1. Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inheritable cardiac disorder characterized by ventricular tachyarrhythmias, progressive loss of cardiomyocytes with fibrofatty replacement and sudden cardiac death (SCD) [
1]. The prevalence of ARVC is about 1:2000–1:5000 and more common in males (2:1–3:1) [
1]. ARVC usually manifests at ages between 12 to 60 years and is a leading cause of SCD due to ventricular tachyarrhythmias in young athletes [
2]. In the most typical form of ARVC, the right ventricle is primarily affected. As the disease progresses, the left ventricle may also be affected [
3].
Most cases of ARVC are attributed to mutations in desmosomal genes, including plakoglobin (JUP), plakophilin-2 (PKP2), desmoplakin (DSP), desmoglein-2 (DSG2) and desmocollin-2 (DSC2) [
4]. However, the exact pathogenic mechanisms by which desmosomal mutations cause life-threatening arrhythmias remain unclear. To date, two hypotheses have been discussed to explain the arrhythmogenic mechanisms in ARVC: (1) structural abnormality-induced conduction-defects; (2) ion channel dysfunction-induced electrophysiological abnormalities. The former is caused mainly by an intercellular fibrofatty deposit, which interrupts the intercellular propagation of electrical pulses. The latter can be caused by ion channel remodeling, leading to a gain-of-function or a loss-of-function of ion channels and subsequent abnormal electrical activity in cardiomyocytes. For example, the peak sodium current (I
Na) has been shown to be reduced in ARVC-cardiomyocytes [
5]. The reduced I
Na may slow the propagation of excitation, leading to a functional conduction defect. El-Battrawy et al. showed that human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) from a patient with ARVC (ARVC-hiPSC-CMs) carrying a mutation in the DSG2 gene displayed multiple ion channel dysfunctions and abnormal electrical activities [
6], pointing to the contribution of ion channel dysfunctions to arrhythmogenesis, independent of structural abnormalities. However, the exact mechanisms by which the conduction defect or ion channel dysfunctions cause tachyarrthmias in patients with ARVC have not been clearly clarified.
Some calcium activated K
+ channels, including small conductance (SK1, SK2, SK3) and intermediate conductance (SK4, also called KCa3.1) calcium-activated K
+ channels, have been linked to arrhythmogenesis in atrial fibrillation (AF) and catecholaminergic polymorphic ventricular tachycardia (CPVT) [
7,
8]. The SK1–3 channel currents can change AP-duration (APD) [
7], whereas SK4 channel currents can influence the pacemaker activity of cells [
9]. Both may enhance the occurrence of arrhythmias in cardiomyocytes. Whether those ion channels, especially the SK4 channels, are also involved in arrhythmogenesis in ARVC, is not known.
It is well-known that besides intracellular Ca
2+ ions, nucleoside diphosphate kinase B (NDPK-B) is an important intracellular regulator of SK4 channels [
10,
11]. NDPK-B can directly phosphorylate SK4 channels at histidine 358 (His358) and enhance channel activity [
12]. In general, nucleoside diphosphate kinases (NDPKs) are ubiquitously expressed nucleoside 5′-triphosphate (NTP)/nucleoside 5′-diphosphate (NDP) transphosphorylases. They are encoded by the NME (nonmetastatic cell) genes, which comprise a family of 10 related genes. Among them, the class I subfamily (consisting of NDPK-A, B, C and D) exerts enzymatic activity [
13]. NDPK-A and NDPK-B are involved in various cellular processes, including proliferation, differentiation, development and metastasis [
14]. In addition, NDPK-B acts as a mammalian protein histidine kinase by transferring the phosphate from its high energy phosphate intermediate to histidine residues on other proteins [
11]. Besides His358 in SK4 channels, another well-characterized NDPK-B substrate is His266 in the β-subunit of heterotrimeric G proteins [
11,
15]. Activation of the G protein by NDPK-B (resulting from phosphorylation of His266) increases intracellular cAMP formation independent of receptor activation. This connects NDPK-B to various physiological and pathophysiological cAMP-related processes in cells such as cardiomyocytes. Indeed NDPK-B has been linked to different diseases, including heart failure [
16]. Whether NDPK-B also plays a role in arrhythmogenic mechanisms in patients with ARVC has not been examined so far. Given that SK4 channels are important for pacemaker activity and are activated by NDPK-B, we designed this study to assess a possible involvement of SK4 channels and NDPK-B in the arrhythmogenesis of ARVC.
