Immune Dysregulation in HFpEF: A Target for Mesenchymal Stem/Stromal Cell Therapy


Abstract
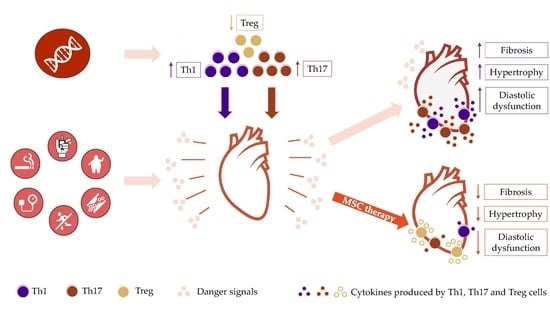
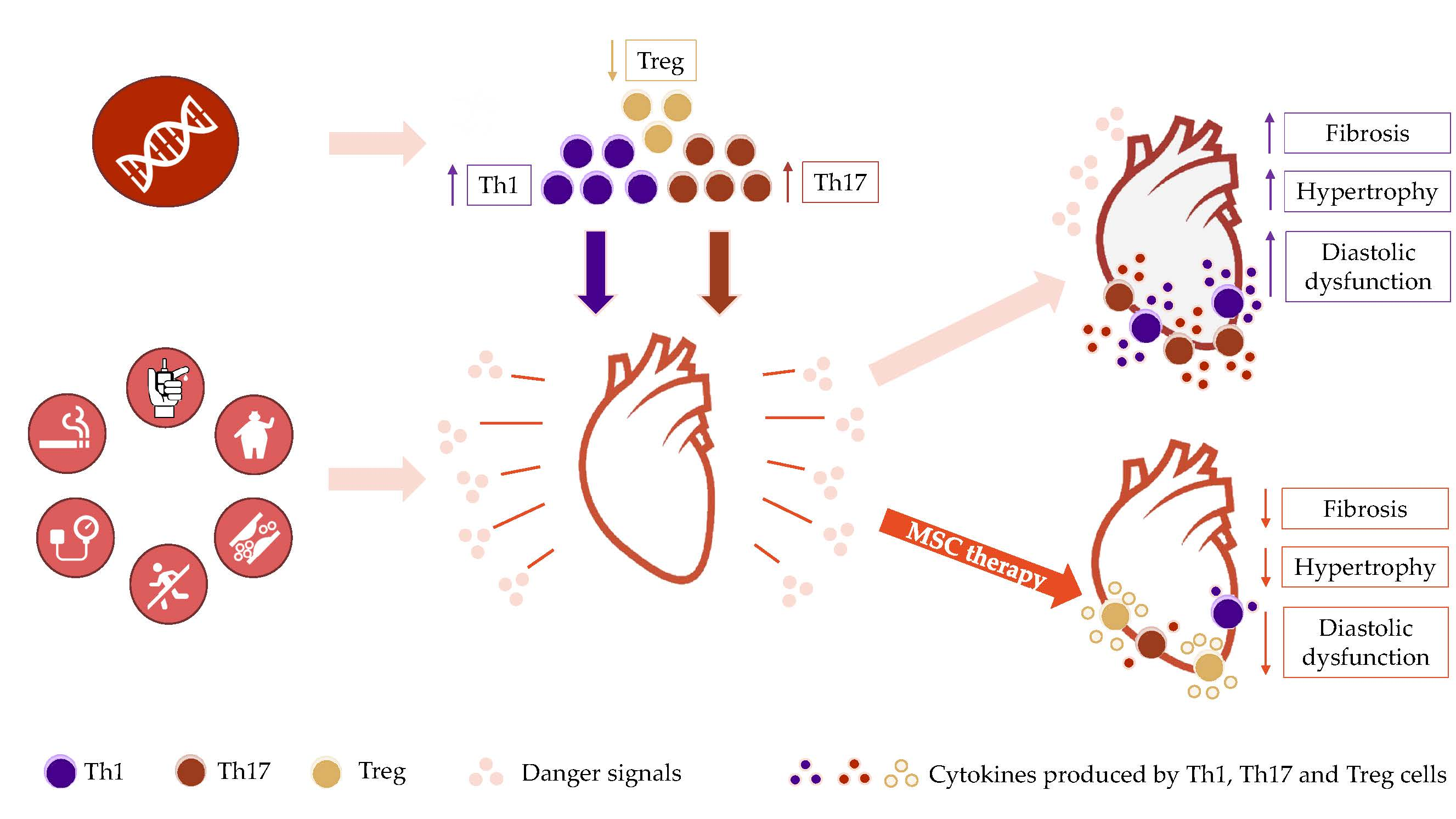
:
