Neuronal and Astrocytic Differentiation from Sanfilippo C Syndrome iPSCs for Disease Modeling and Drug Development

 ,
,  , , ,
, , , 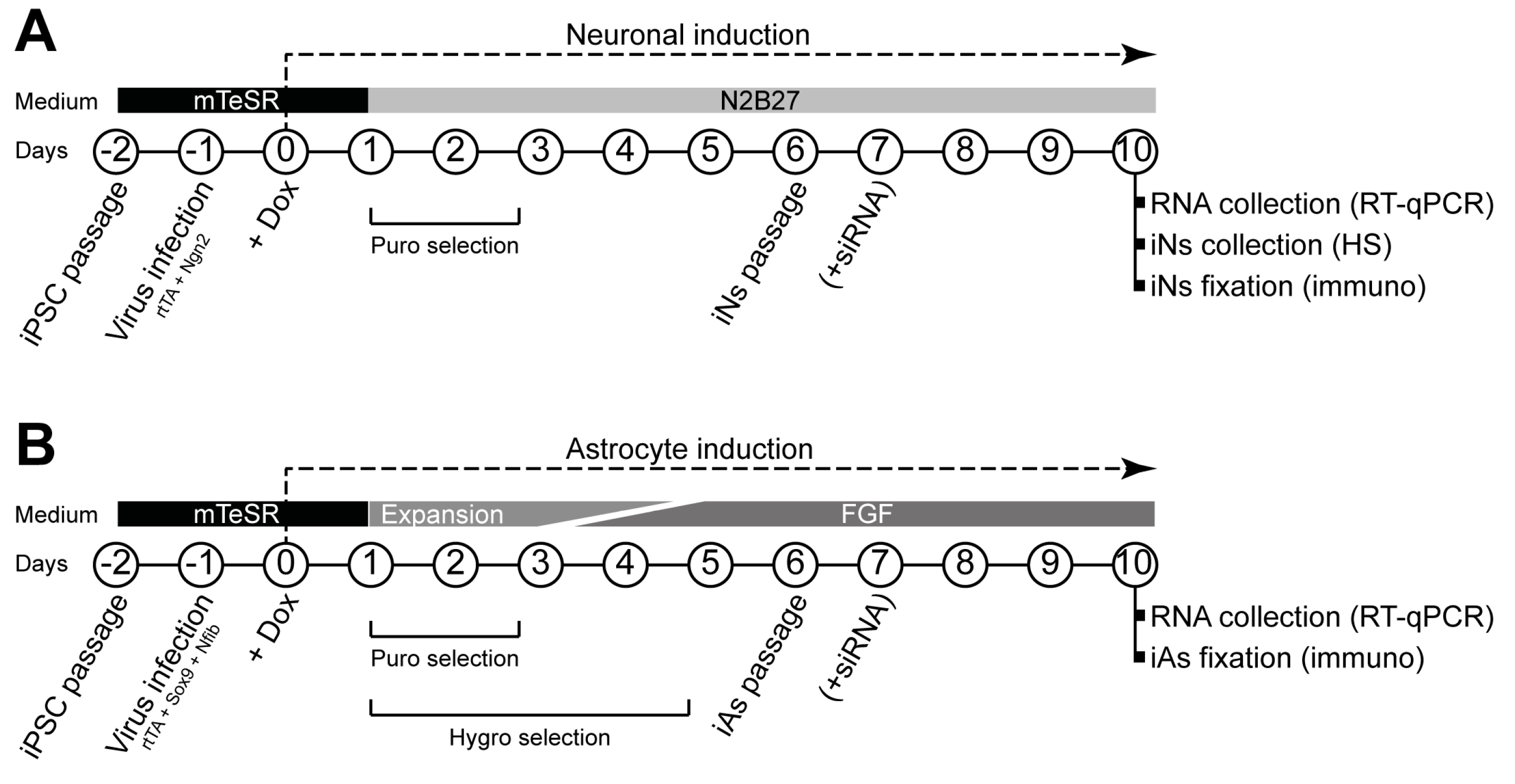{kind=link}
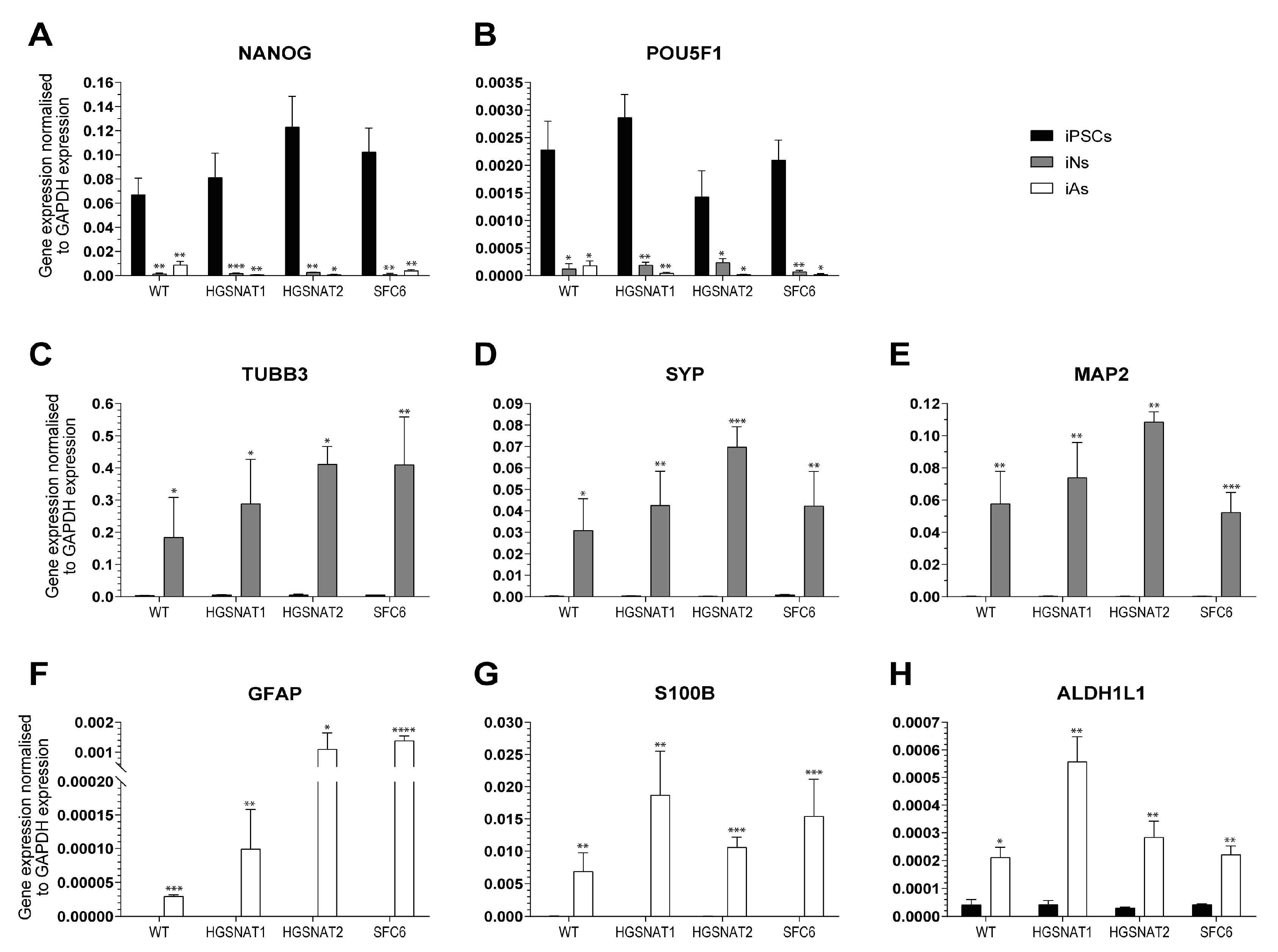{kind=link}
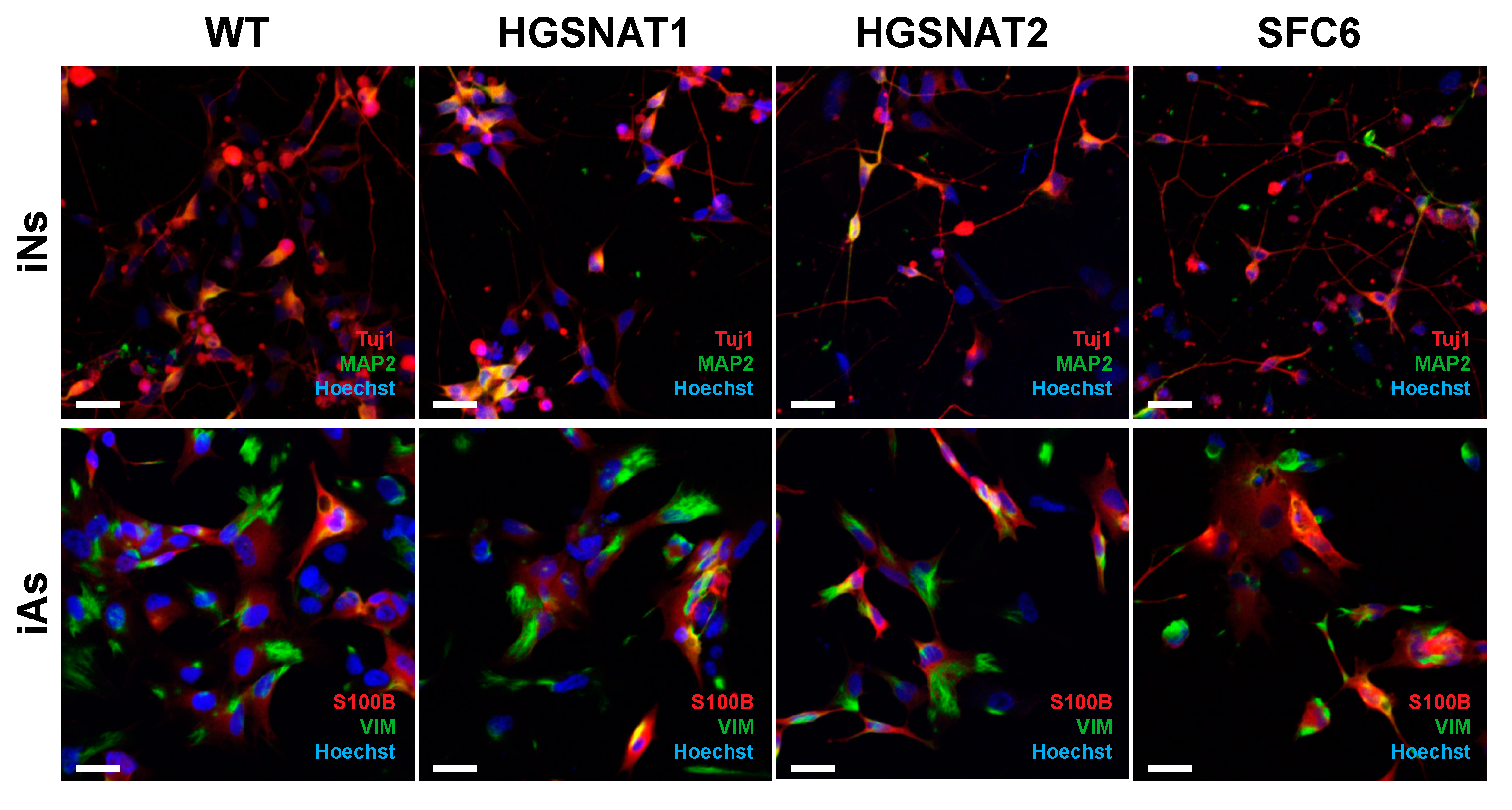{kind=link}
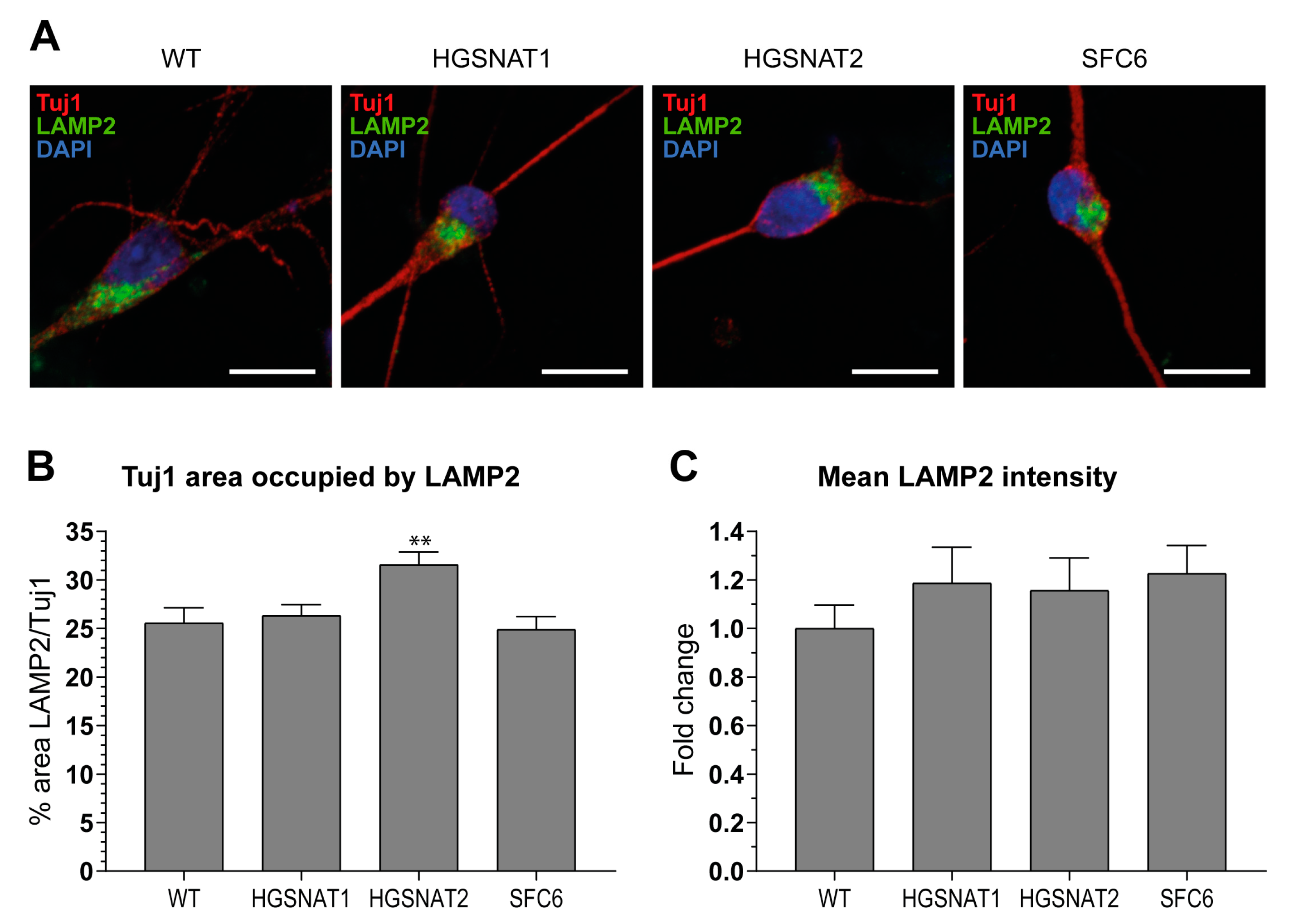{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Human iPSCs
2.2. Lentiviral Production
2.3. Generation of Induced Neurons and Astrocytes from iPSCs
2.4. siRNA Transfection
2.5. Immunofluorescence Staining
2.6. RT-qPCR
2.7. HS Quantity Measurement
2.8. Data Analysis
3. Results
3.1. Generation of iNs and iAs to Model Sanfilippo C Syndrome
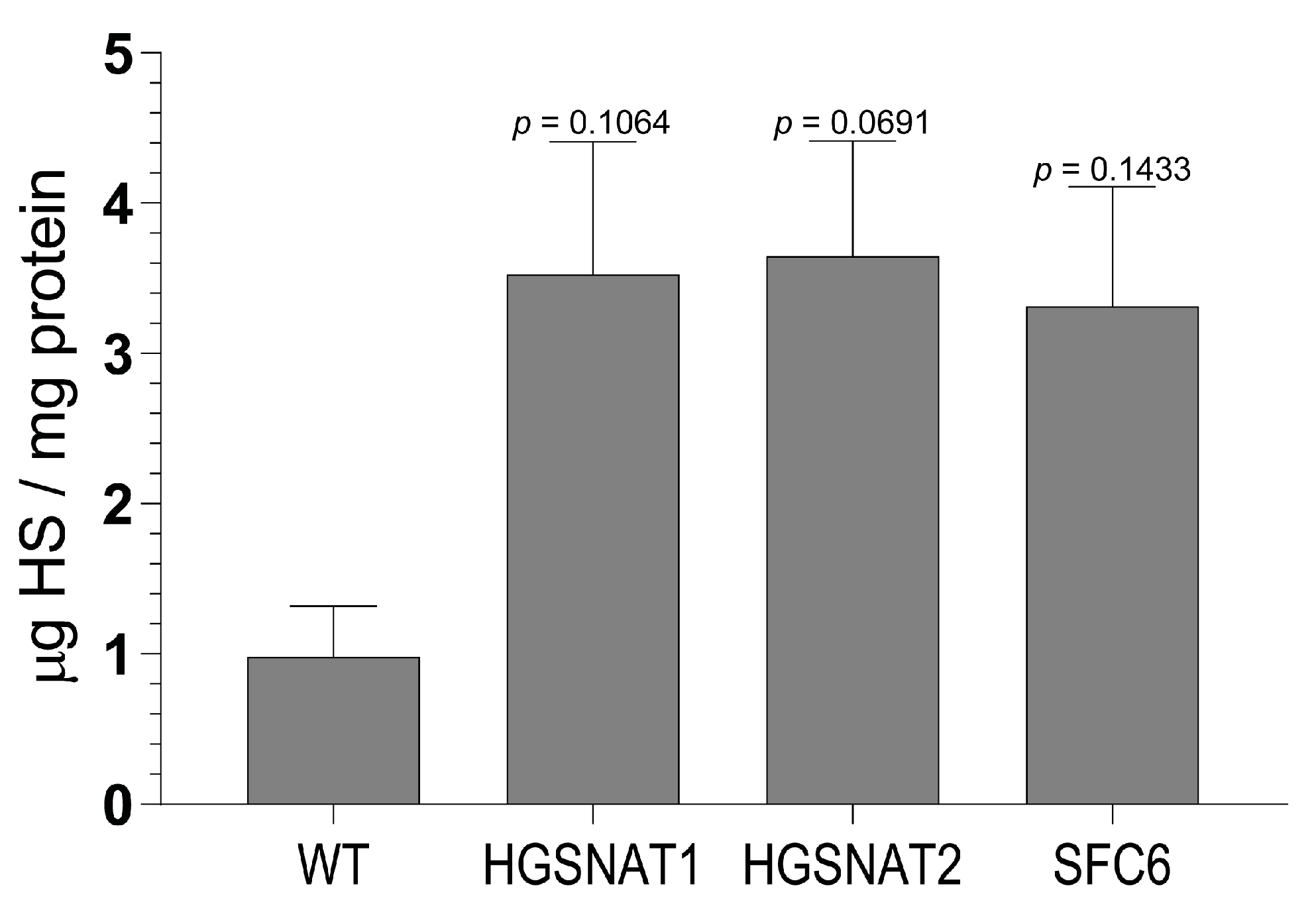
3.2. iNs and iAs Recapitulate Major Sanfilippo C Phenotypes
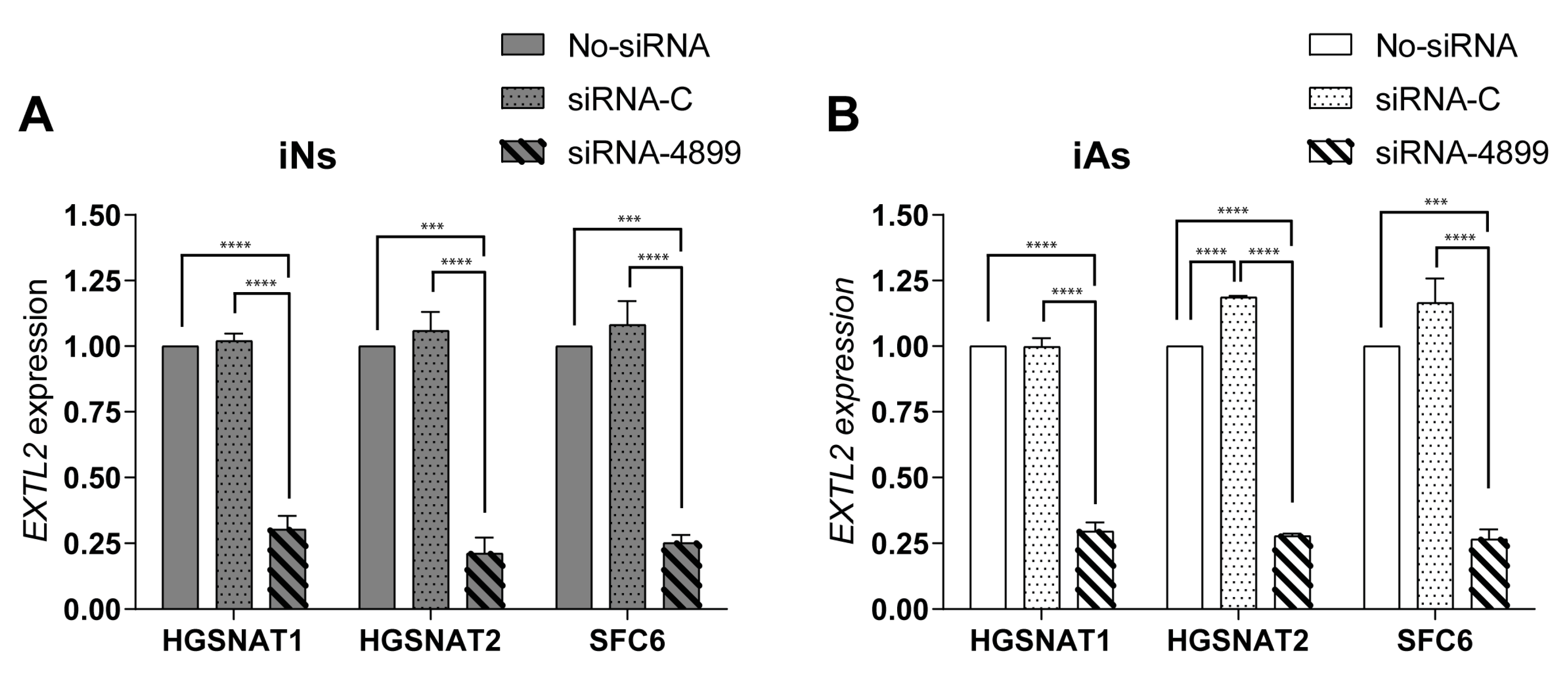
3.3. Short-Term siRNA-Based SRT Is Not Efficient in Disease-Relevant Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Futerman, A.H.; van Meer, G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F.; Muenzer, J. The mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Eds.; McGraw-Hill: New York, NY, USA, 2001; Volume 3, pp. 3421–3452. [Google Scholar]
- Andrade, F.; Aldámiz-Echevarría, L.J.; Llarena, M.; Couce, M.L. Sanfilippo syndrome: Overall review. Pediatr. Int. 2015, 57, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Fedele, A.O. Sanfilippo syndrome: Causes, consequences, and treatments. Appl. Clin. Genet. 2015, 8, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelei, T.; Csetneki, K.; Vokó, Z.; Siffel, C. Epidemiology of Sanfilippo syndrome: Uesults of a systematic literature review. Orphanet. J. Rare Dis. 2018, 13, 53. [Google Scholar] [CrossRef] [Green Version]