2. Materials and Methods
2.1. Ethics Statement
The skin biopsies from two healthy donors and one ARVC patient were obtained with written informed consent. The study was approved by the Ethics Committee of the Medical Faculty Mannheim, University of Heidelberg (approval number: 2018-565N-MA) and by the Ethics Committee of the University Medical Center Göttingen (approval number: 10/9/15). The study was carried out in accordance with the approved guidelines and conducted in accordance with the Helsinki Declaration of 1975 (
https://www.wma.net/what-we-do/medical-ethics/declaration-of-helsinki/), revised in 2013.
2.2. Generation of Human iPS Cells and iPS Cell-Derived Cardiomyocytes (hiPSC-CMs)
The hiPS cells and hiPSC-CMs were generated from the same healthy donors (Donor 1 and Donor 2) and from the same patient with ARVC as described in earlier study [
6]. Briefly, human iPS cells (hiPSCs) were generated from primary human fibroblasts derived from a skin biopsy. The hiPSC line was generated in feeder free culture conditions using the integration-free CytoTune-iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific, Schwerte, Germany #A16517) with the reprogramming factors OCT4, KLF4, SOX2 and c-MYC according to manufacturer’s instructions, with modifications. To prove the success of hiPS cell generation, the generated hiPSCs were characterized for their pluripotency and their in vitro differentiation potential, which have been shown in our recent study [
6].
The generation of hiPSC-CMs has been described in our previous studies. Briefly, culture dishes and wells for hiPSCs were coated with Matrigel (Corning, Kaiserslautern, Germany). The culture medium TeSR-E8 (Stemcell Technologies, Köln, Germany) was used for hiPSCs, and RPMI 1640 Glutamax (Life Technologies, Darmstadt, Germany) containing sodium pyruvate, penicillin/streptomycin, B27 (Life Technologies) and ascorbic acid (Sigma Aldrich, Taufkirchen, Germany) was used for hiPSC-CMs. During the first 3 weeks, CHIR99021 (Miltenyi Biotec, Bergisch Gladbach, Germany ), BMP-4 (R&D Systems, Wiesbaden, Germany), Activin A (R&D Systems), FGF-2 (Miltenyi Biotec) and IWP-4 (Miltenyi Biotec) were added at different time points to induce the cells to differentiate into hiPSC-CMs. During the third week a lactate (Sigma Aldrich) containing RPMI-medium without glucose and glutamine (Biological Industries, Cromwell, IN, USA) was added for selecting cardiomyocytes. At 40 to 60 days of culture with basic culture medium, cardiomyocytes were dissociated from 24 well plates and plated as single cells on matrigel-coated 3.5 cm petri dishes for patch-clamp measurements. The cells from this ARVC patient carried a missense mutation (p.Gly638Arg) in the desmoglein-2 (DSG2) gene.
To prove the successful differentiation of hiPSC-CMs, the expression of different cardiac markers was assessed at mRNA and protein levels, which have been shown in our recent study [
6]. The hiPSC-CMs used in this study were generated by the same differentiation protocol as used in the previous study and displayed similar cardiac features (
Figure S1).
2.3. Polymerase Chain Reaction Assays
The preparation of total RNA using the RNeasy mini kit (Qiagen, Hilden, Germany), including DNAse treatment, was performed by the following protocol. The cDNA was amplified by qPCR on StepOnePlus Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) using a PCR mix with hot start Taq DNA polymerase and SYBR Green (Sibir Rox Hot Mastermix, BIORON, Römerberg, Germany; Cat number 119405) in the presence of sense and antisense primers (400 nM each, RT2 qPCR Primer Assays from Qiagen, Germany). Relative mRNA expression level was calculated as the expression of the mRNA of the gene of interest relative to GAPDH in samples from treated or untreated (control) cells. The expression level was calculated by the ΔΔCT method, based on the threshold cycle (CT), as fold change = 2−Δ(ΔCT), where ΔCT = CTgene of interest − CTGAPDH and Δ(ΔCT) = ΔCTtreated − ΔCTcontrol. Results are shown as means ± SEMs from the measurements of at least 3 biological replicates and 2 technical replicates.
