Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
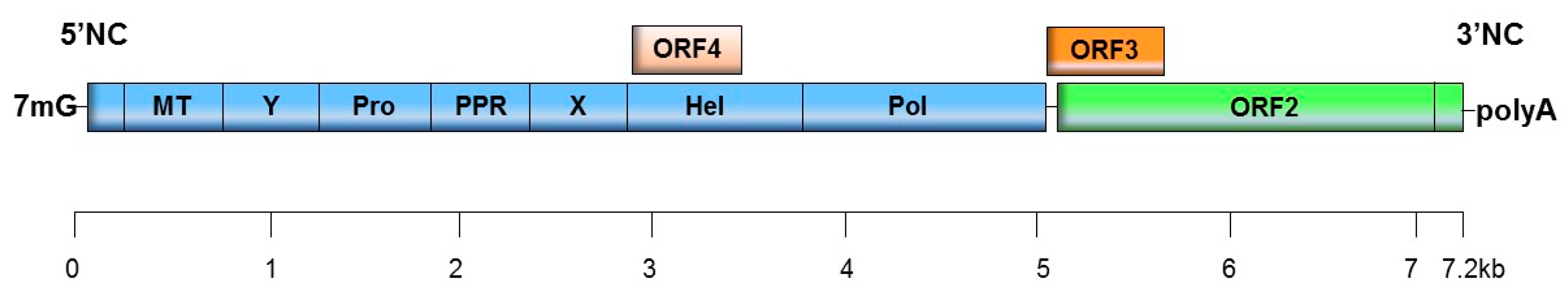
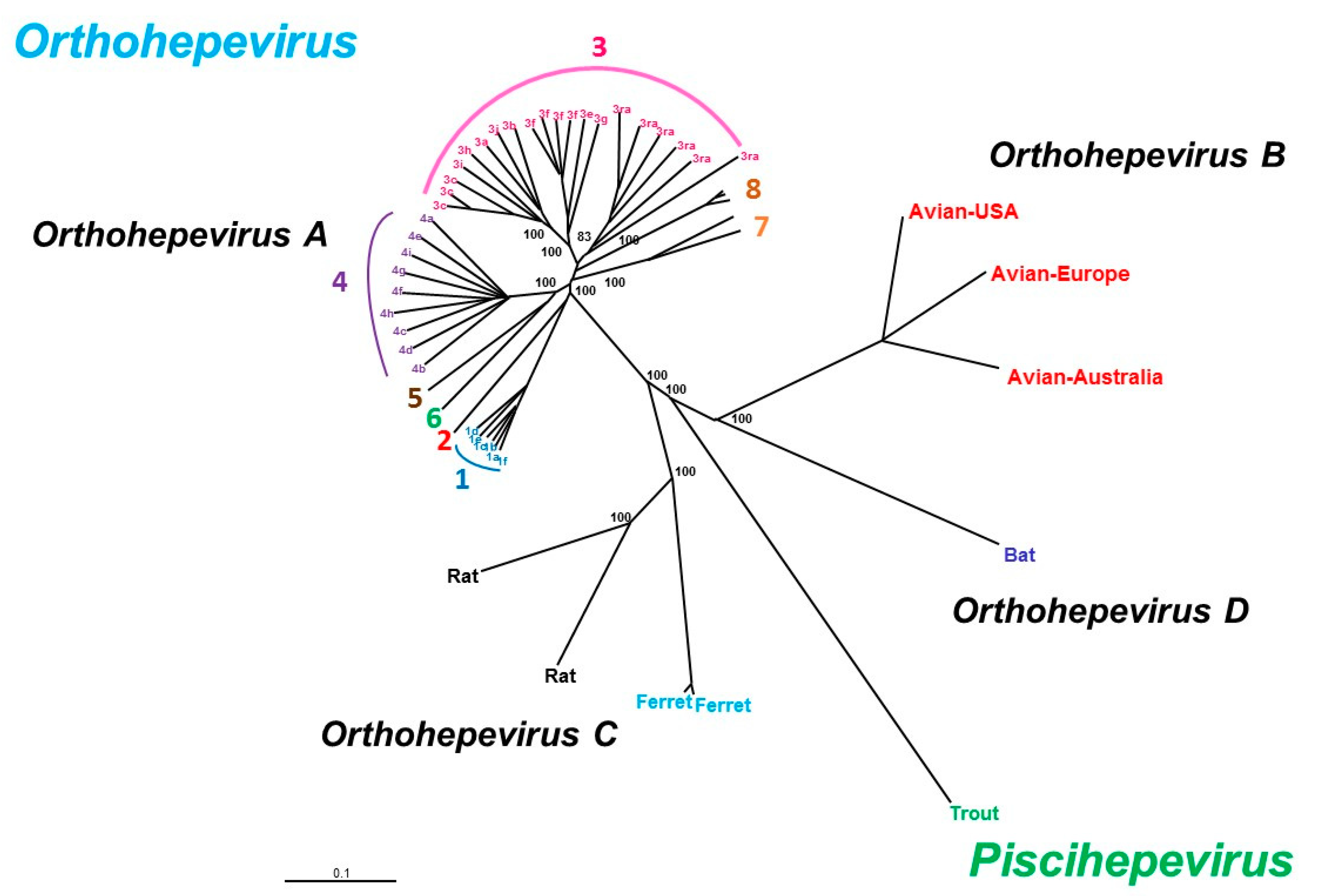
2. HEV Genome and Classification
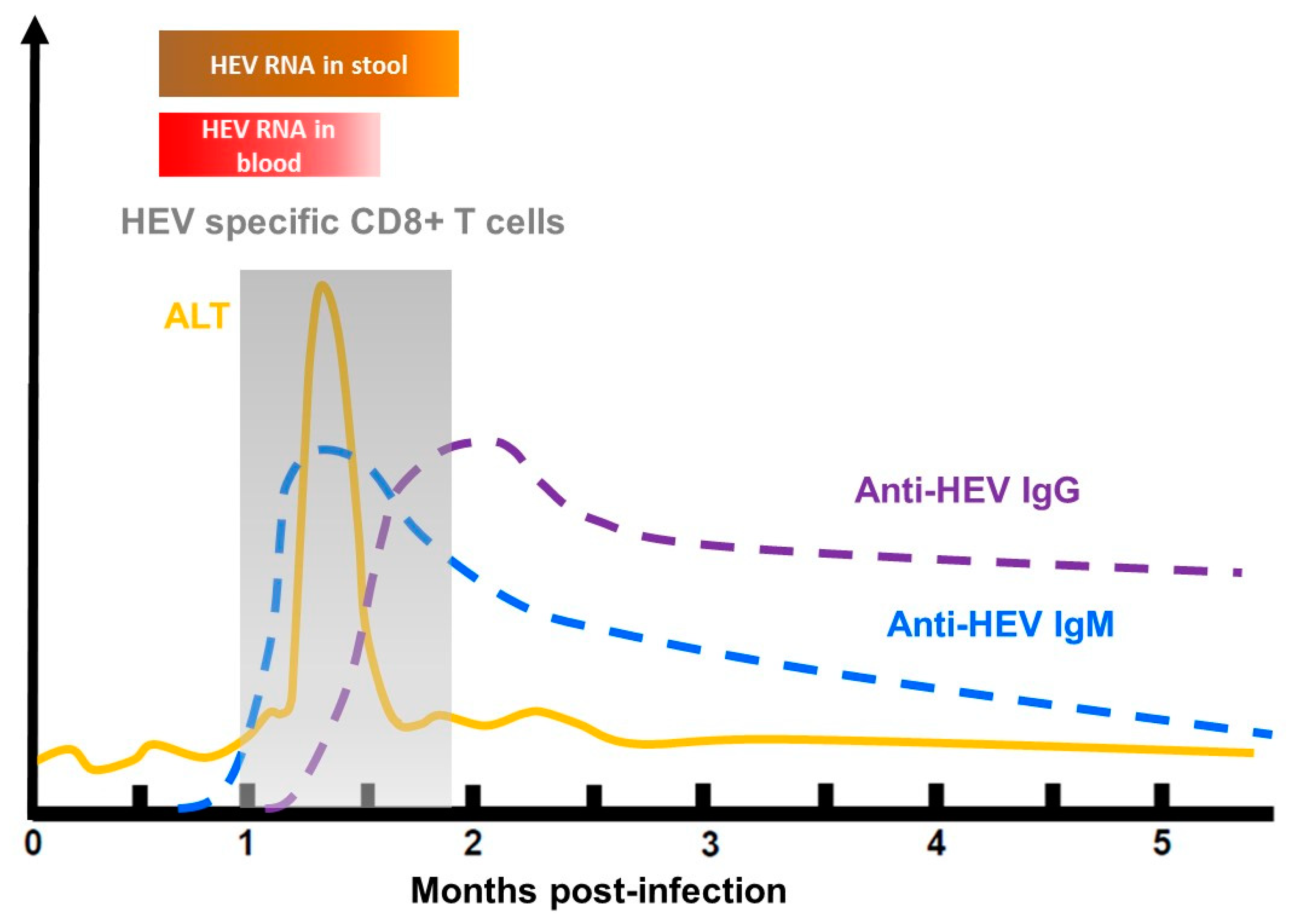
3. Clinical Course of HEV Infection
4. Extra-Hepatic Manifestations
4.1. Neurological Manifestations
4.2. Renal Manifestations
5. Pathogenesis
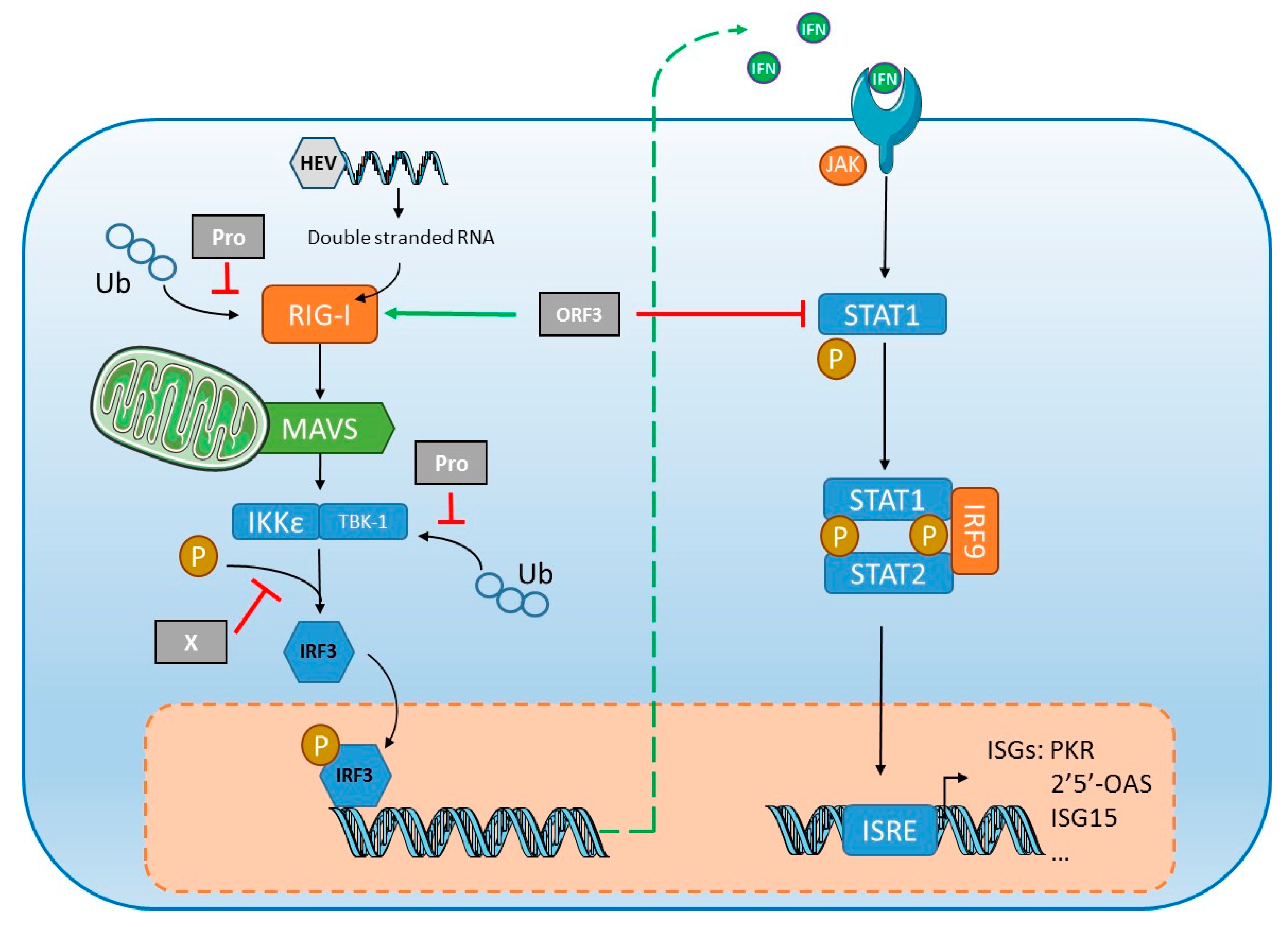
5.1. Innate Immune Response
5.2. Humoral and Cell-Mediated Immune Responses
6. Pathogenesis of Severe Hepatitis
6.1. Severe Hepatitis E in the General Population
6.2. Severe Hepatitis in Pregnant Women
7. Pathogenesis of Chronic Infection in Immunocompromised Patients
8. Treatment
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kamar, N.; Izopet, J.; Pavio, N.; Aggarwal, R.; Labrique, A.; Wedemeyer, H.; Dalton, H.R. Hepatitis E virus infection. Nat. Rev. Dis. Primers 2017, 3, 17086. [Google Scholar] [CrossRef]
- Kenney, S.P.; Meng, X.J. Hepatitis E Virus Genome Structure and Replication Strategy. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Karlin, D.G. Sequence analysis reveals a conserved extension in the capping enzyme of the alphavirus supergroup, and a homologous domain in nodaviruses. Biol. Direct 2015, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar, B.N.; Ranjith Kumar, C.T.; Surjit, M. Endoplasmic Reticulum Stress Induced Synthesis of a Novel Viral Factor Mediates Efficient Replication of Genotype-1 Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ying, D.; Lhomme, S.; Tang, Z.; Walker, C.M.; Xia, N.; Zheng, Z.; Feng, Z. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc. Natl. Acad. Sci. USA 2018, 115, 4773–4778. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Hirai-Yuki, A.; McKnight, K.L.; Lemon, S.M. Naked Viruses that aren’t Always Naked: Quasi-Enveloped Agents of Acute Hepatitis. Annu. Rev. Virol. 2014, 1, 539–560. [Google Scholar] [CrossRef]
- Feng, Z.; Lemon, S.M. Peek-a-boo: Membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol. 2014, 22, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.B.; Simmonds, P. Classification and Genomic Diversity of Enterically Transmitted Hepatitis Viruses. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Nicot, F.; Jeanne, N.; Roulet, A.; Lefebvre, C.; Carcenac, R.; Manno, M.; Dubois, M.; Kamar, N.; Lhomme, S.; Abravanel, F.; et al. Diversity of hepatitis E virus genotype 3. Rev. Med. Virol. 2018, 28, e1987. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Izopet, J.; Oliveira-Filho, E.F.; Ulrich, R.G.; Johne, R.; Koenig, M.; Jameel, S.; Harrison, T.J.; Meng, X.J.; et al. Proposed reference sequences for hepatitis E virus subtypes. J. Gen. Virol. 2016, 97, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.P. The Current Host Range of Hepatitis E Viruses. Viruses 2019, 11, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izopet, J.; Dubois, M.; Bertagnoli, S.; Lhomme, S.; Marchandeau, S.; Boucher, S.; Kamar, N.; Abravanel, F.; Guerin, J.L. Hepatitis E virus strains in rabbits and evidence of a closely related strain in humans, France. Emerg. Infect. Dis. 2012, 18, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Abravanel, F.; Lhomme, S.; El Costa, H.; Schvartz, B.; Peron, J.M.; Kamar, N.; Izopet, J. Rabbit Hepatitis E Virus Infections in Humans, France. Emerg. Infect. Dis. 2017, 23, 1191–1193. [Google Scholar] [CrossRef] [Green Version]
- Sahli, R.; Fraga, M.; Semela, D.; Moradpour, D.; Gouttenoire, J. Rabbit HEV in immunosuppressed patients with hepatitis E acquired in Switzerland. J. Hepatol. 2019, 70, 1023–1025. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.H.; Tan, B.H.; Teo, E.C.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic Infection With Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357. [Google Scholar] [CrossRef] [Green Version]
- Li, T.C.; Bai, H.; Yoshizaki, S.; Ami, Y.; Suzaki, Y.; Doan, Y.H.; Takahashi, K.; Mishiro, S.; Takeda, N.; Wakita, T. Genotype 5 Hepatitis E Virus Produced by a Reverse Genetics System Has the Potential for Zoonotic Infection. Hepatol. Commun. 2019, 3, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Teng, J.L.L.; Lau, S.K.P.; Sridhar, S.; Fu, H.; Gong, W.; Li, M.; Xu, Q.; He, Y.; Zhuang, H.; et al. Transmission of a Novel Genotype of Hepatitis E Virus from Bactrian Camels to Cynomolgus Macaques. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.; Yip, C.C.Y.; Wu, S.; Cai, J.; Zhang, A.J.; Leung, K.H.; Chung, T.W.H.; Chan, J.F.W.; Chan, W.M.; Teng, J.L.L.; et al. Rat Hepatitis E Virus as Cause of Persistent Hepatitis after Liver Transplant. Emerg. Infect. Dis. 2018, 24, 2241–2250. [Google Scholar] [CrossRef] [Green Version]
- Andonov, A.; Robbins, M.; Borlang, J.; Cao, J.; Hattchete, T.; Stueck, A.; Deschaumbault, Y.; Murnaghan, K.; Varga, J.; Johnston, B. Rat hepatitis E virus linked to severe acute hepatitis in an immunocompetent patient. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Pischke, S.; Manns, M.P. Pathogenesis and treatment of hepatitis e virus infection. Gastroenterology 2012, 142, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: Understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuroo, M.S.; Kamili, S.; Khuroo, M.S. Clinical course and duration of viremia in vertically transmitted hepatitis E virus (HEV) infection in babies born to HEV-infected mothers. J. Viral Hepat. 2009, 16, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Singhal, A.; Panda, S.K.; Acharya, S.K. A 20-year single-center experience with acute liver failure during pregnancy: Is the prognosis really worse? Hepatology 2008, 48, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Hazeldine, S.; Banks, M.; Ijaz, S.; Bendall, R. Locally acquired hepatitis E in chronic liver disease. Lancet 2007, 369, 1260. [Google Scholar] [CrossRef]
- Peron, J.M.; Bureau, C.; Poirson, H.; Mansuy, J.M.; Alric, L.; Selves, J.; Dupuis, E.; Izopet, J.; Vinel, J.P. Fulminant liver failure from acute autochthonous hepatitis E in France: Description of seven patients with acute hepatitis E and encephalopathy. J. Viral Hepat. 2007, 14, 298–303. [Google Scholar] [CrossRef]
- Guillois, Y.; Abravanel, F.; Miura, T.; Pavio, N.; Vaillant, V.; Lhomme, S.; Le Guyader, F.S.; Rose, N.; Le Saux, J.C.; King, L.A.; et al. High Proportion of Asymptomatic Infections in an Outbreak of Hepatitis E Associated With a Spit-Roasted Piglet, France, 2013. Clin. Infect. Dis. 2016, 62, 351–357. [Google Scholar] [CrossRef]
- Said, B.; Ijaz, S.; Kafatos, G.; Booth, L.; Thomas, H.L.; Walsh, A.; Ramsay, M.; Morgan, D.; Hepatitis, E.I.I.T. Hepatitis E outbreak on cruise ship. Emerg. Infect. Dis. 2009, 15, 1738–1744. [Google Scholar] [CrossRef]