{kind=link}
1. Introduction
2. Importance of Immune Dysregulation in HFpEF—Key Lines of Evidence
2.1. Animal Models of HFpEF
2.2. The Interplay of CHIP, Immune Dysregulation, and Cardiovascular Disease
2.3. Monocytes: Effectors of Cardiac Remodeling for Better or Worse
2.4. T Cells as Mediators of Chronic Heart Failure Development
3. Complex Therapies for a Complex Disease—Exploring the Potential of Mesenchymal Stem/Stromal Cell Therapies for HFpEF
3.1. In vitro Immunomodulatory Properties of Mesenchymal Stem/Stromal Cells
3.2. Evidence of Cell Therapy in HFpEF Animal Models
3.3. Immunomodulatory Effects of MSCs in Preclinical Models of Th1-Mediated Diseases
3.4. Immunomodulatory Effects of MSCs in Clinical Trials of Th1-Mediated Diseases
3.5. MSC Clinical Trials in HF—Lost in Translation?
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the impact of heart failure in the United States: A policy statement from the American Heart Association. Circ. Heart Fail. 2013, 6, 606–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, Y.; Weston, S.A.; Redfield, M.M.; Chamberlain, A.M.; Manemann, S.M.; Jiang, R.; Killian, J.M.; Roger, V.L. A contemporary appraisal of the heart failure epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern. Med. 2015, 175, 996–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.S.; Fonarow, G.C.; Givertz, M.M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J. Am. Coll. Cardiol. 2017, 70, 776–803. [Google Scholar]
- Shah, S.J.; Katz, D.H.; Deo, R.C. Phenotypic spectrum of heart failure with preserved ejection fraction. Heart Fail. Clin. 2014, 10, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Muñoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef]
- Tromp, J.; Khan, M.A.; Klip, I.T.; Meyer, S.; de Boer, R.A.; Jaarsma, T.; Hillege, H.; van Veldhuisen, D.J.; van der Meer, P.; Voors, A.A. Biomarker Profiles in Heart Failure Patients with Preserved and Reduced Ejection Fraction. J. Am. Heart Assoc. 2017, 6, e003989. [Google Scholar] [CrossRef] [Green Version]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mas-Peiro, S.; Hoffmann, J.; Fichtlscherer, S.; Dorsheimer, L.; Rieger, M.A.; Dimmeler, S.; Vasa-Nicotera, M.; Zeiher, A.M. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, B.; Bansal, S.S.; Ismahil, M.A.; Hamid, T.; Rokosh, G.; Mack, M.; Prabhu, S.D. CCR2+ Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl. Sci. 2018, 3, 230–244. [Google Scholar] [CrossRef]
- Markó, L.; Kvakan, H.; Park, J.K.; Qadri, F.; Spallek, B.; Binger, K.J.; Bowman, E.P.; Kleinewietfeld, M.; Fokuhl, V.; Dechend, R.; et al. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension 2012, 60, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ngwenyama, N.; Salvador, A.M.; Velázquez, F.; Nevers, T.; Levy, A.; Aronovitz, M.; Luster, A.D.; Huggins, G.S.; Alcaide, P. CXCR3 regulates CD4+ T cell cardiotropism in pressure overload-induced cardiac dysfunction. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef]
- Kallikourdis, M.; Martini, E.; Carullo, P.; Sardi, C.; Roselli, G.; Greco, C.M.; Vignali, D.; Riva, F.; Ormbostad Berre, A.M.; Stølen, T.O.; et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat. Commun. 2017, 8, 14680. [Google Scholar] [CrossRef]
- Pilli, D.; Zou, A.; Tea, F.; Dale, R.C.; Brilot, F. Expanding Role of T Cells in Human Autoimmune Diseases of the Central Nervous System. Front. Immunol. 2017, 8, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Passos, G.R.; Sato, D.K.; Becker, J.; Fujihara, K. Th17 Cells Pathways in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders: Pathophysiological and Therapeutic Implications. Mediat. Inflamm. 2016. [Google Scholar] [CrossRef] [PubMed]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. 2017, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Solt, L.A.; Burris, T.P. Th17 cells in Type 1 diabetes: A future perspective. Diabetes Manag. 2015, 5, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-TNF Therapy against Congestive Heart Failure (ATTACH) trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [PubMed] [Green Version]