2.4. Western Blot
The cell lysates of hiPSC-CMs were used for protein isolation. Western blotting was performed using proteins extracted with RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM dithiothreitol, 1% Triton X-100, 1% sodium deoxycholate). The proteins were separated by SDS-PAGE and electrically transferred onto nitrocellulose membranes. After blocking with Roti-block (Roth, Karlsruhe, Germany), membranes were incubated with primary antibodies overnight. Immunocomplexes were incubated with corresponding secondary antibodies and visualized using a chemiluminescent peroxidase substrate (Roche, Mannheim, Germany; or Thermo Scientific, Rockford, IL, USA). Protein expression was quantified using Image J (NIH, Bethesda, MD, USA). Specific primary antibodies used were mouse-anti-NDPK-B (MC-412; Kamiya, Seattle, WA, USA), mouse-anti-γ-tubulin (Sigma-Aldrich) and mouse-anti-SK4 (sc-365265, Santa Cruz Biotechnology, Heidelberg, Germany).The corresponding secondary antibody was rabbit anti-mouse peroxidase (Sigma-Aldrich).
2.5. Recombinant NDPK Isoforms and PHP-1
Expression of His6-tagged NDPK-A, NDPK-B and NDPK-C as well as His6-tagged PHP-1 in and purification from
Escherichia coli was described in detail before [
11].
After the purification of NDPKs, their enzyme activity was confirmed by measuring the transphosphorylase activity (
Figure S2). The transphosphorylase activity of the NDPKs was measured in a reaction mixture containing 50 mM Tris-HCl, pH 7.5, 2 mM MgCl
2, 1 mM DTT and 0.01% BSA (Buffer A). Stock proteins were diluted in Buffer A to a concentration of 300 pM, and 1 volume of the rNDPK solution was mixed with 1 volume of substrate mixture (200 µM GTP and 20 µM ADP in Buffer A) on a 384 well plate. The mixture was incubated at room temperature for 30 min, and 2 volumes of the Kinase-GLO reagent (Promega, Walldorf, Germany, V6711), containing an ATP dependent firefly luciferase, were added. The luminescence was measured using a plate reader (PerkinElmer-EnVision, Baesweiler, Germany). Six different concentrations of ATP (0–10 µM) were used as standards for a calibration curve to calculate the amount of ATP produced by the different NDPKs. All readings shown were obtained under conditions where the ATP formation was still linear with time and enzyme concentration.
The activity of PHP-1 was proven by functional assessments in our previous study, showing that PHP-1 abolished NDPK-B effects, but the enzyme-inactive mutant PHP-1 failed to do so, indicative of the enzyme activity of PHP-1 [
17].
2.6. Patch-Clamp Recordings
The whole-cell patch-clamp recordings (voltage and current-clamp configurations) were carried out at room temperature (22–25 °C). To isolate the I
SK4 TRAM-34 (1 µM) or clotrimazole (3 µM) was added in the bath solution containing 130 mM/L NaCl, 5.9 mM/L KCl, 2.4 mM/L CaCl
2, 1.2 mM/L MgCl
2, 11 mM/L glucose and 10 mM/L HEPES (pH 7.4 (NaOH)). The pipette solution contained 10 mM HEPES, 126 mM KCl, 6 mM NaCl, 1.2 mM MgCl
2, 5 mM EGTA, 11 mM glucose, and 1 mM MgATP (pH 7.4 (KOH)). In addition, appropriate CaCl
2 was added to get the free Ca
2+ concentration of 0.5 μM according to the calculation by the software MAXCHELATOR (
http://web.stanford.edu/~cpatton/downloads.htm). TRAM-34 sensitive currents were evaluated as I
SK4. In experiments, to assess the effects of NDPK-B and PHP-1, NDPK-B (30 ng/mL) alone or with PHP-1 (100 ng/mL), or PHP-1 alone, was added into cells through the recording pipette solution. To minimize the effects of rundown of recorded currents on the results of experiments, we carefully monitored the time-dependent change of currents. Recordings were started when the current became stable, usually within 3 to 5 min. In spontaneous action potential (AP) recordings the extracellular and intracellular solutions were the same as used for current measurements.
