Axonopathy and Reduction of Membrane Resistance: Key Features in a New Murine Model of Human GM1-Gangliosidosis



 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Generation of Transgenic Mice
2.3. Genotyping of Mice
2.4. Cell Culture of Fibroblasts
2.5. Analysis of Glb1 mRNA
2.6. Enzyme Activity and Protein Determination
2.7. Experimental Design and Scoring
2.8. Histology
2.9. Immunohistochemistry
2.10. Transmission Electron Microscopy
2.11. Electrophysiology and Single Cell Electroporation
2.12. Lipid Analysis
2.13. Lipid Raft Isolation and Western Blotting
2.14. Statistical Examination and Design of Figures
3. Results
3.1. Glb1−/− Mice Produced Shortened Dysfunctional β-Galactosidase Due to Skipping of Exon 15
3.2. Glb1−/− Mice Showed Increasing Neurological Disorder Starting at the Age of 3.5 to Four Months
3.3. Significant Neuronal Storage in Glb1−/− Mice
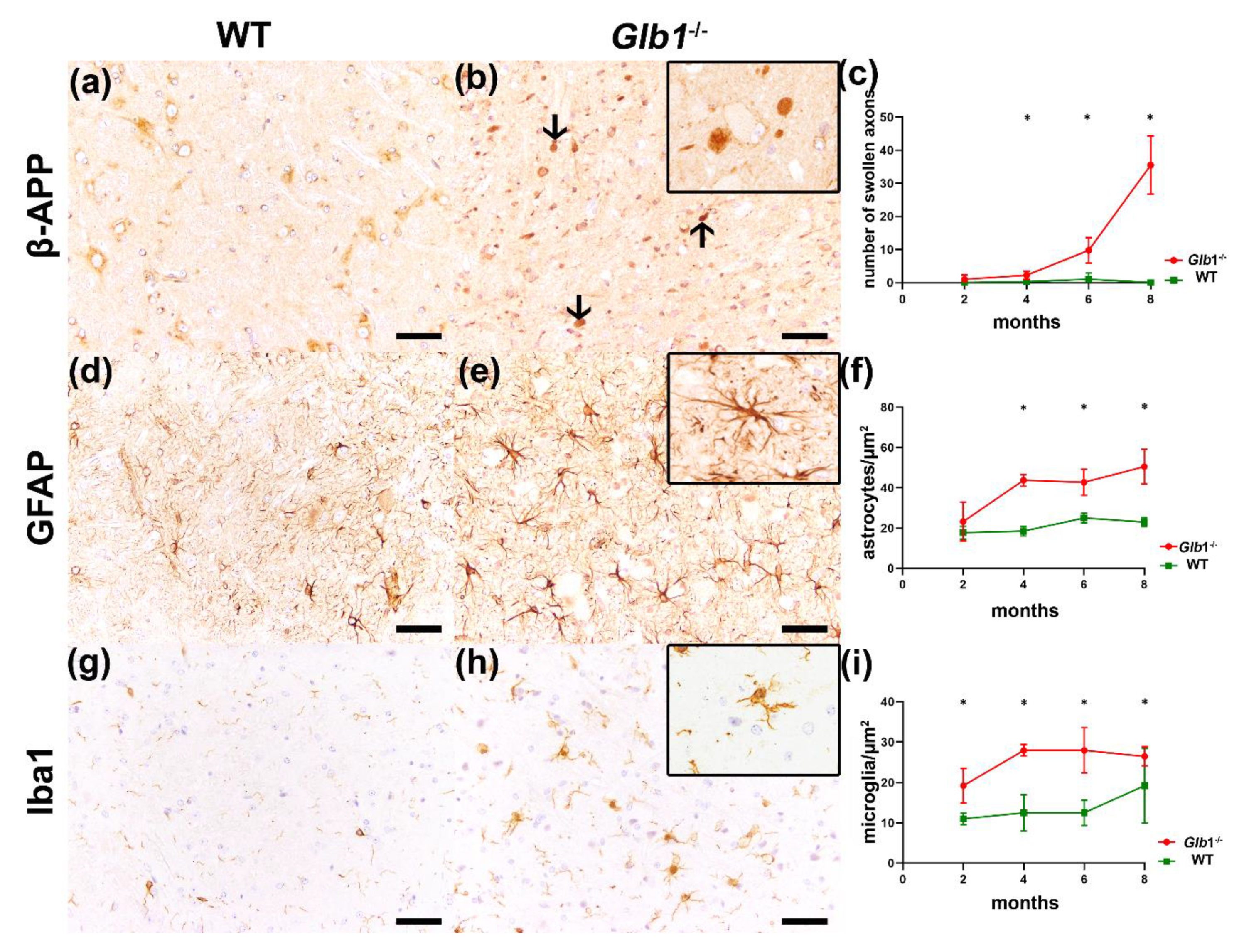
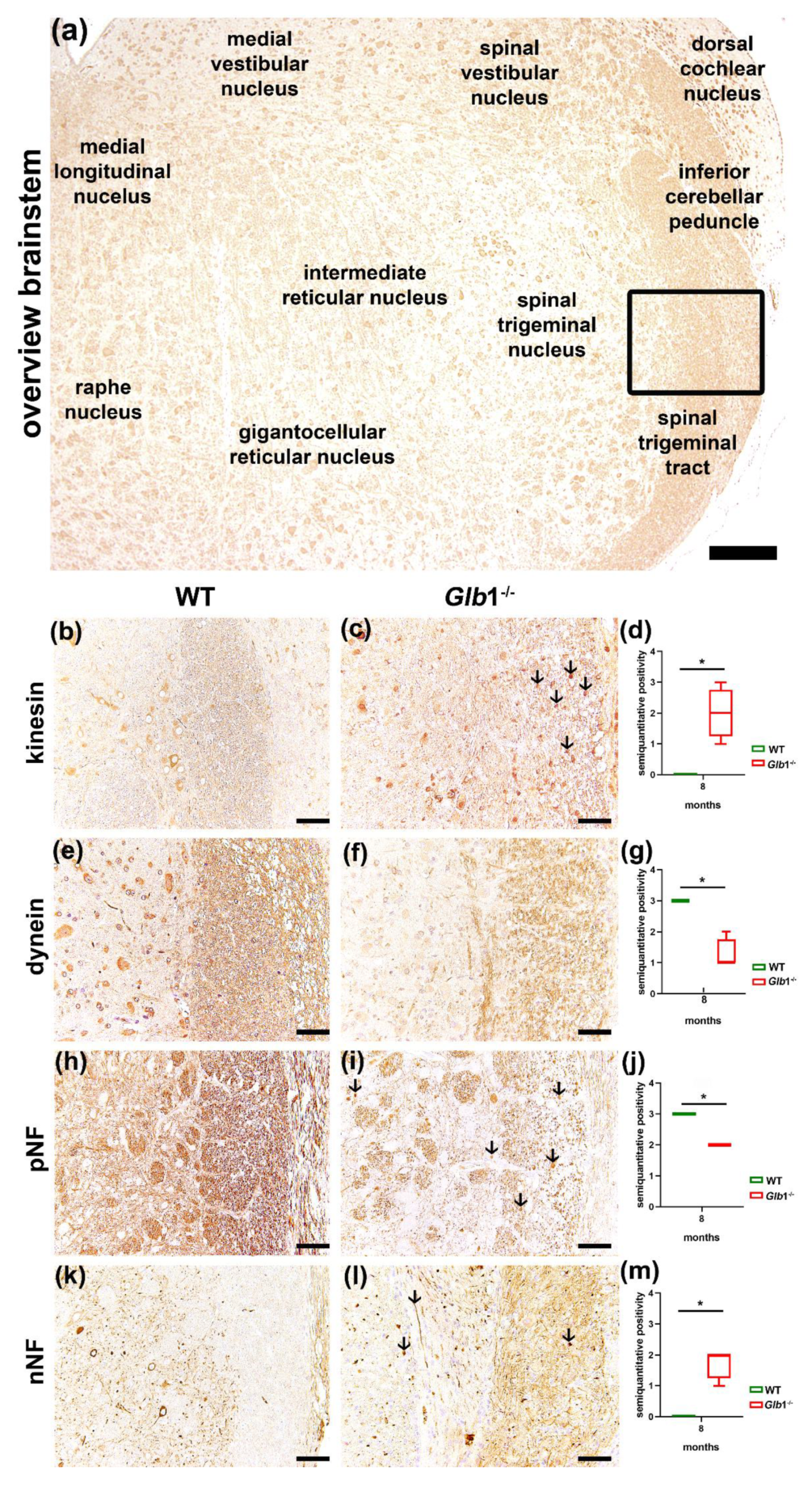
3.4. Detection of Axonal Damage and Myelin Loss
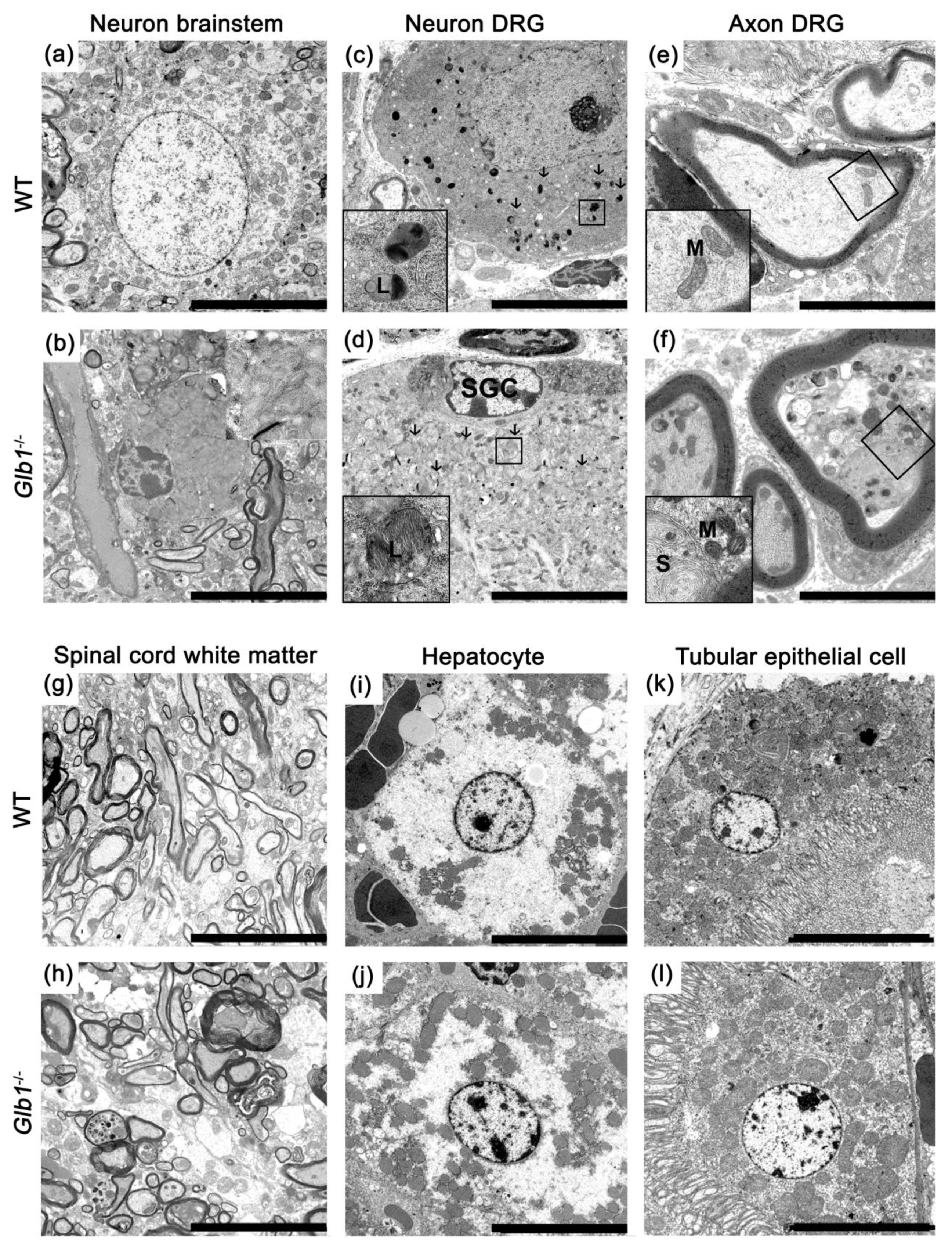
3.5. Glb1−/− Mice Showed Lamellar Storage Material in Lysosomes of Soma and Axons