- Mann, D.L.; McMurray, J.J.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Van Tassell, B.W.; Arena, R.; Biondi-Zoccai, G.; Canada, J.M.; Oddi, C.; Abouzaki, N.A.; Jahangiri, A.; Falcao, R.A.; Kontos, M.C.; Shah, K.B.; et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am. J. Cardiol. 2014, 113, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Van Tassell, B.W.; Buckley, L.F.; Carbone, S.; Trankle, C.R.; Canada, J.M.; Dixon, D.L.; Abouzaki, N.; Oddi-Erdle, C.; Biondi-Zoccai, G.; Arena, R.; et al. Interleukin-1 blockade in heart failure with preserved ejection fraction: Rationale and design of the Diastolic Heart Failure Anakinra Response Trial 2 (D-HART2). Clin. Cardiol. 2017, 40, 626–632. [Google Scholar] [CrossRef]
- Hori, M.; Yamaguchi, O. Is tumor necrosis factor-α friend or foe for chronic heart failure? Circ. Res. 2013, 113, 492–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Linthout, S.; Tschöpe, C. Inflammation—Cause or Consequence of Heart Failure or Both? Curr. Heart Fail. Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traktuev, D.O.; Merfeld-Clauss, S.; Li, J.; Kolonin, M.; Arap, W.; Pasqualini, R.; Johnstone, B.H.; March, K.L. A population of multipotent CD34-positive adipose stromal cells share pericyte and mesenchymal surface markers, reside in a periendothelial location, and stabilize endothelial networks. Circ. Res. 2008, 102, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, A.I. All MSCs are pericytes? Cell Stem Cell 2008, 3, 229–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najar, M.; Bouhtit, F.; Melki, R.; Afif, H.; Hamal, A.; Fahmi, H.; Merimi, M.; Lagneaux, L. Mesenchymal Stromal Cell-Based Therapy: New Perspectives and Challenges. J. Clin. Med. 2019, 8, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganath, S.H.; Levy, O.; Inamdar, M.S.; Karp, J.M. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell 2012, 10, 244–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, H.; Han, Z.; Han, Z.C.; Li, Z. Proangiogenic Features of Mesenchymal Stem Cells and Their Therapeutic Applications. Stem Cells Int. 2016. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Fibbe, W.E. Unraveling mechanisms of mesenchymal stromal cell-mediated immunomodulation through patient monitoring and product characterization. Ann. N. Y. Acad. Sci. 2016, 1370, 15–23. [Google Scholar] [CrossRef]
- Uccelli, A.; de Rosbo, N.K. The immunomodulatory function of mesenchymal stem cells: Mode of action and pathways. Ann. N. Y. Acad. Sci. 2015, 1351, 114–126. [Google Scholar] [CrossRef]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A new mesenchymal stem cell (MSC) paradigm: Polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS ONE 2010, 5, e10088. [Google Scholar] [CrossRef]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal stromal cells: Sensors and switchers of inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, A. Mesenchymal stromal cells: New directions. Cell Stem Cell 2012, 10, 709–716. [Google Scholar] [CrossRef] [Green Version]
- Tokita, Y.; Tang, X.L.; Li, Q.; Wysoczynski, M.; Hong, K.U.; Nakamura, S.; Wu, W.J.; Xie, W.; Li, D.; Hunt, G.; et al. Repeated Administrations of Cardiac Progenitor Cells Are Markedly More Effective Than a Single Administration: A New Paradigm in Cell Therapy. Circ. Res. 2016, 119, 635–651. [Google Scholar] [CrossRef] [Green Version]
- Iso, Y.; Spees, J.L.; Serrano, C.; Bakondi, B.; Pochampally, R.; Song, Y.H.; Sobel, B.E.; Delafontaine, P.; Prockop, D.J. Multipotent human stromal cells improve cardiac function after myocardial infarction in mice without long-term engraftment. Biochem. Biophys. Res. Commun. 2007, 354, 700–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiker, M.; Suzuki, G.; Iyer, V.S.; Canty, J.M.; Lee, T. Assessment of a nuclear affinity labeling method for tracking implanted mesenchymal stem cells. Cell Transpl. 2008, 17, 911–922. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.N.; Cores, J.; Huang, K.; Cui, X.L.; Luo, L.; Zhang, J.Y.; Li, T.S.; Qian, L.; Cheng, K. Concise Review: Is Cardiac Cell Therapy Dead? Embarrassing Trial Outcomes and New Directions for the Future. Stem Cells Transl. Med. 2018, 7, 354–359. [Google Scholar] [CrossRef]