2.7. Drugs
Clotrimazol, ivabradin, mibefradil and nifedipine were purchased from Sigma. TRAM-34 was purchased from Tocris Bioscience.
2.8. Statistical Analysis
If not otherwise indicated, data are shown as means ± SEMs and were analyzed using InStat© (GraphPad, San Diego, CA, USA) and SigmaPlot 11.0 (Systat GmbH, Erkrath Germany). By analyzing the data with the Kolmogorov Smirnov test, it was decided whether parametric or non-parametric tests were to be used for analysis. Student’s t-test and the Mann–Whitney U-test were used to compare continuous variables with normal and non-normal distributions, respectively. To compare categorical variables, the Fisher-test was used. For parametric data of more than two groups, one-way ANOVA with a Bonferroni post-test for multiple comparisons was performed. For non-parametric data, the Kruskal-Wallis test with Dunn’s multiple comparisons post-test was used. An unpaired Student’s t-test was used for comparisons of two independent groups with normal distributions. The paired t-test was used for comparisons of data before and after application of a drug. p <0.05 (two-tailed) was considered significant.
4. Discussion
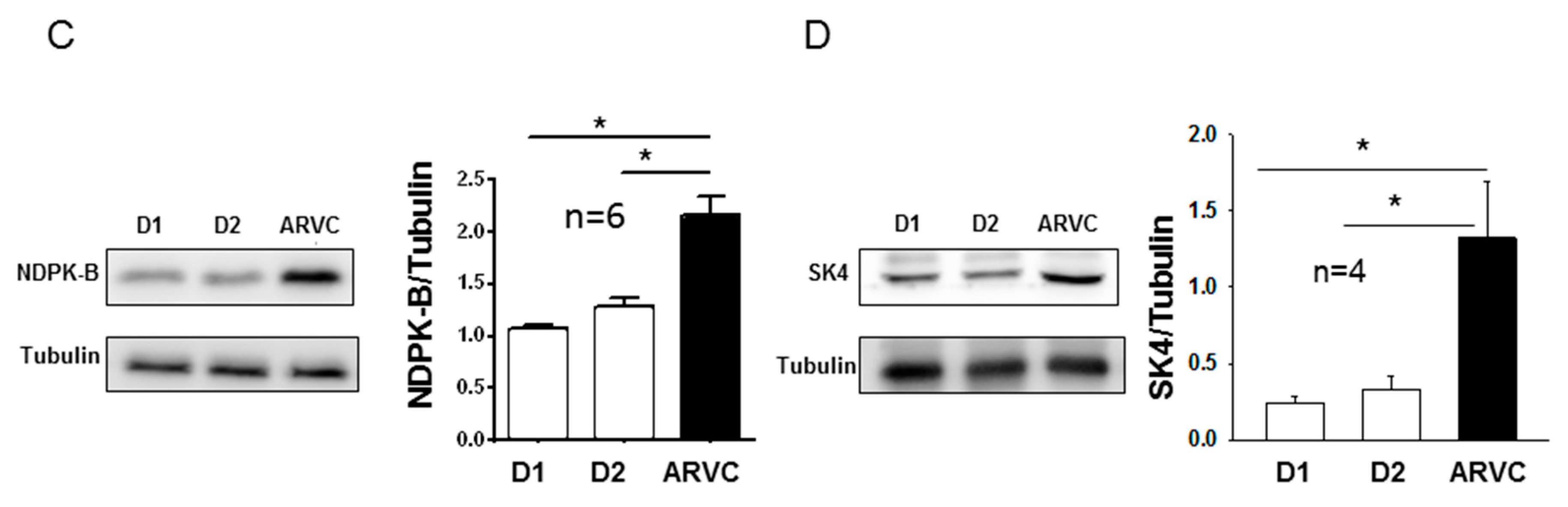
In this study, we investigated the role of NDPK-B and SK4 channels for arrhythmogenesis in ARVC using hiPSC-CMs from a patient with ARVC carrying a missense mutation (p.Gly638Arg) in the DSG2. We demonstrate for the first time that (i) both NDPK-B and SK4 expressions are elevated in ARVC cardiomyocytes; (ii) the application of recombinant NDPK-B into hiPSC-CMs activated SK4 channels enhances both pacemaker activity and arrhythmic events; and (iii) PHP-1 as a phosphohistidine-specific phosphatase acting on SK4 channels suppresses the enhanced pacemaker activity and prevents the occurrence of arrhythmic events in ARVC-hiPSC-CMs.