- Faber, M.; Willrich, N.; Schemmerer, M.; Rauh, C.; Kuhnert, R.; Stark, K.; Wenzel, J.J. Hepatitis E virus seroprevalence, seroincidence and seroreversion in the German adult population. J. Viral Hepat. 2018, 25, 752–758. [Google Scholar] [CrossRef]
- Lhomme, S.; Gallian, P.; Dimeglio, C.; Assal, A.; Abravanel, F.; Tiberghien, P.; Izopet, J. Viral load and clinical manifestations of hepatitis E virus genotype 3 infections. J. Viral Hepat. 2019, 26, 1139–1142. [Google Scholar] [CrossRef]
- Gerolami, R.; Moal, V.; Colson, P. Chronic hepatitis E with cirrhosis in a kidney-transplant recipient. N. Engl. J. Med. 2008, 358, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Haagsma, E.B.; van den Berg, A.P.; Porte, R.J.; Benne, C.A.; Vennema, H.; Reimerink, J.H.; Koopmans, M.P. Chronic hepatitis E virus infection in liver transplant recipients. Liver Transplant. 2008, 14, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Selves, J.; Mansuy, J.M.; Ouezzani, L.; Peron, J.M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colson, P.; Kaba, M.; Moreau, J.; Brouqui, P. Hepatitis E in an HIV-infected patient. J. Clin. Virol. 2009, 45, 269–271. [Google Scholar] [CrossRef]
- Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Tedder, R.S.; Ijaz, S. Persistent carriage of hepatitis E virus in patients with HIV infection. N. Engl. J. Med. 2009, 361, 1025–1027. [Google Scholar] [CrossRef]
- Kenfak-Foguena, A.; Schoni-Affolter, F.; Burgisser, P.; Witteck, A.; Darling, K.E.; Kovari, H.; Kaiser, L.; Evison, J.M.; Elzi, L.; Gurter-De La Fuente, V.; et al. Hepatitis E Virus seroprevalence and chronic infections in patients with HIV, Switzerland. Emerg. Infect. Dis. 2011, 17, 1074–1078. [Google Scholar] [CrossRef]
- Tamura, A.; Shimizu, Y.K.; Tanaka, T.; Kuroda, K.; Arakawa, Y.; Takahashi, K.; Mishiro, S.; Shimizu, K.; Moriyama, M. Persistent infection of hepatitis E virus transmitted by blood transfusion in a patient with T-cell lymphoma. Hepatol. Res. 2007, 37, 113–120. [Google Scholar] [CrossRef]
- Ollier, L.; Tieulie, N.; Sanderson, F.; Heudier, P.; Giordanengo, V.; Fuzibet, J.G.; Nicand, E. Chronic hepatitis after hepatitis E virus infection in a patient with non-Hodgkin lymphoma taking rituximab. Ann. Intern. Med. 2009, 150, 430–431. [Google Scholar] [CrossRef]
- Geng, Y.; Zhang, H.; Huang, W.; Harrison, T.J.; Geng, K.; Li, Z.; Wang, Y.. Persistent hepatitis e virus genotype 4 infection in a child with acute lymphoblastic leukemia. Hepat. Mon. 2014, 14, e15618. [Google Scholar] [CrossRef] [Green Version]
- Peron, J.M.; Mansuy, J.M.; Recher, C.; Bureau, C.; Poirson, H.; Alric, L.; Izopet, J.; Vinel, J.P. Prolonged hepatitis E in an immunocompromised patient. J. Gastroenterol. Hepatol. 2006, 21, 1223–1224. [Google Scholar] [CrossRef]
- Versluis, J.; Pas, S.D.; Agteresch, H.J.; de Man, R.A.; Maaskant, J.; Schipper, M.E.; Osterhaus, A.D.; Cornelissen, J.J.; van der Eijk, A.A. Hepatitis E virus: An underestimated opportunistic pathogen in recipients of allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, H.; Luxembourger, C.; Gottenberg, J.E.; Fournier, S.; Abravanel, F.; Cantagrel, A.; Chatelus, E.; Claudepierre, P.; Hudry, C.; Izopet, J.; et al. Outcome of hepatitis E virus infection in patients with inflammatory arthritides treated with immunosuppressants: A French retrospective multicenter study. Medicine 2015, 94, e675. [Google Scholar] [CrossRef] [PubMed]
- Pischke, S.; Peron, J.M.; von Wulffen, M.; von Felden, J.; Honer Zu Siederdissen, C.; Fournier, S.; Lutgehetmann, M.; Iking-Konert, C.; Bettinger, D.; Par, G.; et al. Chronic Hepatitis E in Rheumatology and Internal Medicine Patients: A Retrospective Multicenter European Cohort Study. Viruses 2019, 11, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robins, A.E.M.; Bowden, D.J.; Gelson, W.T.H. Chronic genotype 1 hepatitis E infection from immunosuppression for ileo-colonic Crohn’s disease. Oxf. Med. Case Rep. 2018, 2018, omy059. [Google Scholar] [CrossRef]
- Ankcorn, M.; Haywood, B.; Tedder, R.; Ijaz, S. Response to: ‘Chronic genotype 1 hepatitis E infection from immunosuppression for ileo-colonic Crohn’s disease’. Oxf. Med. Case Rep. 2019, 2019, omy125. [Google Scholar] [CrossRef]
- Kamar, N.; Rostaing, L.; Legrand-Abravanel, F.; Izopet, J. How should hepatitis E virus infection be defined in organ-transplant recipients? Am. J. Transplant. 2013, 13, 1935–1936. [Google Scholar] [CrossRef]
- Kamar, N.; Mansuy, J.M.; Cointault, O.; Selves, J.; Abravanel, F.; Danjoux, M.; Otal, P.; Esposito, L.; Durand, D.; Izopet, J.; et al. Hepatitis E virus-related cirrhosis in kidney- and kidney-pancreas-transplant recipients. Am. J. Transplant. 2008, 8, 1744–1748. [Google Scholar] [CrossRef]
- Kamar, N.; Garroutste, C.; Haagsma, E.B.; Garrigue, V.; Pischke, S.; Chauvet, C.; Dumortier, J.; Cannesson, A.; Cassuto-Viguier, E.; Thervet, E.; et al. Factors associated with chronic hepatitis in patients with hepatitis E virus infection who have received solid organ transplants. Gastroenterology 2011, 140, 1481–1489. [Google Scholar] [CrossRef]
- Dalton, H.R.; Kamar, N.; van Eijk, J.J.; McLean, B.N.; Cintas, P.; Bendall, R.P.; Jacobs, B.C. Hepatitis E virus and neurological injury. Nat. Rev. Neurol. 2016, 12, 77–85. [Google Scholar] [CrossRef]
- Dalton, H.R.; van Eijk, J.J.J.; Cintas, P.; Madden, R.G.; Jones, C.; Webb, G.W.; Norton, B.; Pique, J.; Lutgens, S.; Devooght-Johnson, N.; et al. Hepatitis E virus infection and acute non-traumatic neurological injury: A prospective multicentre study. J. Hepatol. 2017, 67, 925–932. [Google Scholar] [CrossRef]
- van Eijk, J.J.; Madden, R.G.; van der Eijk, A.A.; Hunter, J.G.; Reimerink, J.H.; Bendall, R.P.; Pas, S.D.; Ellis, V.; van Alfen, N.; Beynon, L.; et al. Neuralgic amyotrophy and hepatitis E virus infection. Neurology 2014, 82, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Eijk, J.J.J.; Dalton, H.R.; Ripellino, P.; Madden, R.G.; Jones, C.; Fritz, M.; Gobbi, C.; Melli, G.; Pasi, E.; Herrod, J.; et al. Clinical phenotype and outcome of hepatitis E virus-associated neuralgic amyotrophy. Neurology 2017, 89, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, M.; Berger, B.; Schemmerer, M.; Endres, D.; Wenzel, J.J.; Stich, O.; Panning, M. Pathological Cerebrospinal Fluid Findings in Patients with Neuralgic Amyotrophy and Acute Hepatitis E Virus Infection. J. Infect. Dis. 2018, 217, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Geurtsvankessel, C.H.; Islam, Z.; Mohammad, Q.D.; Jacobs, B.C.; Endtz, H.P.; Osterhaus, A.D. Hepatitis E and Guillain-Barre syndrome. Clin. Infect. Dis. 2013, 57, 1369–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukae, J.; Tsugawa, J.; Ouma, S.; Umezu, T.; Kusunoki, S.; Tsuboi, Y. Guillain-Barre and Miller Fisher syndromes in patients with anti-hepatitis E virus antibody: A hospital-based survey in Japan. Neurol. Sci. 2016, 37, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, B.; van der Eijk, A.A.; Pas, S.D.; Hunter, J.G.; Madden, R.G.; Tio-Gillen, A.P.; Dalton, H.R.; Jacobs, B.C. Guillain-Barre syndrome associated with preceding hepatitis E virus infection. Neurology 2014, 82, 491–497. [Google Scholar] [CrossRef]