- Klein, U.; Kresse, H.; von Figura, K. Sanfilippo syndrome type C: Deficiency of acetyl-CoA: α-glucosaminide N-acetyltransferase in skin fibroblasts. Proc. Natl. Acad. Sci. USA 1978, 75, 5185–5189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Zhang, H.; Zhang, S.; Bagshaw, R.D.; Tropak, M.B.; Callahan, J.W.; Mahuran, D.J. Identification of the gene encoding the enzyme deficient in mucopolysaccharidosis IIIC (Sanfilippo disease type C). Am. J. Hum. Genet. 2006, 79, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Hřebíček, M.; Mrázová, L.; Seyrantepe, V.; Durand, S.; Roslin, N.M.; Nosková, L.; Hartmannová, H.; Ivánek, R.; Cízkova, A.; Poupetová, H.; et al. Mutations in TMEM76* cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am. J. Hum. Genet. 2006, 79, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Tkachyova, I.; Sinha, A.; Rigat, B.; Mahuran, D. Characterization of the biosynthesis, processing and kinetic mechanism of action of the enzyme deficient in mucopolysaccharidosis IIIC. PLoS ONE 2011, 6, e24951. [Google Scholar] [CrossRef] [Green Version]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Wijburg, F.A.; Whitley, C.B.; Muenzer, J.; Gasperini, S.; Del Toro, M.; Muschol, N.; Cleary, M.; Sevin, C.; Shapiro, E.; Bhargava, P.; et al. Intratechal heparan-N-sulfatase in patients with Sanfilippo syndrome type A: A phase IIb randomized trial. Mol. Genet. Metab. 2019, 126, 121–130. [Google Scholar] [CrossRef]