The frequently discussed mechanism underlying tachyarrhythmias in ARVC is the conduction slowing between cells caused by cell-detaching or intercellular fibrosis plus fat-deposit. However, some studies demonstrated that electrophysiological changes precede structural changes [
18], suggesting that other possible arrhythmogenic mechanisms are involved in ARVC. We observed reduced sodium channel currents and abnormal APs with reduced Vmaxs in cells from the ARVC patient [
6]. Vmax is important for the conduction of the excitation in and between cells. Therefore, reduced Vmax can cause a slowing down of the conduction and may lead to tachyarrhythmias in patients with ARVC. However, mechanisms other than the conduction defect might contribute to the phenotype.
In the current study, we detected enhanced expression and currents of SK4 channels in ARVC-hiPSC-CMs, which have been shown before to contribute to pacemaker activity in embryonic stem cell derived cardiomyocytes and hiPSC-CMs as well as mouse cardiomyocytes [
8,
9]. In addition, we also observed higher automaticity and occurrence rate of arrhythmic events, which obviously involve I
SK4 in ARVC-hiPSC-CMs. The contribution of SK4 channels to pacemaker activity and arrhythmogenesis was also observed in CPVT-hiPSC-CMs [
8]. Considering that SK4 channel activity is related to the occurrence of arrhythmic events in both CPVT- and ARVC-hiPSC-CMs and important for cell automaticity, we speculate that the elevated pacemaker activity in cardiomyocytes can contribute to arrhythmogenesis in patients with ARVC. It is well-known that SK4 channels exist in embryonic stem-derived cardiomyocytes, iPSC-CMs and mouse pacemaker cells [
8]. Whether SK4 channels are expressed in human ventricular cardiomyocytes, especially in “diseased” cardiomyocytes in patients with ARVC, is not known. If the SK4 channel expression in diseased cardiomyocytes is upregulated together with NDPK-B, as shown here for ARVC-hiPSC-CMs, the automaticity of the cells will be enhanced and the likelihood of ectopic beats will be increased. Therefore, both the enhanced ectopic excitations and conduction defect caused by structural abnormalities or ion channel dysfunctions might contribute to the occurrence of tachyarrhythmias in ARVC.
How SK4 channels influence pacemaker activity is still not fully explored. It has been hypothesized that activation of SK4 channels leads to an enhanced K
+ efflux resulting in hyperpolarization of cell membrane. The hyperpolarization increases the driving force for influx of positively charged ions conducted by hyperpolarization activated channels (HCN, also called I
f channels) or T-type or L-type calcium channels. The enhanced inward current accelerates the diastolic depolarization and enhances automaticity (pacemaker activity). In hiPSC-CMs, however, I
f channel expression is low and I
f current is small [
19]. More strikingly, an I
f blocker (ivarbradine), a T-type calcium channel blocker (mibefradil) and an L-type calcium channel blocker (nifedipine) all failed to largely reduce the automaticity, suggesting that those ion channels are, alone, not critical for the pacemaker activity in hiPSC-CMs. Another study showed that HERG channel currents determined the MDP (maximum diastolic potential) but not the pacemaker activity in hiPSC-CMs [
20]. Nevertheless, the current study demonstrates that SK4 currents are important for the pacemaker activity in hiPSC-CMs as the SK4 ion channel blocker TRAM-34 largely or completely inhibited the automaticity of both donor and ARVC-hiPSC-CMs.