- Stevens, O.; Claeys, K.G.; Poesen, K.; Saegeman, V.; Van Damme, P. Diagnostic Challenges and Clinical Characteristics of Hepatitis E Virus-Associated Guillain-Barre Syndrome. JAMA Neurol. 2017, 74, 26–33. [Google Scholar] [CrossRef]
- Abravanel, F.; Pique, J.; Couturier, E.; Nicot, F.; Dimeglio, C.; Lhomme, S.; Chiabrando, J.; Saune, K.; Peron, J.M.; Kamar, N.; et al. Acute hepatitis E in French patients and neurological manifestations. J. Infect. 2018, 77, 220–226. [Google Scholar] [CrossRef]
- Kamar, N.; Izopet, J.; Cintas, P.; Garrouste, C.; Uro-Coste, E.; Cointault, O.; Rostaing, L. Hepatitis E virus-induced neurological symptoms in a kidney-transplant patient with chronic hepatitis. Am. J. Transplant. 2010, 10, 1321–1324. [Google Scholar] [CrossRef]
- Shi, R.; Soomro, M.H.; She, R.; Yang, Y.; Wang, T.; Wu, Q.; Li, H.; Hao, W. Evidence of Hepatitis E virus breaking through the blood-brain barrier and replicating in the central nervous system. J. Viral Hepat. 2016, 23, 930–939. [Google Scholar] [CrossRef]
- Tian, J.; Shi, R.; Liu, T.; She, R.; Wu, Q.; An, J.; Hao, W.; Soomro, M.H. Brain Infection by Hepatitis E Virus Probably via Damage of the Blood-Brain Barrier Due to Alterations of Tight Junction Proteins. Front. Cell. Infect. Microbiol. 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, F.; Xu, L.; Lin, Z.; de Vrij, F.M.S.; Ayo-Martin, A.C.; van der Kroeg, M.; Zhao, M.; Yin, Y.; Wang, W.; et al. Hepatitis E Virus Infects Neurons and Brains. J. Infect. Dis. 2017, 215, 1197–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drave, S.A.; Debing, Y.; Walter, S.; Todt, D.; Engelmann, M.; Friesland, M.; Wedemeyer, H.; Neyts, J.; Behrendt, P.; Steinmann, E. Extra-hepatic replication and infection of hepatitis E virus in neuronal-derived cells. J. Viral Hepat. 2016, 23, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Helsen, N.; Debing, Y.; Paeshuyse, J.; Dallmeier, K.; Boon, R.; Coll, M.; Sancho-Bru, P.; Claes, C.; Neyts, J.; Verfaillie, C.M. Stem cell-derived hepatocytes: A novel model for hepatitis E virus replication. J. Hepatol. 2016, 64, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Li, T.C.; Suzaki, Y.; Ami, Y.; Tsunemitsu, H.; Miyamura, T.; Takeda, N. Mice are not susceptible to hepatitis E virus infection. J. Vet. Med. Sci. 2008, 70, 1359–1362. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Mansuy, J.M.; Esposito, L.; Legrand-Abravanel, F.; Peron, J.M.; Durand, D.; Rostaing, L.; Izopet, J. Acute hepatitis and renal function impairment related to infection by hepatitis E virus in a renal allograft recipient. Am. J. Kidney Dis. 2005, 45, 193–196. [Google Scholar] [CrossRef]
- Kamar, N.; Weclawiak, H.; Guilbeau-Frugier, C.; Legrand-Abravanel, F.; Cointault, O.; Ribes, D.; Esposito, L.; Cardeau-Desangles, I.; Guitard, J.; Sallusto, F.; et al. Hepatitis E virus and the kidney in solid-organ transplant patients. Transplantation 2012, 93, 617–623. [Google Scholar] [CrossRef]
- Taton, B.; Moreau, K.; Lepreux, S.; Bachelet, T.; Trimoulet, P.; De Ledinghen, V.; Pommereau, A.; Ronco, P.; Kamar, N.; Merville, P.; et al. Hepatitis E virus infection as a new probable cause of de novo membranous nephropathy after kidney transplantation. Transpl. Infect. Dis. 2013, 15, E211–E215. [Google Scholar] [CrossRef]
- Guinault, D.; Ribes, D.; Delas, A.; Milongo, D.; Abravanel, F.; Puissant-Lubrano, B.; Izopet, J.; Kamar, N. Hepatitis E Virus-Induced Cryoglobulinemic Glomerulonephritis in a Nonimmunocompromised Person. Am. J. Kidney Dis. 2016, 67, 660–663. [Google Scholar] [CrossRef]
- Del Bello, A.; Guilbeau-Frugier, C.; Josse, A.G.; Rostaing, L.; Izopet, J.; Kamar, N. Successful treatment of hepatitis E virus-associated cryoglobulinemic membranoproliferative glomerulonephritis with ribavirin. Transpl. Infect. Dis. 2015, 17, 279–283. [Google Scholar] [CrossRef]
- Marion, O.; Abravanel, F.; Del Bello, A.; Esposito, L.; Lhomme, S.; Puissant-Lubrano, B.; Alric, L.; Faguer, S.; Izopet, J.; Kamar, N. Hepatitis E virus-associated cryoglobulinemia in solid-organ-transplant recipients. Liver Int. 2018, 38, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G. Renal involvement in hepatitis C infection: Cryoglobulinemic glomerulonephritis. Kidney Int. 1998, 54, 650–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Zhao, C.; Huang, W.; Harrison, T.J.; Zhang, H.; Geng, K.; Wang, Y. Detection and assessment of infectivity of hepatitis E virus in urine. J. Hepatol. 2016, 64, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Marion, O.; Capelli, N.; Lhomme, S.; Dubois, M.; Pucelle, M.; Abravanel, F.; Kamar, N.; Izopet, J. Hepatitis E virus genotype 3 and capsid protein in the blood and urine of immunocompromised patients. J. Infect. 2019, 78, 232–240. [Google Scholar] [CrossRef]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.C.; Saliou, J.M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F.; et al. Hepatitis E Virus Lifecycle and Identification of 3 Forms of the ORF2 Capsid Protein. Gastroenterology 2018, 154, 211–223. [Google Scholar] [CrossRef]
- Robinson, R.A.; Burgess, W.H.; Emerson, S.U.; Leibowitz, R.S.; Sosnovtseva, S.A.; Tsarev, S.; Purcell, R.H. Structural characterization of recombinant hepatitis E virus ORF2 proteins in baculovirus-infected insect cells. Protein Expr. Purif. 1998, 12, 75–84. [Google Scholar] [CrossRef]
- Williams, T.P.; Kasorndorkbua, C.; Halbur, P.G.; Haqshenas, G.; Guenette, D.K.; Toth, T.E.; Meng, X.J. Evidence of extrahepatic sites of replication of the hepatitis E virus in a swine model. J. Clin. Microbiol. 2001, 39, 3040–3046. [Google Scholar] [CrossRef] [Green Version]
- Soomro, M.H.; Shi, R.; She, R.; Yang, Y.; Hu, F.; Li, H. Antigen detection and apoptosis in Mongolian gerbil’s kidney experimentally intraperitoneally infected by swine hepatitis E virus. Virus Res. 2016, 213, 343–352. [Google Scholar] [CrossRef]
- Han, J.; Lei, Y.; Liu, L.; Liu, P.; Xia, J.; Zhang, Y.; Zeng, H.; Wang, L.; Wang, L.; Zhuang, H. SPF rabbits infected with rabbit hepatitis E virus isolate experimentally showing the chronicity of hepatitis. PLoS ONE 2014, 9, e99861. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xia, J.; Wang, L.; Wang, Y. Experimental infection of rabbits with genotype 3 hepatitis E virus produced both chronicity and kidney injury. Gut 2017, 66, 561–562. [Google Scholar] [CrossRef]