- Banerjee, M.N.; Bolli, R.; Hare, J.M. Clinical Studies of Cell Therapy in Cardiovascular Medicine: Recent Developments and Future Directions. Circ. Res. 2018, 123, 266–287. [Google Scholar] [CrossRef] [PubMed]
- Valero-Muñoz, M.; Backman, W.; Sam, F. Murine Models of Heart Failure with Preserved Ejection Fraction: A “Fishing Expedition”. JACC Basic Transl. Sci. 2017, 2, 770–789. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Lio, C.J.; Samaniego-Castruita, D.; Li, X.; Rao, A. Loss of TET2 and TET3 in regulatory T cells unleashes effector function. Nat. Commun. 2019, 10, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damsker, J.M.; Hansen, A.M.; Caspi, R.R. Th1 and Th17 cells: Adversaries and collaborators. Ann. N. Y. Acad. Sci. 2010, 1183, 211–221. [Google Scholar] [CrossRef]
- Blanton, R.M.; Carrillo-Salinas, F.J.; Alcaide, P. T-cell recruitment to the heart: Friendly guests or unwelcome visitors? Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H124–H140. [Google Scholar] [CrossRef]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brüne, B.; Wagner, S.; Serve, H.; et al. Association of Mutations Contributing to Clonal Hematopoiesis with Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Glezeva, N.; Voon, V.; Watson, C.; Horgan, S.; McDonald, K.; Ledwidge, M.; Baugh, J. Exaggerated inflammation and monocytosis associate with diastolic dysfunction in heart failure with preserved ejection fraction: Evidence of M2 macrophage activation in disease pathogenesis. J. Card. Fail. 2015, 21, 167–177. [Google Scholar] [CrossRef]
- Belperio, J.A.; Dy, M.; Murray, L.; Burdick, M.D.; Xue, Y.Y.; Strieter, R.M.; Keane, M.P. The role of the Th2 CC chemokine ligand CCL17 in pulmonary fibrosis. J. Immunol. 2004, 173, 4692–4698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.T.; Hsu, H.; Lin, C.C.; Pan, S.Y.; Liu, S.Y.; Wu, C.F.; Tsai, P.Z.; Liao, C.T.; Cheng, H.T.; Chiang, W.C.; et al. Inflammatory macrophages switch to CCL17-expressing phenotype and promote peritoneal fibrosis. J. Pathol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgadari, C.; Angiolillo, A.L.; Tosato, G. Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. Blood 1996, 87, 3877–3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, M.; Pasini, E.; Chen-Scarabelli, C.; Corsetti, G.; Patel, H.; Linardi, D.; Onorati, F.; Faggian, G.; Scarabelli, T.; Saravolatz, L. Elevated Cardiac Troponin in Clinical Scenarios Beyond Obstructive Coronary Artery Disease. Med. Sci. Monit. 2019, 25, 7115–7125. [Google Scholar] [CrossRef]
- Cavender, M.A.; White, W.B.; Jarolim, P.; Bakris, G.L.; Cushman, W.C.; Kupfer, S.; Gao, Q.; Mehta, C.R.; Zannad, F.; Cannon, C.P.; et al. Serial Measurement of High-Sensitivity Troponin I and Cardiovascular Outcomes in Patients with Type 2 Diabetes Mellitus in the EXAMINE Trial (Examination of Cardiovascular Outcomes with Alogliptin Versus Standard of Care). Circulation 2017, 135, 1911–1921. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Zhao, M.; Khaldoyanidi, S.K.; Discipio, R.G. The chemokine CCL18 causes maturation of cultured monocytes to macrophages in the M2 spectrum. Immunology 2012, 135, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Murooka, T.T.; Rahbar, R.; Platanias, L.C.; Fish, E.N. CCL5-mediated T-cell chemotaxis involves the initiation of mRNA translation through mTOR/4E-BP1. Blood 2008, 111, 4892–4901. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Vishwanath, R.P.; Aguilar, B.; Starr, R.; Najbauer, J.; Aboody, K.S.; Jensen, M.C. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J. Immunol. 2007, 179, 3332–3341. [Google Scholar] [CrossRef]
- Laroumanie, F.; Douin-Echinard, V.; Pozzo, J.; Lairez, O.; Tortosa, F.; Vinel, C.; Delage, C.; Calise, D.; Dutaur, M.; Parini, A.; et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014, 129, 2111–2124. [Google Scholar] [CrossRef] [Green Version]