The next question needing to be addressed is how the SK4 channel expression and current are enhanced in ARVC-cells. Our recent study showed that the intracellular Ca
2+-concentration in ARVC-hiPSC-CMs was the same as that in donor cells [
6], suggesting that the increased SK4 currents resulted from a regulation other than an elevated intracellular Ca
2+ concentration required for the calmodulin-dependent regulation of SK4. Besides Ca
2+, several protein kinases, including PKA, PKG, PKC and NDPK-B, have been shown to regulate SK4 channels. Among them, NDPK-B seems to be the most important regulator for SK4 channel activity. The activation of SK4 channels by NDPK-B has been well established in different cell systems, including HEK cells, lymphocytes and smooth muscle cells [
10,
11,
12,
17]. It is known that NDPK-B can phosphorylate the histidine residue at the position 358 (His358) in the SK4 channel [
12]. In the presence of Ca
2+, upon phosphorylation of His358, copper ion binding in the channel is abrogated, and the calcium-induced conformational changes in the calmodulin-binding domain lead to channel opening [
10]. The histidine phosphatase PHP-1 can dephosphorylate His358 and counteract the effects of NDPK-B in all cell types analyzed so far. In our ARVC-hiPSC-CMs, NDPK-B expression at both mRNA and protein levels was upregulated, suggesting the possibility that the increased I
SK4 in ARVC-hiPSC-CMs may result from the enhanced histidine phosphorylation in SK4 channels by NDPK-B, aside from an increased channel density. Indeed, recombinant NDPK-B but bot NDPK-A or NDPK-C, applied into hiPSC-CMs through the patch pipette, enhanced I
SK4, and additional application of PHP-1 abolished the effect of NDPK-B. Importantly, application of PHP-1 alone also reduced I
SK4 in hiPSC-CMs, indicating a contribution of endogenous histidine phosphorylation by NDPK-B to I
SK4. These data demonstrate that SK4 channels in hiPSC-CMs can be activated by NDPK-B. Considering that NDPK-B is an upstream regulator of SK4 channels, and both the SK4 blocker TRAM-34 and NDPK-B counter-actor PHP-1 reduced pacemaker activity and occurrence of arrhythmias in ARVC cells, we speculate that NDPK-B/SK4 upregulation might be a reason for arrhythmogenesis in ARVC. However it remains to be clarified (i) how NDPK-B expression was upregulated based on the desmosomal gene mutation occurring in ARVC, (ii) whether the observed changes in NDPK-B and SK4 are general in all ARVC patients with different gene mutations and (iii) why application of NDPK-B into a cell caused different forms of arrhythmias. Additionally, the possibility that the elevated expression level of NDPK-B may enhance the pacemaker activity and arrhythmias through non-SK4 involving signaling events cannot be excluded. Nevertheless, our data indicate that the SK4-activating effect of NDPK-B can contribute to ectopic activity and the occurrence of arrhythmias in ARVC, at least with the DSG2 mutation (p.Gly638Arg).
In summary, this study revealed that in hiPSC-CMs from an ARVC patient, (i) NDPK-B and SK4 channels were upregulated, (ii) SK4 channels were activated, (iii) pacemaker activity was enhanced and (iv) the occurrence of arrhythmias was increased. We therefore speculate that upregulation of NDPK-B and SK4 can contribute to arrhythmogenesis in ARVC through enhanced histidine phosphorylation of SK4. Thus, NDPK-B may possibly be a potential therapeutic target for treating arrhythmias in ARVC-patients.
5. Study Limitations
Some limitations should be considered in extrapolating the data from the current study. The hiPSC-CMs from only two healthy donors and one ARVC-patient were used for this study. Therefore, we cannot rule out the differences among individuals. The results from the patient of this study should, from a statistical point of view, not be interpreted as that from the whole population of ARVC-patients. From a practical point of view, it is difficult to include several patients with the same mutation in the same gene, and hence, for the present study it was not feasible to include cells from second and third ARVC patients to increase the power. Patients with ARVC carrying other DSG2 mutations or mutations in other genes were not recruited for this study, and therefore, whether NDPK-B and SK4 upregulation exist in other ARVC-patients with different gene mutations as well needs to be clarified in further studies. In addition, immaturity is a well-known limitation of hiPSC-CMs. The differences of cell properties, including electrical activities between hiPSC-CMs and native cardiomyocytes, should be also considered in interpreting the data of this study. Furthermore, due to the limitation of availability, the native cardiac cells from ARVC-patients were not investigated for this study. Whether the upregulation of NDPK-B and SK4 is the phenotypic characteristic\in ARVC-patients, which is the most important question for this study, cannot be addressed, although it may be the case, at least in some ARVC-patients with a certain gene mutation. Nevertheless, this study may trigger further studies in this area.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