- Welling, L.; Marchal, J.P.; van Hasselt, P.; van der Ploeg, A.T.; Wijburg, F.A.; Boelens, J.J. Early Umbilical Cord Blood-Derived Stem Cell Transplantation Does Not Prevent Neurological Deterioration in Mucopolysaccharidosis Type III. JIMD. Rep. 2015, 18, 63–68. [Google Scholar] [PubMed] [Green Version]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological chaperone therapy: Preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldhammer, M.; Durand, S.; Pshezhetsky, A.V. Protein misfolding as an underlying molecular defect in mucopolysaccharidosis III type C. PLoS One 2009, 4, e7434. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, M.; Zérah, M.; Gougeon, M.L.; Ausseil, J.; de Bournonville, S.; Husson, B.; Zafeiriou, D.; Parenti, G.; Bourget, P.; Poirier, B.; et al. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: An uncontrolled phase 1/2 clinical trial. Lancet Neurol. 2017, 16, 712–720. [Google Scholar] [CrossRef]
- Tardieu, M.; Zérah, M.; Husson, B.; de Bournonville, S.; Deiva, K.; Adamsbaum, C.; Vincent, F.; Hocquemiller, M.; Broissand, C.; Furlan, V.; et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cdnas in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum. Gene Ther. 2014, 25, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, L.; Lotfi, P.; Pal, R.; di Ronza, A.; Sharma, J.; Sardiello, M. Lysosome biogenesis in health and disease. J. Neurochem. 