- Altara, R.; Manca, M.; Hessel, M.H.; Gu, Y.; van Vark, L.C.; Akkerhuis, K.M.; Staessen, J.A.; Struijker-Boudier, H.A.; Booz, G.W.; Blankesteijn, W.M. CXCL10 is a Circulating Inflammatory Marker in Patients with Advanced Heart Failure: A Pilot Study. J. Cardiovasc. Transl. Res. 2016, 9, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Mian, M.O.; Barhoumi, T.; Briet, M.; Paradis, P.; Schiffrin, E.L. Deficiency of T-regulatory cells exaggerates angiotensin II-induced microvascular injury by enhancing immune responses. J. Hypertens. 2016, 34, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Chenivesse, C.; Chang, Y.; Azzaoui, I.; Ait Yahia, S.; Morales, O.; Plé, C.; Foussat, A.; Tonnel, A.B.; Delhem, N.; Yssel, H.; et al. Pulmonary CCL18 recruits human regulatory T cells. J. Immunol. 2012, 189, 128–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevers, T.; Salvador, A.M.; Grodecki-Pena, A.; Knapp, A.; Velázquez, F.; Aronovitz, M.; Kapur, N.K.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ. Heart Fail. 2015, 8, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevers, T.; Salvador, A.M.; Velazquez, F.; Ngwenyama, N.; Carrillo-Salinas, F.J.; Aronovitz, M.; Blanton, R.M.; Alcaide, P. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J. Exp. Med. 2017, 214, 3311–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fudim, M.; Ambrosy, A.P.; Sun, J.L.; Anstrom, K.J.; Bart, B.A.; Butler, J.; AbouEzzeddine, O.; Greene, S.J.; Mentz, R.J.; Redfield, M.M.; et al. High-Sensitivity Troponin I in Hospitalized and Ambulatory Patients with Heart Failure with Preserved Ejection Fraction: Insights From the Heart Failure Clinical Research Network. J. Am. Heart Assoc. 2018, 7, e010364. [Google Scholar] [CrossRef]
- Giamouzis, G.; Schelbert, E.B.; Butler, J. Growing Evidence Linking Microvascular Dysfunction with Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2016, 5, e003259. [Google Scholar] [CrossRef] [Green Version]
- Gomberg-Maitland, M.; Shah, S.J.; Guazzi, M. Inflammation in Heart Failure with Preserved Ejection Fraction: Time to put out the Fire. JACC Heart Fail. 2016, 4, 325–328. [Google Scholar] [CrossRef]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.J.; Koo, J.B.; Kim, H.Y.; Seo, J.W.; Lee, E.J.; Kim, W.R.; Cho, J.Y.; Hahm, K.B.; Hong, S.P.; Kim, D.H.; et al. Umbilical cord/placenta-derived mesenchymal stem cells inhibit fibrogenic activation in human intestinal myofibroblasts via inhibition of myocardin-related transcription factor A. Stem Cell Res. Ther. 2019, 10, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcayaga-Miranda, F.; Cuenca, J.; Khoury, M. Antimicrobial Activity of Mesenchymal Stem Cells: Current Status and New Perspectives of Antimicrobial Peptide-Based Therapies. Front. Immunol. 2017, 8, 339. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Wu, W.; Xu, X.; Liao, L.; Zheng, F.; Messinger, S.; Sun, X.; Chen, J.; Yang, S.; Cai, J.; et al. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: A randomized controlled trial. JAMA 2012, 307, 1169–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.A.; Cameron, C.; Noorbaloochi, S.; Cullis, T.; Tucker, M.; Christensen, R.; Ghogomu, E.T.; Coyle, D.; Clifford, T.; Tugwell, P.; et al. Risk of serious infection in biological treatment of patients with rheumatoid arthritis: A systematic review and meta-analysis. Lancet 2015, 386, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Rigato, M.; Monami, M.; Fadini, G.P. Autologous Cell Therapy for Peripheral Arterial Disease: Systematic Review and Meta-Analysis of Randomized, Nonrandomized, and Noncontrolled Studies. Circ. Res. 2017, 120, 1326–1340. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef]
- Sun, L.; Louie, M.C.; Vannella, K.M.; Wilke, C.A.; LeVine, A.M.; Moore, B.B.; Shanley, T.P. New concepts of IL-10-induced lung fibrosis: Fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L341–L353. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K. Abandoning M1/M2 for a Network Model of Macrophage Function. Circ. Res. 2016, 119, 414–417. [Google Scholar] [CrossRef] [Green Version]