- Zamvar, V.; McClean, P.; Odeka, E.; Richards, M.; Davison, S. Hepatitis E virus infection with nonimmune hemolytic anemia. J. Pediatr. Gastroenterol. Nutr. 2005, 40, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Abid, S.; Khan, A.H. Severe hemolysis and renal failure in glucose-6-phosphate dehydrogenase deficient patients with hepatitis E. Am. J. Gastroenterol. 2002, 97, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.; Pramanik, S.; Biswas, B.; Mallick, D. Hepatitis E virus infection in a 7-year-old boy with glucose 6-phosphate dehydrogenase deficiency. J. Pediatr. Hematol. Oncol. 2009, 31, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Monga, A.; Makkar, R.P.; Arora, A.; Mukhopadhyay, S.; Gupta, A.K. Case report: Acute hepatitis E infection with coexistent glucose-6-phosphate dehydrogenase deficiency. Can. J. Infect. Dis. 2003, 14, 230–231. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.A.; Lal, A.; Idrees, M.; Hussain, A.; Jeet, C.; Malik, F.A.; Iqbal, Z.; Rehman, H. Hepatitis E virus-associated aplastic anaemia: The first case of its kind. J. Clin. Virol. 2012, 54, 96–97. [Google Scholar] [CrossRef]
- Woolson, K.L.; Forbes, A.; Vine, L.; Beynon, L.; McElhinney, L.; Panayi, V.; Hunter, J.G.; Madden, R.G.; Glasgow, T.; Kotecha, A.; et al. Extra-hepatic manifestations of autochthonous hepatitis E infection. Aliment. Pharmacol. Ther. 2014, 40, 1282–1291. [Google Scholar] [CrossRef]
- Jaroszewicz, J.; Flisiak, R.; Kalinowska, A.; Wierzbicka, I.; Prokopowicz, D. Acute hepatitis E complicated by acute pancreatitis: A case report and literature review. Pancreas 2005, 30, 382–384. [Google Scholar] [CrossRef]
- Makharia, G.K.; Garg, P.K.; Tandon, R.K. Acute pancreatitis associated with acute hepatitis E infection. Trop. Gastroenterol. 2003, 24, 200–201. [Google Scholar]
- Bhagat, S.; Wadhawan, M.; Sud, R.; Arora, A. Hepatitis viruses causing pancreatitis and hepatitis: A case series and review of literature. Pancreas 2008, 36, 424–427. [Google Scholar] [CrossRef]
- Nayak, H.K.; Kamble, N.L.; Raizada, N.; Garg, S.; Daga, M.K. Acute pancreatitis complicating acute hepatitis e virus infection: A case report and review. Case Rep. Hepatol. 2013, 2013, 531235. [Google Scholar] [CrossRef] [Green Version]
- Deniel, C.; Coton, T.; Brardjanian, S.; Guisset, M.; Nicand, E.; Simon, F. Acute pancreatitis: A rare complication of acute hepatitis E. J. Clin. Virol. 2011, 51, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Raj, M.; Kumar, K.; Ghoshal, U.C.; Saraswat, V.A.; Aggarwal, R.; Mohindra, S. Acute Hepatitis E-Associated Acute Pancreatitis: A Single Center Experience and Literature Review. Pancreas 2015, 44, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Marion, O.; Lhomme, S.; Nayrac, M.; Dubois, M.; Pucelle, M.; Requena, M.; Migueres, M.; Abravanel, F.; Peron, J.M.; Carrere, N.; et al. Hepatitis E virus replication in human intestinal cells. Gut 2019. [Google Scholar] [CrossRef] [PubMed]
- Capelli, N.; Marion, O.; Dubois, M.; Allart, S.; Bertrand-Michel, J.; Lhomme, S.; Abravanel, F.; Izopet, J.; Chapuy-Regaud, S. Vectorial Release of Hepatitis E Virus in Polarized Human Hepatocytes. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, S.U.; Purcell, R.H. Fields Virology 2013, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2242–2258. [Google Scholar]
- Yu, C.; Boon, D.; McDonald, S.L.; Myers, T.G.; Tomioka, K.; Nguyen, H.; Engle, R.E.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H. Pathogenesis of hepatitis E virus and hepatitis C virus in chimpanzees: Similarities and differences. J. Virol. 2010, 84, 11264–11278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanford, R.E.; Feng, Z.; Chavez, D.; Guerra, B.; Brasky, K.M.; Zhou, Y.; Yamane, D.; Perelson, A.S.; Walker, C.M.; Lemon, S.M. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 11223–11228. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.H.; Zhang, X.; Tran, C.; Skinner, B. Expression profiles of host immune response-related genes against HEV genotype 3 and genotype 1 infections in rhesus macaques. J. Viral Hepat. 2018. [Google Scholar] [CrossRef]
- Dong, C.; Zafrullah, M.; Mixson-Hayden, T.; Dai, X.; Liang, J.; Meng, J.; Kamili, S. Suppression of interferon-alpha signaling by hepatitis E virus. Hepatology 2012, 55, 1324–1332. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, L.; Wang, W.; Watashi, K.; Wang, Y.; Sprengers, D.; de Ruiter, P.E.; van der Laan, L.J.; Metselaar, H.J.; Kamar, N.; et al. Disparity of basal and therapeutically activated interferon signalling in constraining hepatitis E virus infection. J. Viral Hepat. 2016, 23, 294–304. [Google Scholar] [CrossRef]
- Nan, Y.; Ma, Z.; Wang, R.; Yu, Y.; Kannan, H.; Fredericksen, B.; Zhang, Y.J. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J. Virol. 2014, 88, 8696–8705. [Google Scholar] [CrossRef] [Green Version]
- Nan, Y.; Yu, Y.; Ma, Z.; Khattar, S.K.; Fredericksen, B.; Zhang, Y.J. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J. Virol. 2014, 88, 11924–11932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devhare, P.B.; Chatterjee, S.N.; Arankalle, V.A.; Lole, K.S. Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS ONE 2013, 8, e63793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Li, X.; Ambardekar, C.; Hu, Z.; Lhomme, S.; Feng, Z. Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog. 2017, 13, e1006417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, I.M.; Verhoye, L.; Cocquerel, L.; Abravanel, F.; Foquet, L.; Montpellier, C.; Debing, Y.; Farhoudi, A.; Wychowski, C.; Dubuisson, J.; et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut 2017, 66, 920–929. [Google Scholar] [CrossRef] [PubMed]
- van de Garde, M.D.; Pas, S.D.; van der Net, G.; de Man, R.A.; Osterhaus, A.D.; Haagmans, B.L.; Boonstra, A.; Vanwolleghem, T. Hepatitis E Virus (HEV) Genotype 3 Infection of Human Liver Chimeric Mice as a Model for Chronic HEV Infection. J. Virol. 2016, 90, 4394–4401. [Google Scholar] [CrossRef] [Green Version]
- Murata, K.; Kang, J.H.; Nagashima, S.; Matsui, T.; Karino, Y.; Yamamoto, Y.; Atarashi, T.; Oohara, M.; Uebayashi, M.; Sakata, H.; et al. IFN-lambda3 as a host immune response in acute hepatitis E virus infection. Cytokine 2019, 125, 154816. [Google Scholar] [CrossRef]
- Srivastava, R.; Aggarwal, R.; Jameel, S.; Puri, P.; Gupta, V.K.; Ramesh, V.S.; Bhatia, S.; Naik, S. Cellular immune responses in acute hepatitis E virus infection to the viral open reading frame 2 protein. Viral Immunol. 2007, 20, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.; Aggarwal, R.; Bhagat, M.R.; Chowdhury, A.; Naik, S. Alterations in natural killer cells and natural killer T cells during acute viral hepatitis E. J. Viral Hepat. 2008, 15, 910–916. [Google Scholar] [CrossRef]