3.6. Neurons of Glb1−/− Mice Showed Abundant Vacuolar Structures in Their Soma and Dendrites
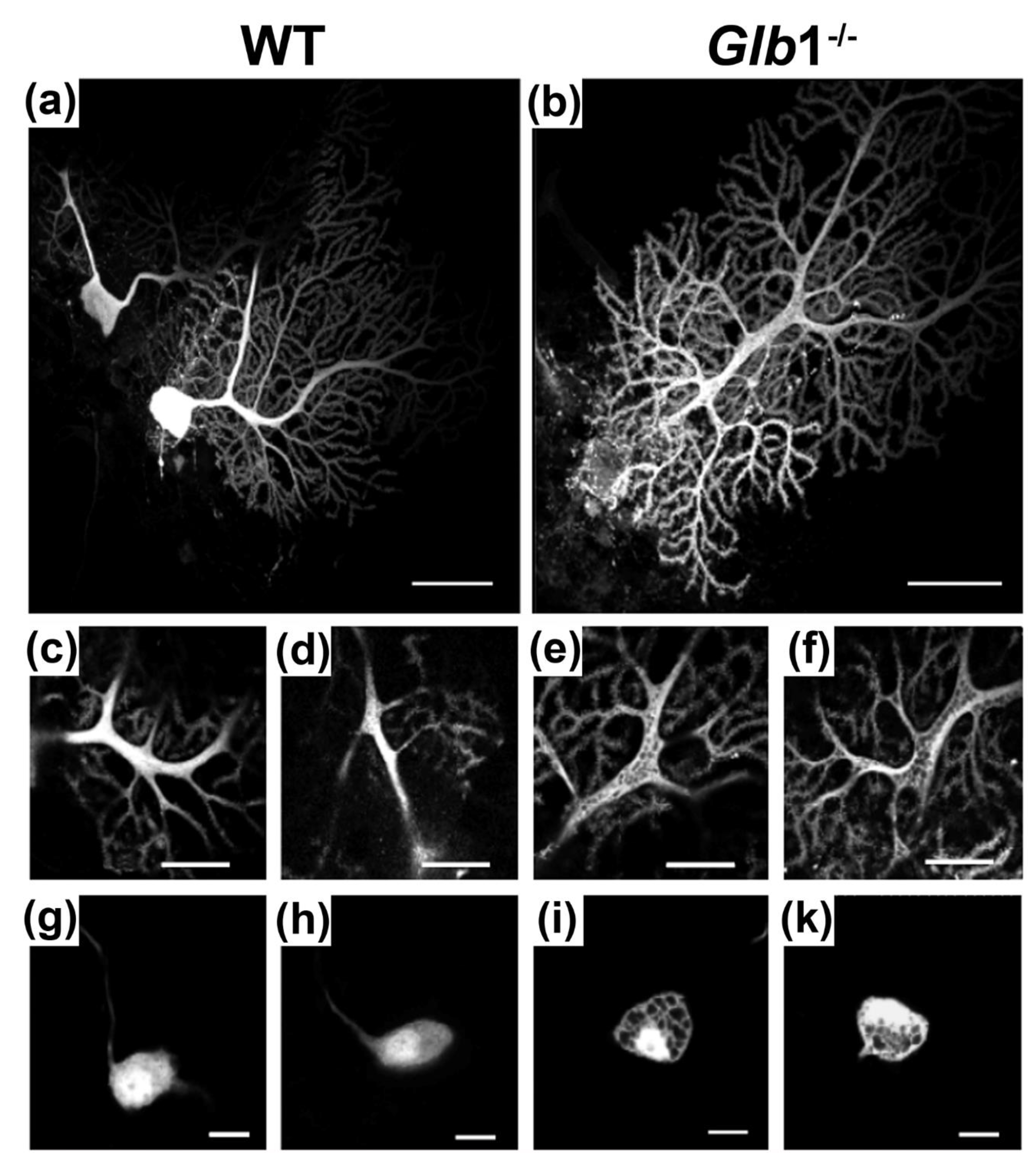
3.7. Biophysical Consequences of Loss of β-Galactosidase Function
3.8. Glb1−/− Fibroblasts Show Lipid Accumulations and Membrane Alterations
3.9. Tissue-Specific Phospho- and Glycolipid Accumulations Present in Glb1−/− Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Okada, S.; O’Brien, J.S. Generalized Gangliosidosis: Beta-Galactosidase Deficiency. Science 1968, 160, 1002–1004. [Google Scholar] [CrossRef] [PubMed]
- Baker, H.J.; Lindsey, J.R. Animal Model: Feline Gm1 Gangliosidosis. Am. J. Pathol. 1974, 74, 649–652. [Google Scholar] [PubMed]
- Barker, C.G.; Blakemore, W.F.; Dell, A.; Palmer, A.C.; Tiller, P.R.; Winchester, B.G. Gm1 Gangliosidosis (Type 1) in a Cat. Biochem. J. 1986, 235, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, H.J., Jr.; Lindsey, J.R.; McKhann, G.M.; Farrell, D.F. Neuronal Gm1 Gangliosidosis in a Siamese Cat with Beta-Galactosidase Deficiency. Science 1971, 174, 838–839. [Google Scholar] [CrossRef]