- Krampera, M.; Glennie, S.; Dyson, J.; Scott, D.; Laylor, R.; Simpson, E.; Dazzi, F. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 2003, 101, 3722–3729. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Augello, A.; Tasso, R.; Negrini, S.M.; Amateis, A.; Indiveri, F.; Cancedda, R.; Pennesi, G. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur. J. Immunol. 2005, 35, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Krampera, M.; Cosmi, L.; Angeli, R.; Pasini, A.; Liotta, F.; Andreini, A.; Santarlasci, V.; Mazzinghi, B.; Pizzolo, G.; Vinante, F.; et al. Role for Interferon-γ in the Immunomodulatory Activity of Human Bone Marrow Mesenchymal Stem Cells. Stem Cells 2006, 24, 386–398. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T. Regulatory T cells: How do they suppress immune responses? Int. Immunol. 2009, 21, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Hamdani, N.; Miteva, K.; Koschel, A.; Müller, I.; Pinzur, L.; Aberman, Z.; Pappritz, K.; Linke, W.A.; Tschöpe, C. Placenta-Derived Adherent Stromal Cells Improve Diabetes Mellitus-Associated Left Ventricular Diastolic Performance. Stem Cells Transl. Med. 2017, 6, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Gallet, R.; de Couto, G.; Simsolo, E.; Valle, J.; Sun, B.; Liu, W.; Tseliou, E.; Zile, M.R.; Marbán, E. Cardiosphere-derived cells reverse heart failure with preserved ejection fraction (HFpEF) in rats by decreasing fibrosis and inflammation. JACC Basic Transl. Sci. 2016, 1, 14–28. [Google Scholar] [CrossRef] [Green Version]
- Kelm, N.Q.; Beare, J.E.; Yuan, F.; George, M.; Shofner, C.M.; Keller, B.B.; Hoying, J.B.; LeBlanc, A.J. Adipose-derived cells improve left ventricular diastolic function and increase microvascular perfusion in advanced age. PLoS ONE 2018, 13, e0202934. [Google Scholar] [CrossRef]
- Dardalhon, V.; Korn, T.; Kuchroo, V.K.; Anderson, A.C. Role of Th1 and Th17 cells in organ-specific autoimmunity. J. Autoimmun. 2008, 31, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Shigemoto-Kuroda, T.; Oh, J.Y.; Kim, D.K.; Jeong, H.J.; Park, S.Y.; Lee, H.J.; Park, J.W.; Kim, T.W.; An, S.Y.; Prockop, D.J.; et al. MSC-derived Extracellular Vesicles Attenuate Immune Responses in Two Autoimmune Murine Models: Type 1 Diabetes and Uveoretinitis. Stem Cell Rep. 2017, 8, 1214–1225. [Google Scholar] [CrossRef] [Green Version]
- Kota, D.J.; Wiggins, L.L.; Yoon, N.; Lee, R.H. TSG-6 produced by hMSCs delays the onset of autoimmune diabetes by suppressing Th1 development and enhancing tolerogenicity. Diabetes 2013, 62, 2048–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurte, M.; Bravo-Alegría, J.; Torres, A.; Carrasco, V.; Ibáñez, C.; Vega-Letter, A.M.; Fernández-O’Ryan, C.; Irarrázabal, C.E.; Figueroa, F.E.; Fuentealba, R.A.; et al. Intravenous administration of bone marrow-derived mesenchymal stem cells induces a switch from classical to atypical symptoms in experimental autoimmune encephalomyelitis. Stem Cells Int. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahfouz, M.M.; Abdelsalam, R.M.; Masoud, M.A.; Mansour, H.A.; Ahmed-Farid, O.A.; Kenawy, S.A. The neuroprotective effect of mesenchymal stem cells on an experimentally induced model for multiple sclerosis in mice. J. Biochem. Mol. Toxicol. 2017, 31, e21936. [Google Scholar] [CrossRef]
- Semon, J.A.; Maness, C.; Zhang, X.; Sharkey, S.A.; Beuttler, M.M.; Shah, F.S.; Pandey, A.C.; Gimble, J.M.; Zhang, S.; Scruggs, B.A.; et al. Comparison of human adult stem cells from adipose tissue and bone marrow in the treatment of experimental autoimmune encephalomyelitis. Stem Cell Res. Ther. 2014, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Laso-García, F.; Ramos-Cejudo, J.; Carrillo-Salinas, F.J.; Otero-Ortega, L.; Feliú, A.; Gómez-de Frutos, M.; Mecha, M.; Díez-Tejedor, E.; Guaza, C.; Gutiérrez-Fernández, M. Therapeutic potential of extracellular vesicles derived from human mesenchymal stem cells in a model of progressive multiple sclerosis. PLoS ONE 2018, 