- Prabhu, S.B.; Gupta, P.; Durgapal, H.; Rath, S.; Gupta, S.D.; Acharya, S.K.; Panda, S.K. Study of cellular immune response against Hepatitis E virus (HEV). J. Viral Hepat. 2011, 18, 587–594. [Google Scholar] [CrossRef]
- Liu, T.; Xiao, P.; Li, R.; She, R.; Tian, J.; Wang, J.; Mao, J.; Yin, J.; Shi, R. Increased Mast Cell Activation in Mongolian Gerbils Infected by Hepatitis E Virus. Front. Microbiol. 2018, 9, 2226. [Google Scholar] [CrossRef]
- Lhomme, S.; Legrand-Abravanel, F.; Kamar, N.; Izopet, J. Screening, diagnosis and risks associated with Hepatitis E virus infection. Expert Rev. Anti Infect. Ther. 2019, 17, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, M.S.; Kamili, S.; Dar, M.Y.; Moecklii, R.; Jameel, S. Hepatitis E and long-term antibody status. Lancet 1993, 341, 1355. [Google Scholar] [PubMed]
- Tang, X.; Yang, C.; Gu, Y.; Song, C.; Zhang, X.; Wang, Y.; Zhang, J.; Hew, C.L.; Li, S.; Xia, N.; et al. Structural basis for the neutralization and genotype specificity of hepatitis E virus. Proc. Natl. Acad. Sci. USA 2011, 108, 10266–10271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.C.; Zhang, J.; Zhang, X.F.; Zhou, C.; Wang, Z.Z.; Huang, S.J.; Wang, H.; Yang, C.L.; Jiang, H.M.; Cai, J.P.; et al. Efficacy and safety of a recombinant hepatitis E vaccine in healthy adults: A large-scale, randomised, double-blind placebo-controlled, phase 3 trial. Lancet 2010, 376, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shih, J.W.; Xia, N.S. Long-term efficacy of a hepatitis E vaccine. N. Engl. J. Med. 2015, 372, 2265–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, S.U.; Purcell, R.H. Recombinant vaccines for hepatitis E. Trends Mol. Med. 2001, 7, 462–466. [Google Scholar] [CrossRef]
- Bryan, J.P.; Tsarev, S.A.; Iqbal, M.; Ticehurst, J.; Emerson, S.; Ahmed, A.; Duncan, J.; Rafiqui, A.R.; Malik, I.A.; Purcell, R.H.; et al. Epidemic hepatitis E in Pakistan: Patterns of serologic response and evidence that antibody to hepatitis E virus protects against disease. J. Infect. Dis. 1994, 170, 517–521. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.F.; Zhou, C.; Wang, Z.Z.; Huang, S.J.; Yao, X.; Liang, Z.L.; Wu, T.; Li, J.X.; Yan, Q.; et al. Protection against hepatitis E virus infection by naturally acquired and vaccine-induced immunity. Clin. Microbiol. Infect. 2014, 20, O397–O405. [Google Scholar] [CrossRef] [Green Version]
- Shata, M.T.; Daef, E.A.; Zaki, M.E.; Abdelwahab, S.F.; Marzuuk, N.M.; Sobhy, M.; Rafaat, M.; Abdelbaki, L.; Nafeh, M.A.; Hashem, M.; et al. Protective role of humoral immune responses during an outbreak of hepatitis E in Egypt. Trans. R. Soc. Trop. Med. Hyg. 2012, 106, 613–618. [Google Scholar] [CrossRef]
- Shrestha, M.P.; Scott, R.M.; Joshi, D.M.; Mammen, M.P., Jr.; Thapa, G.B.; Thapa, N.; Myint, K.S.; Fourneau, M.; Kuschner, R.A.; Shrestha, S.K.; et al. Safety and efficacy of a recombinant hepatitis E vaccine. N. Engl. J. Med. 2007, 356, 895–903. [Google Scholar] [CrossRef]
- Abravanel, F.; Lhomme, S.; Chapuy-Regaud, S.; Mansuy, J.M.; Muscari, F.; Sallusto, F.; Rostaing, L.; Kamar, N.; Izopet, J. Hepatitis E virus reinfections in solid-organ-transplant recipients can evolve into chronic infections. J. Infect. Dis. 2014, 209, 1900–1906. [Google Scholar] [CrossRef]
- Chapuy-Regaud, S.; Dubois, M.; Plisson-Chastang, C.; Bonnefois, T.; Lhomme, S.; Bertrand-Michel, J.; You, B.; Simoneau, S.; Gleizes, P.E.; Flan, B.; et al. Characterization of the lipid envelope of exosome encapsulated HEV particles protected from the immune response. Biochimie 2017, 141, 70–79. [Google Scholar] [CrossRef]
- Husain, M.M.; Aggarwal, R.; Kumar, D.; Jameel, S.; Naik, S. Effector T cells immune reactivity among patients with acute hepatitis E. J. Viral Hepat. 2011, 18, e603–e608. [Google Scholar] [CrossRef]
- TrehanPati, N.; Sukriti, S.; Geffers, R.; Hissar, S.; Riese, P.; Toepfer, T.; Guzman, C.A.; Sarin, S.K. Gene expression profiles of T cells from hepatitis E virus infected patients in acute and resolving phase. J. Clin. Immunol. 2011, 31, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, A.S.; Das, R.; Rathod, S.B.; Arankalle, V.A. Cytokine profiles, CTL response and T cell frequencies in the peripheral blood of acute patients and individuals recovered from hepatitis E infection. PLoS ONE 2012, 7, e31822. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Halliday, J.S.; Swadling, L.; Madden, R.G.; Bendall, R.; Hunter, J.G.; Maggs, J.; Simmonds, P.; Smith, D.B.; Vine, L.; et al. Characterization of the Specificity, Functionality, and Durability of Host T-Cell Responses Against the Full-Length Hepatitis E Virus. Hepatology 2016, 64, 1934–1950. [Google Scholar] [CrossRef]
- Al-Ayoubi, J.; Behrendt, P.; Bremer, B.; Suneetha, P.V.; Gisa, A.; Rinker, F.; Manns, M.P.; Cornberg, M.; Wedemeyer, H.; Kraft, A.R.M. Hepatitis E virus ORF 1 induces proliferative and functional T-cell responses in patients with ongoing and resolved hepatitis E. Liver Int. 2018, 38, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Gisa, A.; Suneetha, P.V.; Behrendt, P.; Pischke, S.; Bremer, B.; Falk, C.S.; Manns, M.P.; Cornberg, M.; Wedemeyer, H.; Kraft, A.R. Cross-genotype-specific T-cell responses in acute hepatitis E virus (HEV) infection. J. Viral Hepat. 2016, 23, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Aggarwal, R.; Naik, S.R.; Saraswat, V.; Ghoshal, U.C.; Naik, S. Hepatitis E virus is responsible for decompensation of chronic liver disease in an endemic region. Indian J. Gastroenterol. 2004, 23, 59–62. [Google Scholar] [PubMed]
- Saravanabalaji, S.; Tripathy, A.S.; Dhoot, R.R.; Chadha, M.S.; Kakrani, A.L.; Arankalle, V.A. Viral load, antibody titers and recombinant open reading frame 2 protein-induced TH1/TH2 cytokines and cellular immune responses in self-limiting and fulminant hepatitis e. Intervirology 2009, 52, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Aggarwal, R.; Sachdeva, S.; Alam, M.I.; Jameel, S.; Naik, S. Adaptive immune responses during acute uncomplicated and fulminant hepatitis E. J. Gastroenterol. Hepatol. 2011, 26, 306–311. [Google Scholar] [CrossRef] [PubMed]
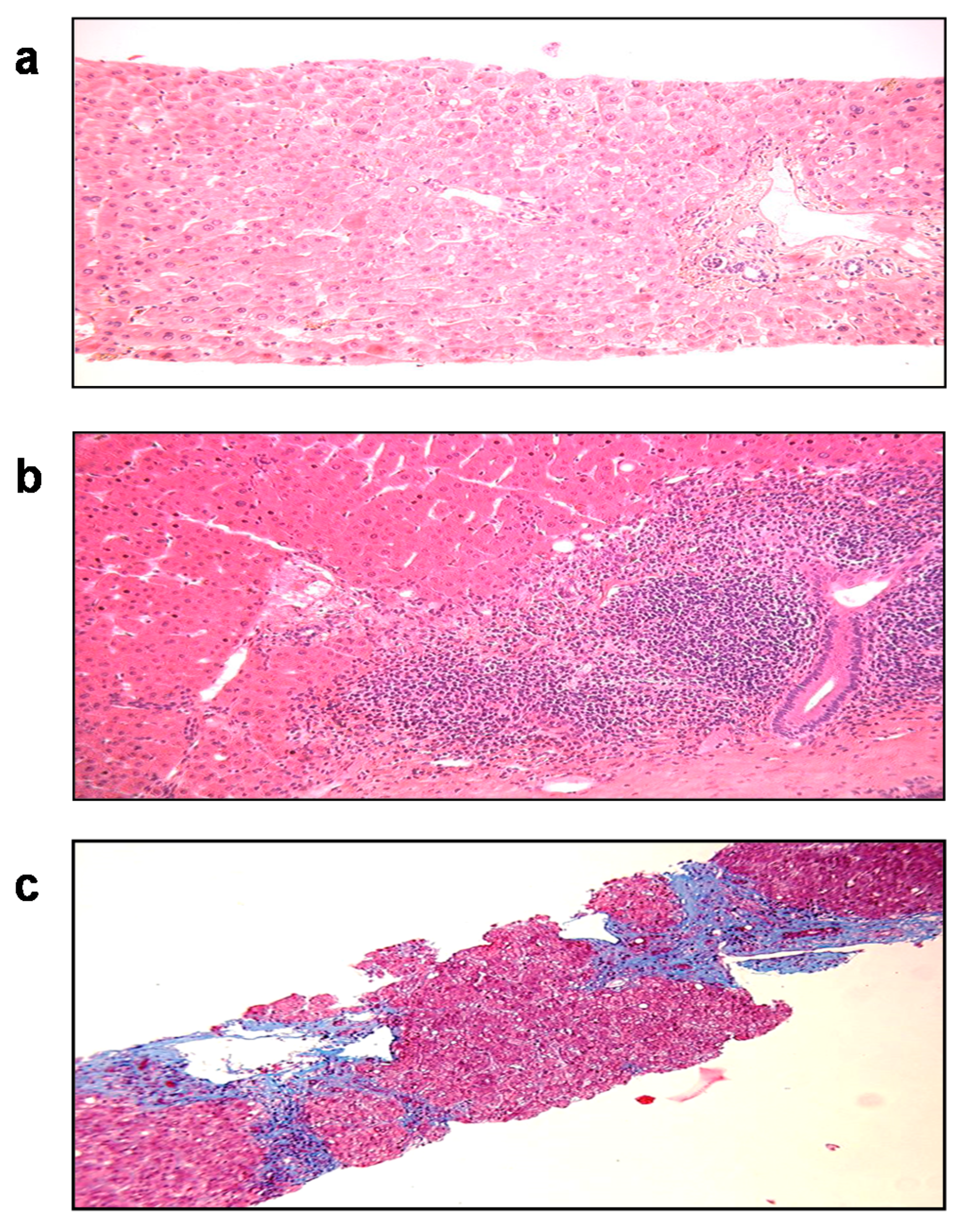
