitumor activity mediated by interleukin-15 in a murine metastatic colon carcinoma model. Clin. Cancer Res. 2010, 16, 6019–6028. [Google Scholar] [CrossRef]
- Xu, W.; Jones, M.; Liu, B.; Zhu, X.; Johnson, C.B.; Edwards, A.C.; Kong, L.; Jeng, E.K.; Han, K.; Marcus, W.D.; et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: Interleukin-15 receptor alphaSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res. 2013, 73, 3075–3086. [Google Scholar] [CrossRef]
- Rosario, M.; Liu, B.; Kong, L.; Collins, L.I.; Schneider, S.E.; Chen, X.; Han, K.; Jeng, E.K.; Rhode, P.R.; Leong, J.W.; et al. The IL-15-Based ALT-803 Complex Enhances FcgammaRIIIa-Triggered NK Cell Responses and In Vivo Clearance of B Cell Lymphomas. Clin. Cancer Res. 2016, 22, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Rhode, P.R.; Egan, J.O.; Xu, W.; Hong, H.; Webb, G.M.; Chen, X.; Liu, B.; Zhu, X.; Wen, J.; You, L.; et al. Comparison of the Superagonist Complex, ALT-803, to IL15 as Cancer Immunotherapeutics in Animal Models. Cancer Immunol. Res. 2016, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Knudson, K.M.; Hicks, K.C.; Alter, S.; Schlom, J.; Gameiro, S.R. Mechanisms involved in IL-15 superagonist enhancement of anti-PD-L1 therapy. J. Immunother. Cancer 2019, 7, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Fallon, J.; Tighe, R.; Kradjian, G.; Guzman, W.; Bernhardt, A.; Neuteboom, B.; Lan, Y.; Sabzevari, H.; Schlom, J.; Greiner, J.W. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget 2014, 5, 1869–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, J.; Heery, C.R.; Kim, J.W.; Jochems, C.; Donahue, R.N.; Montgomery, A.S.; McMahon, S.; Lamping, E.; Marté, J.L.; Madan, R.A.; et al. First-in-Human Phase I Trial of a Tumor-Targeted Cytokine (NHS-IL12) in Subjects with Metastatic Solid Tumors. Clin. Cancer Res. 2019, 25, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kondadasula, S.V.; Roda, J.M.; Parihar, R.; Yu, J.; Lehman, A.; Caligiuri, M.A.; Tridandapani, S.; Burry, R.W.; Carson, W.E. Colocalization of the IL-12 receptor and FcgammaRIIIa to natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-gamma. Blood 2008, 111, 4173–4183. [Google Scholar] [CrossRef]
- Jaime-Ramirez, A.C.; Mundy-Bosse, B.L.; Kondadasula, S.; Jones, N.B.; Roda, J.M.; Mani, A.; Parihar, R.; Karpa, V.; Papenfuss, T.L.; LaPerle, K.M.; et al. IL-12 enhances the antitumor actions of trastuzumab via NK cell IFN-gamma production. J. Immunol. 2011, 186, 3401–3409. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, Y.; Rolfe, P.A.; Hernández, V.M.; Guzman, W.; Kradjian, G.; Marelli, B.; Qin, G.; Qi, J.; Wang, H.; et al. Combination Therapy with NHS-muIL12 and Avelumab (anti-PD-L1) Enhances Antitumor Efficacy in Preclinical Cancer Models. Clin. Cancer Res. 2017, 23, 5869–5880. [Google Scholar] [CrossRef]
- Molgora, M.; Bonavita, E.; Ponzetta, A.; Riva, F.; Barbagallo, M.; Jaillon, S.; Popović, B.; Bernardini, G.; Magrini, E.; Gianni, F.; et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017, 551, 110–114. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanseviero, E. NK Cell-Fc Receptors Advance Tumor Immunotherapy. J. Clin. Med. 2019, 8, 1667. https://doi.org/10.3390/jcm8101667
Sanseviero E. NK Cell-Fc Receptors Advance Tumor Immunotherapy. Journal of Clinical Medicine. 2019; 8(10):1667. https://doi.org/10.3390/jcm8101667
Chicago/Turabian StyleSanseviero, Emilio. 2019. "NK Cell-Fc Receptors Advance Tumor Immunotherapy" Journal of Clinical Medicine 8, no. 10: 1667. https://doi.org/10.3390/jcm8101667
APA StyleSanseviero, E. (2019). NK Cell-Fc Receptors Advance Tumor Immunotherapy. Journal of Clinical Medicine, 8(10), 1667. https://doi.org/10.3390/jcm8101667