- De Maria, R.; Divari, S.; Bo, S.; Sonnio, S.; Lotti, D.; Capucchio, M.T.; Castagnaro, M. Β-Galactosidase Deficiency in a Korat Cat: A New Form of Feline Gm1-Gangliosidosis. Acta Neuropathol. 1998, 96, 307–314. [Google Scholar] [CrossRef]
- Uddin, M.M.; Hossain, M.A.; Rahman, M.M.; Chowdhury, M.A.; Tanimoto, T.; Yabuki, A.; Hye-Sook, C.; Osamu, Y. Identification of Bangladeshi Domestic Cats with Gm1 Gangliosidosis Caused by the c.1448G<C Mutation of the Feline Glb1 Gene: Case Study. J. Vet. Med. Sci. 2013, 75, 395–397. [Google Scholar]
- Uddin, M.M.; Tanimoto, T.; Yabuki, A.; Kotani, T.; Kuwamura, M.; Chang, H.S.; Yamato, O. Mutation Analysis of Gm1 Gangliosidosis in a Siamese Cat from Japan in the 1960s. J. Feline Med. Surg. 2012, 14, 900–902. [Google Scholar] [CrossRef]
- Ueno, H.; Yamato, O.; Sugiura, T.; Kohyama, M.; Yabuki, A.; Miyoshi, K.; Matsuda, K.; Uchide, T. Gm1 Gangliosidosis in a Japanese Domestic Cat: A New Variant Identified in Hokkaido, Japan. J. Vet. Med. Sci. 2016, 78, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Alroy, J.; Orgad, U.; DeGasperi, R.; Richard, R.; Warren, C.D.; Knowles, K.; Thalhammer, J.G.; Raghavan, S.S. Canine