- Agrawal, V.; Goel, A.; Rawat, A.; Naik, S.; Aggarwal, R. Histological and immunohistochemical features in fatal acute fulminant hepatitis E. Indian J. Pathol. Microbiol. 2012, 55, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Chang, D.Y.; Lee, H.W.; Lee, H.; Kim, J.H.; Sung, P.S.; Kim, K.H.; Hong, S.H.; Kang, W.; Lee, J.; et al. Innate-like Cytotoxic Function of Bystander-Activated CD8(+) T Cells Is Associated with Liver Injury in Acute Hepatitis, A. Immunity 2018, 48, 161–173.e165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeblaoui, A.; Haim-Boukobza, S.; Marchadier, E.; Mokhtari, C.; Roque-Afonso, A.M. Genotype 4 hepatitis e virus in france: An autochthonous infection with a more severe presentation. Clin. Infect. Dis. 2013, 57, e122–e126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subissi, L.; Peeters, M.; Lamoral, S.; Klamer, S.; Suin, V.; Van Gucht, S. Subtype-specific differences in the risk of hospitalisation among patients infected with hepatitis E virus genotype 3 in Belgium, 2010–2018. Epidemiol. Infect. 2019, 147, e224. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Walimbe, A.M.; Arankalle, V.A. Hepatitis E virus from India exhibits significant amino acid mutations in fulminant hepatic failure patients. Virus Genes 2013, 46, 47–53. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P. Hepatitis E virus and fulminant hepatitis-A virus or host-specific pathology? Liver Int. 2015, 35, 1334–1340. [Google Scholar] [CrossRef] [Green Version]
- Borkakoti, J.; Ahmed, G.; Kar, P. Report of a novel C1483W mutation in the hepatitis E virus polymerase in patients with acute liver failure. Infect. Genet. Evol. 2016, 44, 51–54. [Google Scholar] [CrossRef]
- Borkakoti, J.; Ahmed, G.; Rai, A.; Kar, P. Report of novel H105R, D29N, V27A mutations in the methyltransferase region of the HEV genome in patients with acute liver failure. J. Clin. Virol. 2017, 91, 1–4. [Google Scholar] [CrossRef]
- Anty, R.; Ollier, L.; Peron, J.M.; Nicand, E.; Cannavo, I.; Bongain, A.; Giordanengo, V.; Tran, A. First case report of an acute genotype 3 hepatitis E infected pregnant woman living in South-Eastern France. J. Clin. Virol. 2012, 54, 76–78. [Google Scholar] [CrossRef]
- Tabatabai, J.; Wenzel, J.J.; Soboletzki, M.; Flux, C.; Navid, M.H.; Schnitzler, P. First case report of an acute hepatitis E subgenotype 3c infection during pregnancy in Germany. J. Clin. Virol. 2014, 61, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Bouthry, E.; Benachi, A.; Vivanti, A.J.; Letamendia, E.; Vauloup-Fellous, C.; Roque-Afonso, A.M. Autochthonous Hepatitis E during Pregnancy, France. Emerg. Infect. Dis. 2018, 24, 1586–1587. [Google Scholar] [CrossRef] [PubMed]
- Gouilly, J.; Chen, Q.; Siewiera, J.; Cartron, G.; Levy, C.; Dubois, M.; Al-Daccak, R.; Izopet, J.; Jabrane-Ferrat, N.; El Costa, H. Genotype specific pathogenicity of hepatitis E virus at the human maternal-fetal interface. Nat. Commun. 2018, 9, 4748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Devi, S.G.; Kar, P.; Agarwal, S.; Husain, S.A.; Gupta, R.K.; Sharma, S. Association of cytokines in hepatitis E with pregnancy outcome. Cytokine 2014, 65, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S. The Th1/Th2 paradigm. Immunol. Today 1997, 18, 263–266. [Google Scholar] [CrossRef]
- Pal, R.; Aggarwal, R.; Naik, S.R.; Das, V.; Das, S.; Naik, S. Immunological alterations in pregnant women with acute hepatitis E. J. Gastroenterol. Hepatol. 2005, 20, 1094–1101. [Google Scholar] [CrossRef]
- Sehgal, R.; Patra, S.; David, P.; Vyas, A.; Khanam, A.; Hissar, S.; Gupta, E.; Kumar, G.; Kottilil, S.; Maiwall, R.; et al. Impaired monocyte-macrophage functions and defective Toll-like receptor signaling in hepatitis E virus-infected pregnant women with acute liver failure. Hepatology 2015, 62, 1683–1696. [Google Scholar] [CrossRef]
- Kmush, B.L.; Labrique, A.; Li, W.; Klein, S.L.; Schulze, K.; Shaikh, S.; Ali, H.; Engle, R.E.; Wu, L.; Purcell, R.H.; et al. The Association of Cytokines and Micronutrients with Hepatitis E Virus Infection During Pregnancy and the Postpartum Period in Rural Bangladesh. Am. J. Trop. Med. Hyg. 2016, 94, 203–211. [Google Scholar] [CrossRef]
- Stoszek, S.K.; Abdel-Hamid, M.; Saleh, D.A.; El Kafrawy, S.; Narooz, S.; Hawash, Y.; Shebl, F.M.; El Daly, M.; Said, A.; Kassem, E.; et al. High prevalence of hepatitis E antibodies in pregnant Egyptian women. Trans R. Soc. Trop. Med. Hyg. 2006, 100, 95–101. [Google Scholar] [CrossRef]
- Jilani, N.; Das, B.C.; Husain, S.A.; Baweja, U.K.; Chattopadhya, D.; Gupta, R.K.; Sardana, S.; Kar, P. Hepatitis E virus infection and fulminant hepatic failure during pregnancy. J. Gastroenterol. Hepatol. 2007, 22, 676–682. [Google Scholar] [CrossRef]
- Bi, Y.; Yang, C.; Yu, W.; Zhao, X.; Zhao, C.; He, Z.; Jing, S.; Wang, H.; Huang, F. Pregnancy serum facilitates hepatitis E virus replication in vitro. J. Gen. Virol. 2015, 96, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Jilani, N.; Husain, S.A.; Pasha, S.T.; Anand, R.; Rai, A.; Das, B.C. Does hepatitis E viral load and genotypes influence the final outcome of acute liver failure during pregnancy? Am. J. Gastroenterol. 2008, 103, 2495–2501. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.D.; Das, B.C.; Kumar, A.; Gondal, R.; Kumar, D.; Kar, P. High viral load and deregulation of the progesterone receptor signaling pathway: Association with hepatitis E-related poor pregnancy outcome. J. Hepatol. 2011, 54, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Borkakoti, J.; Hazam, R.K.; Mohammad, A.; Kumar, A.; Kar, P. Does high viral load of hepatitis E virus influence the severity and prognosis of acute liver failure during pregnancy? J. Med. Virol. 2013, 85, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Legrand-Abravanel, F.; Kamar, N.; Sandres-Saune, K.; Lhomme, S.; Mansuy, J.M.; Muscari, F.; Sallusto, F.; Rostaing, L.; Izopet, J. Hepatitis E virus infection without reactivation in solid-organ transplant recipients, France. Emerg. Infect. Dis. 2011, 17, 30–37. [Google Scholar] [CrossRef]
- Pischke, S.; Stiefel, P.; Franz, B.; Bremer, B.; Suneetha, P.V.; Heim, A.; Ganzenmueller, T.; Schlue, J.; Horn-Wichmann, R.; Raupach, R.; et al. Chronic hepatitis E in heart transplant recipients. Am. J. Transplant. 2012, 12, 3128–3133. [Google Scholar] [CrossRef]
- Pischke, S.; Hardtke, S.; Bode, U.; Birkner, S.; Chatzikyrkou, C.; Kauffmann, W.; Bara, C.L.; Gottlieb, J.; Wenzel, J.; Manns, M.P.; et al. Ribavirin treatment of acute and chronic hepatitis E: A single-centre experience. Liver Int. 2013, 33, 722–726. [Google Scholar] [CrossRef]
- Moal, V.; Textoris, J.; Ben Amara, A.; Mehraj, V.; Berland, Y.; Colson, P.; Mege, J.L. Chronic hepatitis E virus infection is specifically associated with an interferon-related transcriptional program. J. Infect. Dis. 2013, 207, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Rostaing, L.; Kamar, N.; Izopet, J. Hepatitis E virus quasispecies and the outcome of acute hepatitis E in solid-organ transplant patients. J. Virol. 2012, 86, 10006–10014. [Google Scholar] [CrossRef] [Green Version]
- Suneetha, P.V.; Pischke, S.; Schlaphoff, V.; Grabowski, J.; Fytili, P.; Gronert, A.; Bremer, B.; Markova, A.; Jaroszewicz, J.; Bara, C.; et al. Hepatitis E virus (HEV)-specific T-cell responses are associated with control of HEV infection. Hepatology 2012, 55, 695–708. [Google Scholar] [CrossRef]