13, e0202590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, P.O.; Schwarcz, E.; Korsgren, O.; Le Blanc, K. Preserved β-cell function in type 1 diabetes by mesenchymal stromal cells. Diabetes 2015, 64, 587–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Wu, Z.; Xu, X.; Liao, L.; Chen, J.; Huang, L.; Wu, W.; Luo, F.; Wu, C.; Pugliese, A.; et al. Umbilical Cord Mesenchymal Stromal Cell with Autologous Bone Marrow Cell Transplantation in Established Type 1 Diabetes: A Pilot Randomized Controlled Open-Label Clinical Study to Assess Safety and Impact on Insulin Secretion. Diabetes Care 2016, 39, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Jiang, Z.; Zhao, T.; Ye, M.; Hu, C.; Yin, Z.; Li, H.; Zhang, Y.; Diao, Y.; Li, Y.; et al. Reversal of type 1 diabetes via islet β cell regeneration following immune modulation by cord blood-derived multipotent stem cells. BMC Med. 2012, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Li, L.; Wan, B.; Yang, M.; Hong, J.; Gu, W.; Wang, W.; Ning, G. Immune response after autologous hematopoietic stem cell transplantation in type 1 diabetes mellitus. Stem Cell Res. Ther. 2017, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Llufriu, S.; Sepúlveda, M.; Blanco, Y.; Marín, P.; Moreno, B.; Berenguer, J.; Gabilondo, I.; Martínez-Heras, E.; Sola-Valls, N.; Arnaiz, J.A.; et al. Randomized placebo-controlled phase II trial of autologous mesenchymal stem cells in multiple sclerosis. PLoS ONE 2014, 9, e113936. [Google Scholar] [CrossRef]
- Bonab, M.M.; Sahraian, M.A.; Aghsaie, A.; Karvigh, S.A.; Hosseinian, S.M.; Nikbin, B.; Lotfi, J.; Khorramnia, S.; Motamed, M.R.; Togha, M.; et al. Autologous mesenchymal stem cell therapy in progressive multiple sclerosis: An open label study. Curr. Stem Cell Res. Ther. 2012, 7, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Laroni, A.; Brundin, L.; Clanet, M.; Fernandez, O.; Nabavi, S.M.; Muraro, P.A.; Oliveri, R.S.; Radue, E.W.; Sellner, J.; et al. MEsenchymal StEm cells for Multiple Sclerosis (MESEMS): A randomized, double blind, cross-over phase I/II clinical trial with autologous mesenchymal stem cells for the therapy of multiple sclerosis. Trials 2019, 20, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmeler, S.; Zeiher, A.M.; Schneider, M.D. Unchain my heart: The scientific foundations of cardiac repair. J. Clin. Investig. 2005, 115, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Traverse, J.H.; Henry, T.D.; Pepine, C.J.; Willerson, J.T.; Zhao, D.X.; Ellis, S.G.; Forder, J.R.; Anderson, R.D.; Hatzopoulos, A.K.; Penn, M.S.; et al. Effect of the use and timing of bone marrow mononuclear cell delivery on left ventricular function after acute myocardial infarction: The TIME randomized trial. JAMA 2012, 308, 2380–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hare, J.M.; DiFede, D.L.; Rieger, A.C.; Florea, V.; Landin, A.M.; El-Khorazaty, J.; Khan, A.; Mushtaq, M.; Lowery, M.H.; Byrnes, J.J.; et al. Randomized Comparison of Allogeneic Versus Autologous Mesenchymal Stem Cells for Nonischemic Dilated Cardiomyopathy: POSEIDON-DCM Trial. J. Am. Coll. Cardiol. 2017, 69, 526–537. [Google Scholar] [CrossRef]
- Perin, E.C.; Borow, K.M.; Silva, G.V.; DeMaria, A.N.; Marroquin, O.C.; Huang, P.P.; Traverse, J.H.; Krum, H.; Skerrett, D.; Zheng, Y.; et al. A Phase II Dose-Escalation Study of Allogeneic Mesenchymal Precursor Cells in Patients with Ischemic or Nonischemic Heart Failure. Circ. Res. 2015, 117, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.; Epstein, S.E.; Greene, S.J.; Quyyumi, A.A.; Sikora, S.; Kim, R.J.; Anderson, A.S.; Wilcox, J.E.; Tankovich, N.I.; Lipinski, M.J.; et al. Intravenous Allogeneic Mesenchymal Stem Cells for Nonischemic Cardiomyopathy: Safety and Efficacy Results of a Phase II-A Randomized Trial. Circ. Res. 2017, 120, 332–340. [Google Scholar] [CrossRef]
- Yao, K.; Huang, R.; Qian, J.; Cui, J.; Ge, L.; Li, Y.; Zhang, F.; Shi, H.; Huang, D.; Zhang, S.; et al. Administration of intracoronary bone marrow mononuclear cells on chronic myocardial infarction improves diastolic function. Heart 2008, 94, 1147–1153. [Google Scholar] [CrossRef] [Green Version]