Gm1-Gangliosidosis. A Clinical, Morphologic, Histochemical, and Biochemical Comparison of Two Different Models. Am. J. Pathol. 1992, 140, 675–689. [Google Scholar]
- Uddin, M.M.; Arata, S.; Takeuchi, Y.; Chang, H.S.; Mizukami, K.; Yabuki, A.; Rahman, M.M.; Kohyama, M.; Hossain, M.A.; Takayama, K.; et al. Molecular Epidemiology of Canine Gm1 Gangliosidosis in the Shiba Inu Breed in Japan: Relationship between Regional Prevalence and Carrier Frequency. BMC Vet. Res. 2013, 9, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Kreutzer, R.; Leeb, T.; Müller, G.; Moritz, A.; Baumgärtner, W. A Duplication in the Canine Beta-Galactosidase Gene Glb1 Causes Exon Skipping and Gm1-Gangliosidosis in Alaskan Huskies. Genetics 2005, 170, 1857–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, D.H.; Harrington, D.D.; Keenana, T.W.; Hinsman, E.J. Neuronal-Visceral Gm1 Gangliosidosis in a Dog with Β-Galactosidase Deficiency. Science 1976, 194, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Ahern-Rindell, A.J.; Prieur, D.J.; Murnane, R.D.; Raghavan, S.S.; Daniel, P.F.; McCluer, R.H.; Walkley, S.U.; Parish, S.M. Inherited Lysosomal Storage Disease Associated with Deficiencies of Beta-Galactosidase and Alpha-Neuraminidase in Sheep. Am. J. Med. Genet. 1988, 31, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Murnane, R.D.; Prieur, D.J.; Ahern-Rindell, A.J.; Holler, L.D.; Parish, S.M. Clinical and Clinicopathologic Characteristics of Ovine Gm-1 Gangliosidosis. J. Vet. Intern. Med. 1994, 8, 221–223. [Google Scholar] [CrossRef]
- Skelly, B.J.; Jeffrey, M.; Franklin, R.J.; Winchester, B.G. A New Form of Ovine Gm1-Gangliosidosis. Acta Neuropathol. 1995, 89, 374–379. [Google Scholar] [CrossRef]
- Ryder, S.J.; Simmons, M.M. A Lysosomal Storage Disease of Romney Sheep That Resembles Human Type 3 Gm1 Gangliosidosis. Acta Neuropathol. 2001, 101, 225–228. [Google Scholar] [CrossRef]
- Donnelly, W.J.; Sheahan, B.J.; Kelly, M. Β-Galactosidase Deficiency in Gm1 Gangliosidosis of Friesian Calves. Res. Vet. Sci. 1973, 15, 139–141. [Google Scholar] [CrossRef]