- Kamar, N.; Legrand-Abravanel, F.; Dalton, H.R.; Izopet, J. Hepatitis E virus-specific T-cell response after transplantation. Hepatology 2012, 55, 1643. [Google Scholar] [CrossRef] [PubMed]
- Abravanel, F.; Barrague, H.; Dorr, G.; Saune, K.; Peron, J.M.; Alric, L.; Kamar, N.; Izopet, J.; Champagne, E. Conventional and innate lymphocytes response at the acute phase of HEV infection in transplanted patients. J. Infect. 2016, 72, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Cao, Q.M.; Subramaniam, S.; Yugo, D.M.; Heffron, C.L.; Rogers, A.J.; Kenney, S.P.; Tian, D.; Matzinger, S.R.; Overend, C.; et al. Pig model mimicking chronic hepatitis E virus infection in immunocompromised patients to assess immune correlates during chronicity. Proc. Natl. Acad. Sci. USA 2017, 114, 6914–6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlister, V.C.; Haddad, E.; Renouf, E.; Malthaner, R.A.; Kjaer, M.S.; Gluud, L.L. Cyclosporin versus tacrolimus as primary immunosuppressant after liver transplantation: A meta-analysis. Am. J. Transplant. 2006, 6, 1578–1585. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, X.; Debing, Y.; Chen, K.; Van Der Laan, L.J.; Neyts, J.; Janssen, H.L.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Calcineurin inhibitors stimulate and mycophenolic acid inhibits replication of hepatitis E virus. Gastroenterology 2014, 146, 1775–1783. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Metselaar, H.J.; Janssen, H.L.; Peppelenbosch, M.P.; Pan, Q. Rapamycin and everolimus facilitate hepatitis E virus replication: Revealing a basal defense mechanism of PI3K-PKB-mTOR pathway. J. Hepatol. 2014, 61, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Lhomme, S.; Abravanel, F.; Cointault, O.; Esposito, L.; Cardeau-Desangles, I.; Del Bello, A.; Dorr, G.; Lavayssiere, L.; Nogier, M.B.; et al. An Early Viral Response Predicts the Virological Response to Ribavirin in Hepatitis E Virus Organ Transplant Patients. Transplantation 2015, 99, 2124–2131. [Google Scholar] [CrossRef]
- Lhomme, S.; Garrouste, C.; Kamar, N.; Saune, K.; Abravanel, F.; Mansuy, J.M.; Dubois, M.; Rostaing, L.; Izopet, J. Influence of polyproline region and macro domain genetic heterogeneity on HEV persistence in immunocompromised patients. J. Infect. Dis. 2014, 209, 300–303. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, Y.; Oshiro, Y.; Tanaka, T.; Yoshizumi, T.; Okajima, H.; Ishiyama, K.; Nakanishi, C.; Hidaka, M.; Wada, H.; Hibi, T.; et al. A Nationwide Survey of Hepatitis E Virus Infection and Chronic Hepatitis E in Liver Transplant Recipients in Japan. EBioMedicine 2015, 2, 1607–1612. [Google Scholar] [CrossRef] [Green Version]
- Pischke, S.; Wedemeyer, H. Hepatitis E In Transplant Recipients: Why Is This Not A Problem In Japan? EBioMedicine 2015, 2, 1564–1565. [Google Scholar] [CrossRef] [Green Version]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Mansuy, J.M.; Rostaing, L.; Kamar, N.; Izopet, J. Characterization of the polyproline region of the hepatitis E virus in immunocompromised patients. J. Virol. 2014, 88, 12017–12025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A naturally occurring human/hepatitis E recombinant virus predominates in serum but not in faeces of a chronic hepatitis E patient and has a growth advantage in cell culture. J. Gen. Virol. 2012, 93, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenroder, J.; Nickel, P.; Hofmann, J. An ORF1-rearranged hepatitis E virus derived from a chronically infected patient efficiently replicates in cell culture. J. Viral Hepat. 2014, 21, 447–456. [Google Scholar] [CrossRef]
- Peron, J.M.; Dalton, H.; Izopet, J.; Kamar, N. Acute autochthonous hepatitis E in western patients with underlying chronic liver disease: A role for ribavirin? J. Hepatol. 2011, 54, 1323–1324. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Abravanel, F.; Selves, J.; Garrouste, C.; Esposito, L.; Lavayssiere, L.; Cointault, O.; Ribes, D.; Cardeau, I.; Nogier, M.B.; et al. Influence of immunosuppressive therapy on the natural history of genotype 3 hepatitis-E virus infection after organ transplantation. Transplantation 2010, 89, 353–360. [Google Scholar] [CrossRef]
- Kamar, N.; Rostaing, L.; Abravanel, F.; Garrouste, C.; Esposito, L.; Cardeau-Desangles, I.; Mansuy, J.M.; Selves, J.; Peron, J.M.; Otal, P.; et al. Pegylated interferon-alpha for treating chronic hepatitis E virus infection after liver transplantation. Clin. Infect. Dis. 2010, 50, e30–e33. [Google Scholar] [CrossRef] [Green Version]
- Haagsma, E.B.; Riezebos-Brilman, A.; van den Berg, A.P.; Porte, R.J.; Niesters, H.G. Treatment of chronic hepatitis E in liver transplant recipients with pegylated interferon alpha-2b. Liver Transplant. 2010, 16, 474–477. [Google Scholar] [CrossRef]
- Kamar, N.; Abravanel, F.; Garrouste, C.; Cardeau-Desangles, I.; Mansuy, J.M.; Weclawiak, H.; Izopet, J.; Rostaing, L. Three-month pegylated interferon-alpha-2a therapy for chronic hepatitis E virus infection in a haemodialysis patient. Nephrol. Dial. Transplant. 2010, 25, 2792–2795. [Google Scholar] [CrossRef] [Green Version]
- Mallet, V.; Nicand, E.; Sultanik, P.; Chakvetadze, C.; Tesse, S.; Thervet, E.; Mouthon, L.; Sogni, P.; Pol, S. Brief communication: Case reports of ribavirin treatment for chronic hepatitis E. Ann. Intern. Med. 2010, 153, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Rostaing, L.; Abravanel, F.; Garrouste, C.; Lhomme, S.; Esposito, L.; Basse, G.; Cointault, O.; Ribes, D.; Nogier, M.B.; et al. Ribavirin therapy inhibits viral replication on patients with chronic hepatitis e virus infection. Gastroenterology 2010, 139, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Tripon, S.; Bismuth, M.; Hillaire, S.; Dumortier, J.; Radenne, S.; Coilly, A.; Garrigue, V.; D’Alteroche, L.; et al. Ribavirin for chronic hepatitis E virus infection in transplant recipients. N. Engl. J. Med. 2014, 370, 1111–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, O.; Lhomme, S.; Del Bello, A.; Abravanel, F.; Esposito, L.; Hebral, A.L.; Lavayssiere, L.; Cointault, O.; Ribes, D.; Izopet, J.; et al. Monitoring hepatitis E virus fecal shedding to optimize ribavirin treatment duration in chronically infected transplant patients. J. Hepatol. 2019, 70, 206–209. [Google Scholar] [CrossRef] [Green Version]
- Debing, Y.; Emerson, S.U.; Wang, Y.; Pan, Q.; Balzarini, J.; Dallmeier, K.; Neyts, J. Ribavirin inhibits in vitro hepatitis E virus replication through depletion of cellular GTP pools and is moderately synergistic with alpha interferon. Antimicrob. Agents Chemother. 2014, 58, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todt, D.; Gisa, A.; Radonic, A.; Nitsche, A.; Behrendt, P.; Suneetha, P.V.; Pischke, S.; Bremer, B.; Brown, R.J.; Manns, M.P.; et al. In vivo evidence for ribavirin-induced mutagenesis of the hepatitis E virus genome. Gut 2016, 65, 1733–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotty, S.; Cameron, C.E.; Andino, R. RNA virus error catastrophe: Direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 2001, 98, 6895–6900. [Google Scholar] [CrossRef] [Green Version]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147, 1008–1011. [Google Scholar] [CrossRef]