2019, 148, 573–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tordo, J.; O’Leary, C.; Antunes, A.S.L.M.; Palomar, N.; Aldrin-Kirk, P.; Basche, M.; Bennett, A.; D’Souza, Z.; Gleitz, H.; Godwin, A.; et al. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain 2018, 141, 2014–2031. [Google Scholar] [CrossRef]
- Roberts, A.L.; Rees, M.H.; Klebe, S.; Fletcher, J.M.; Byers, S. Improvement in behaviour after substrate deprivation therapy with rhodamine B in a mouse model of MPS IIIA. Mol. Genet. Metab. 2007, 92, 115–121. [Google Scholar] [CrossRef]
- Jakóbkiewicz-Banecka, J.; Piotrowska, E.; Narajczyk, M.; Barańska, S.; Wegrzyn, G. Genistein-mediated inhibition of glycosaminoglycan synthesis, which corrects storage in cells of patients suffering from mucopolysaccharidoses, acts by influencing an epidermal growth factor-dependent pathway. J. Biomed. Sci. 2009, 16, 26. [Google Scholar] [CrossRef] [Green Version]
- Malinowska, M.; Wilkinson, F.L.; Langford-Smith, K.J.; Langford-Smith, A.; Brown, J.R.; Crawford, B.E.; Vanier, M.T.; Grynkiewicz, G.; Wynn, R.F.; Wraith, J.E.; et al. Genistein improves neuropathology and corrects behaviour in a mouse model of neurodegenerative metabolic disease. PLoS One 2010, 5, e14192. [Google Scholar] [CrossRef] [Green Version]
- Delgadillo, V.; O’Callaghan, M.M.; Artuch, R.; Montero, R.; Pineda, M. Genistein supplementation in patients affected by Sanfilippo disease. J. Inherit. Metab. Dis. 2011, 34, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Kaidonis, X.; Liaw, W.C.; Roberts, A.D.; Ly, M.; Anson, D.; Byers, S. Gene silencing of EXTL2 and EXTL3 as a substrate deprivation therapy for heparan sulphate storing mucopolysaccharidoses. Eur. J. Hum. Genet. 2010, 18, 194–199. [Google Scholar] [CrossRef]