- Kanelidis, A.J.; Premer, C.; Lopez, J.; Balkan, W.; Hare, J.M. Route of Delivery Modulates the Efficacy of Mesenchymal Stem Cell Therapy for Myocardial Infarction: A Meta-Analysis of Preclinical Studies and Clinical Trials. Circ. Res. 2017, 120, 1139–1150. [Google Scholar] [CrossRef]
- Leuschner, F.; Nahrendorf, M. Novel functions of macrophages in the heart: Insights into electrical conduction, stress, and diastolic dysfunction. Eur. Heart J. 2019. [Google Scholar] [CrossRef]
- Rodríguez-Perea, A.L.; Arcia, E.D.; Rueda, C.M.; Velilla, P.A. Phenotypical characterization of regulatory T cells in humans and rodents. Clin. Exp. Immunol. 2016, 185, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bervar, M.; Kozelj, M.; Poglajen, G.; Sever, M.; Zemljic, G.; Frljak, S.; Cukjati, M.; Cernelc, P.; Haddad, F.; Vrtovec, B. Effects of Transendocardial CD34+ Cell Transplantation on Diastolic Parameters in Patients with Nonischemic Dilated Cardiomyopathy. Stem Cells Transl. Med. 2017, 6, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Diederichsen, A.C.; Møller, J.E.; Thayssen, P.; Videbaek, L.; Saekmose, S.G.; Barington, T.; Kassem, M. Changes in left ventricular filling patterns after repeated injection of autologous bone marrow cells in heart failure patients. Scand. Cardiovasc. J. 2010, 44, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Beeres, S.L.; Lamb, H.J.; Roes, S.D.; Holman, E.R.; Kaandorp, T.A.; Fibbe, W.E.; de Roos, A.; van der Wall, E.E.; Schalij, M.J.; Bax, J.J.; et al. Effect of intramyocardial bone marrow cell injection on diastolic function in patients with chronic myocardial ischemia. J. Magn. Reson. Imaging 2008, 27, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Vrtovec, B.; Poglajen, G.; Lezaic, L.; Sever, M.; Socan, A.; Domanovic, D.; Cernelc, P.; Torre-Amione, G.; Haddad, F.; Wu, J.C. Comparison of transendocardial and intracoronary CD34+ cell transplantation in patients with nonischemic dilated cardiomyopathy. Circulation 2013, 128, S42–S49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezaic, L.; Socan, A.; Poglajen, G.; Peitl, P.K.; Sever, M.; Cukjati, M.; Cernelc, P.; Wu, J.C.; Haddad, F.; Vrtovec, B. Intracoronary transplantation of CD34+ cells is associated with improved myocardial perfusion in patients with nonischemic dilated cardiomyopathy. J. Card. Fail. 2015, 21, 145–152. [Google Scholar] [CrossRef]
- Mazo, M.; Gavira, J.J.; Abizanda, G.; Moreno, C.; Ecay, M.; Soriano, M.; Aranda, P.; Collantes, M.; Alegría, E.; Merino, J.; et al. Transplantation of mesenchymal stem cells exerts a greater long-term effect than bone marrow mononuclear cells in a chronic myocardial infarction model in rat. Cell Transpl. 2010, 19, 313–328. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Chen, B.; Liang, Z.; Deng, W.; Jiang, Y.; Li, S.; Xu, J.; Wu, Q.; Zhang, Z.; Xie, B.; et al. Comparison of bone marrow mesenchymal stem cells with bone marrow-derived mononuclear cells for treatment of diabetic critical limb ischemia and foot ulcer: A double-blind, randomized, controlled trial. Diabetes Res. Clin. Pract. 2011, 92, 26–36. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sava, R.I.; Pepine, C.J.; March, K.L. Immune Dysregulation in HFpEF: A Target for Mesenchymal Stem/Stromal Cell Therapy. J. Clin. Med. 2020, 9, 241. https://doi.org/10.3390/jcm9010241
Sava RI, Pepine CJ, March KL. Immune Dysregulation in HFpEF: A Target for Mesenchymal Stem/Stromal Cell Therapy. Journal of Clinical Medicine. 2020; 9(1):241. https://doi.org/10.3390/jcm9010241
Chicago/Turabian StyleSava, Ruxandra I., Carl J. Pepine, and Keith L. March. 2020. "Immune Dysregulation in HFpEF: A Target for Mesenchymal Stem/Stromal Cell Therapy" Journal of Clinical Medicine 9, no. 1: 241. https://doi.org/10.3390/jcm9010241
APA StyleSava, R. I., Pepine, C. J., & March, K. L. (2020). Immune Dysregulation in HFpEF: A Target for Mesenchymal Stem/Stromal Cell Therapy. Journal of Clinical Medicine, 9(1), 241. https://doi.org/10.3390/jcm9010241