- Lhomme, S.; Kamar, N.; Nicot, F.; Ducos, J.; Bismuth, M.; Garrigue, V.; Petitjean-Lecherbonnier, J.; Ollivier, I.; Alessandri-Gradt, E.; Goria, O.; et al. Mutation in the Hepatitis E Virus Polymerase and Outcome of Ribavirin Therapy. Antimicrob. Agents Chemother. 2015, 60, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Debing, Y.; Ramiere, C.; Dallmeier, K.; Piorkowski, G.; Trabaud, M.A.; Lebosse, F.; Scholtes, C.; Roche, M.; Legras-Lachuer, C.; de Lamballerie, X.; et al. Hepatitis E virus mutations associated with ribavirin treatment failure result in altered viral fitness and ribavirin sensitivity. J. Hepatol. 2016, 65, 499–508. [Google Scholar] [CrossRef]
- Kamar, N.; Abravanel, F.; Behrendt, P.; Hofmann, J.; Pageaux, G.P.; Barbet, C.; Moal, V.; Couzi, L.; Horvatits, T.; De Man, R.A.; et al. Ribavirin for Hepatitis E Virus Infection After Organ Transplantation: A Large European Retrospective Multicenter Study. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Dao Thi, V.L.; Debing, Y.; Wu, X.; Rice, C.M.; Neyts, J.; Moradpour, D.; Gouttenoire, J. Sofosbuvir Inhibits Hepatitis E Virus Replication In Vitro and Results in an Additive Effect When Combined With Ribavirin. Gastroenterology 2016, 150, 82–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wezel, E.M.; de Bruijne, J.; Damman, K.; Bijmolen, M.; van den Berg, A.P.; Verschuuren, E.A.M.; Ruigrok, G.A.; Riezebos-Brilman, A.; Knoester, M. Sofosbuvir Add-on to Ribavirin Treatment for Chronic Hepatitis E Virus Infection in Solid Organ Transplant Recipients Does Not Result in Sustained Virological Response. Open Forum Infect. Dis. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Drinane, M.; Jing Wang, X.; Watt, K. Sofosbuvir and Ribavirin Eradication of Refractory Hepatitis E in an Immunosuppressed Kidney Transplant Recipient. Hepatology 2019, 69, 2297–2299. [Google Scholar] [CrossRef] [PubMed]
- van der Valk, M.; Zaaijer, H.L.; Kater, A.P.; Schinkel, J. Sofosbuvir shows antiviral activity in a patient with chronic hepatitis E virus infection. J. Hepatol. 2017, 66, 242–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todesco, E.; Demeret, S.; Calin, R.; Roque-Afonso, A.M.; Thibault, V.; Mallet, V.; Akhavan, S.; Jaspard, M.; Peytavin, G.; Poynard, T.; et al. Chronic hepatitis E in HIV/HBV coinfected patient: Lack of power of sofosbuvir-ribavirin. AIDS 2017, 31, 1346–1348. [Google Scholar] [CrossRef] [PubMed]
- Todesco, E.; Mazzola, A.; Akhavan, S.; Abravanel, F.; Poynard, T.; Roque-Afonso, A.M.; Peytavin, G.; Marcelin, A.G.; Calmus, Y.; Lecuyer, L.; et al. Chronic hepatitis E in a heart transplant patient: Sofosbuvir and ribavirin regimen not fully effective. Antivir. Ther. 2018. [Google Scholar] [CrossRef]
- Donnelly, M.C.; Imlach, S.N.; Abravanel, F.; Ramalingam, S.; Johannessen, I.; Petrik, J.; Fraser, A.R.; Campbell, J.D.; Bramley, P.; Dalton, H.R.; et al. Sofosbuvir and Daclatasvir Anti-Viral Therapy Fails to Clear HEV Viremia and Restore Reactive T Cells in a HEV/HCV Co-Infected Liver Transplant Recipient. Gastroenterology 2017, 152, 300–301. [Google Scholar] [CrossRef] [Green Version]
- De Martin, E.; Antonini, T.M.; Coilly, A.; Pittau, G.; Vibert, E.; Duclos-Vallee, J.C.; Samuel, D.; Roque-Afonso, A.M. HCV and HEV recurrence after liver transplantation: One antiviral therapy for two viruses. Transpl. Int. 2017, 30, 318–319. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Pan, Q. No Clear Evidence for an Effect of Sofosbuvir against Hepatitis E Virus in Organ Transplant Patients. Hepatology 2019, 69, 1846–1847. [Google Scholar] [CrossRef]
- Cornberg, M.; Pischke, S.; Müller, T.; Behrendt, P.; Piecha, F.; Benckert, J.; Smith, A.; Koch, A.; Lohse, A.; Hardtke, S.; et al. LBO-04; Efficacy and safety of sofosbuvir monotherapy in patients with chronic hepatitis E-The HepNet SofE pilot study. J. Hepatol. 2019, 70, e129–e130. [Google Scholar] [CrossRef]
- Kaushik, N.; Subramani, C.; Anang, S.; Muthumohan, R.; Shalimar, B.N.; Ranjith-Kumar, C.T.; Surjit, M. Zinc Salts Block Hepatitis E Virus Replication by Inhibiting the Activity of Viral RNA-Dependent RNA Polymerase. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, O.; Abravanel, F.; Izopet, J.; Kamar, N. Failure to respond to ribavirin despite elevated intra-erythrocyte zinc level in transplant-patients with chronic hepatitis E virus infection. Transpl. Infect. Dis. 2019, 21, e13050. [Google Scholar] [CrossRef]
- Todt, D.; Moeller, N.; Praditya, D.; Kinast, V.; Friesland, M.; Engelmann, M.; Verhoye, L.; Sayed, I.M.; Behrendt, P.; Dao Thi, V.L.; et al. The natural compound silvestrol inhibits hepatitis E virus (HEV) replication in vitro and in vivo. Antivir. Res. 2018, 157, 151–158. [Google Scholar] [CrossRef]
- Netzler, N.E.; Enosi Tuipulotu, D.; Vasudevan, S.G.; Mackenzie, J.M.; White, P.A. Antiviral Candidates for Treating Hepatitis E Virus Infection. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiyama, T.; Kobayashi, T.; Jirintai, S.; Kii, I.; Nagashima, S.; Prathiwi Primadharsini, P.; Nishizawa, T.; Okamoto, H. Screening of novel drugs for inhibiting hepatitis E virus replication. J. Virol. Methods 2019, 270, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Soon, C.F.; Behrendt, P.; Todt, D.; Manns, M.P.; Wedemeyer, H.; Sallberg Chen, M.; Cornberg, M. Defining virus-specific CD8+ TCR repertoires for therapeutic regeneration of T cells against chronic hepatitis E. J. Hepatol. 2019, 71, 673–684. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lhomme, S.; Marion, O.; Abravanel, F.; Izopet, J.; Kamar, N. Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections. J. Clin. Med. 2020, 9, 331. https://doi.org/10.3390/jcm9020331
Lhomme S, Marion O, Abravanel F, Izopet J, Kamar N. Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections. Journal of Clinical Medicine. 2020; 9(2):331. https://doi.org/10.3390/jcm9020331
Chicago/Turabian StyleLhomme, Sébastien, Olivier Marion, Florence Abravanel, Jacques Izopet, and Nassim Kamar. 2020. "Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections" Journal of Clinical Medicine 9, no. 2: 331. https://doi.org/10.3390/jcm9020331
APA StyleLhomme, S., Marion, O., Abravanel, F., Izopet, J., & Kamar, N. (2020). Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections. Journal of Clinical Medicine, 9(2), 331. https://doi.org/10.3390/jcm9020331