- Dziedzic, D.; Wegrzyn, G.; Jakóbkiewicz-Banecka, J. Impairment of glycosaminoglycan synthesis in mucopolysaccharidosis type IIIA cells by using siRNA: A potential therapeutic approach for Sanfilippo disease. Eur. J. Hum. Genet. 2010, 18, 200–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canals, I.; Benetó, N.; Cozar, M.; Vilageliu, L.; Grinberg, D. EXTL2 and EXTL3 inhibition with siRNAs as a promising substrate reduction therapy for Sanfilippo C syndrome. Sci. Rep. 2015, 5, 13654. [Google Scholar] [CrossRef] [PubMed]
- Zunke, F.; Mazzulli, J.R. Modeling neuronopathic storage diseases with patient-derived culture systems. Neurobiol. Dis. 2019, 127, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Zhang, S.C. Neural Subtype Specification from Human Pluripotent Stem Cells. Cell Stem Cell 2016, 19, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2019, 98, 239–389. [Google Scholar] [CrossRef]
- Oh, Y.; Jang, J. Directed Differentiation of Pluripotent Stem Cells by Transcription Factors. Mol. Cells 2019, 42, 200–209. [Google Scholar]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neurone 2013, 78, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Canals, I.; Ginisty, A.; Quist, E.; Timmerman, R.; Fritze, J.; Miskinyte, G.; Monni, E.; Hansen, M.G.; Hidalgo, I.; Bryder, D.; et al. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat. Methods 2018, 15, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Li, X.F.; Zhou, Y.W.; Cai, P.F.; Fu, W.C.; Wang, J.H.; Chen, J.Y.; Yang, Q.N. CRISPR/Cas9 facilitates genomic editing for large-scale functional studies in pluripotent stem cell cultures. Hum. Genet. 2019, 138, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, T.; Blanchard, S.; Toli, D.; Roy, E.; Bigou, S.; Froissart, R.; Rouvet, I.; Vitry, S.; Heard, J.M.; Bohl, D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum. Mol Genet. 2011, 20, 3653–3666. [Google Scholar] [CrossRef] [Green Version]
- Canals, I.; Soriano, J.; Orlandi, J.G.; Torrent, R.; Richaud-Patin, Y.; Jiménez-Delgado, S.; Merlin, S.; Follenzi, A.; Consiglio, A.; Vilageliu, L.; et al. Activity and High-Order Effective Connectivity Alterations in Sanfilippo C Patient-Specific Neuronal Networks. Stem Cell Reports 2015, 5, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Benetó, N.; Cozar, M.; García-Morant, M.; Creus-Bachiller, E.; Vilageliu, L.; Grinberg, D.; Canals, I. Generation of two compound heterozygous HGSNAT-mutated lines from healthy induced pluripotent stem cells using CRISPR/Cas9 to model Sanfilippo C syndrome. Stem Cell Res. 2019, 41, 101616. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Oguma, T.; Tomatsu, S.; Montano, A.M.; Okazaki, O. Analytical method for the determination of disaccharides derived from keratan, heparan, and dermatan sulfates in human serum and plasma by high-performance liquid chromatography/turbo ionspray ionization tandem mass spectrometry. Anal. Biochem. 2007, 368, 79–86. [Google Scholar] [CrossRef]
- Martins, C.; Hůlková, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef] [Green Version]
- Marcó, S.; Pujol, A.; Roca, C.; Motas, S.; Ribera, A.; Garcia, M.; Molas, M.; Villacampa, P.; Melia, C.S.; Sánchez, V.; et al. Progressive neurologic and somatic disease in a novel mouse model of human mucopolysaccharidosis type IIIC. Dis. Model Mech. 2016, 9, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Kreuger, J.; Kjellén, L. Heparan sulfate biosynthesis: Regulation and variability. J. Histochem. Cytochem. 2012, 60, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Wuyts, W.; Van Hul, W.; De Boulle, K.; Hendrickx, J.; Bakker, E.; Vanhoenacker, F.; Mollica, F.; Lüdecke, H.J.; Sayli, B.S.; Pazzaglia, U.E.; et al. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am. J. Hum. Genet. 1998, 62, 346–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadanaka, S.; Zhou, S.; Kagiyama, S.; Shoji, N.; Sugahara, K.; Sugihara, K.; Asano, M.; Kitagawa, H. EXTL2, a member of the EXT family of tumor suppressors, controls glycosaminoglycan biosynthesis in a xylose kinase-dependent manner. J. Biol. Chem. 2013, 288, 9321–9333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadanaka, S.; Kagiyama, S.; Kitagawa, H. Roles of EXTL2, a member of the EXT family of tumour suppressors, in liver injury and regeneration processes. Biochem. J. 2013, 454, 133–145. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benetó, N.; Cozar, M.; Castilla-Vallmanya, L.; Zetterdahl, O.G.; Sacultanu, M.; Segur-Bailach, E.; García-Morant, M.; Ribes, A.; Ahlenius, H.; Grinberg, D.; et al. Neuronal and Astrocytic Differentiation from Sanfilippo C Syndrome iPSCs for Disease Modeling and Drug Development. J. Clin. Med. 2020, 9, 644. https://doi.org/10.3390/jcm9030644
Benetó N, Cozar M, Castilla-Vallmanya L, Zetterdahl OG, Sacultanu M, Segur-Bailach E, García-Morant M, Ribes A, Ahlenius H, Grinberg D, et al. Neuronal and Astrocytic Differentiation from Sanfilippo C Syndrome iPSCs for Disease Modeling and Drug Development. Journal of Clinical Medicine. 2020; 9(3):644. https://doi.org/10.3390/jcm9030644
Chicago/Turabian StyleBenetó, Noelia, Monica Cozar, Laura Castilla-Vallmanya, Oskar G. Zetterdahl, Madalina Sacultanu, Eulalia Segur-Bailach, María García-Morant, Antonia Ribes, Henrik Ahlenius, Daniel Grinberg, and et al. 2020. "Neuronal and Astrocytic Differentiation from Sanfilippo C Syndrome iPSCs for Disease Modeling and Drug Development" Journal of Clinical Medicine 9, no. 3: 644. https://doi.org/10.3390/jcm9030644
APA StyleBenetó, N., Cozar, M., Castilla-Vallmanya, L., Zetterdahl, O. G., Sacultanu, M., Segur-Bailach, E., García-Morant, M., Ribes, A., Ahlenius, H., Grinberg, D., Vilageliu, L., & Canals, I. (2020). Neuronal and Astrocytic Differentiation from Sanfilippo C Syndrome iPSCs for Disease Modeling and Drug Development. Journal of Clinical Medicine, 9(3), 644. https://doi.org/10.3390/jcm9030644