- Sheahan, B.J.; Donnelly, W.J. Enzyme Histochemical and Ultrastructural Alterations in the Brains of Friesian Calves with Gm1 Gangliosidosis. Acta Neuropathol. 1974, 30, 73–84. [Google Scholar] [CrossRef]
- Bermudez, A.J.; Johnson, G.C.; Vanier, M.T.; Schroder, M.; Suzuki, K.; Stogsdill, P.L.; Johnson, G.S.; O’Brien, D.; Moore, C.P.; Fry, W.W. Gangliosidosis in Emus (Dromaius Novaehollandiae). Avian Dis. 1995, 39, 292–303. [Google Scholar] [CrossRef]
- Bermudez, A.J.; Freischütz, B.; Yu, R.K.; Nonneman, D.; Johnson, G.S.; Boon, G.D.; Stogsdill, P.L.; Ledoux, D.R. Heritability and Biochemistry of Gangliosidosis in Emus (Dromaius Novaehollandiae). Avian Dis. 1997, 41, 838–849. [Google Scholar] [CrossRef]
- Muthupalani, S.; Torres, P.A.; Wang, B.C.; Zeng, B.J.; Eaton, S.; Erdelyi, I.; Ducore, R.; Maganti, R.; Keating, J.; Perry, B.J.; et al. Gm1-Gangliosidosis in American Black Bears: Clinical, Pathological, Biochemical and Molecular Genetic Characterization. Mol. Genet. Metab. 2014, 111, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.N.; Martin, M.d.; Schroder, M.; Vanier, M.T.; Hara, Y.; Suzuki, K.; Suzuki, K.; d’Azzo, A. Generalized Cns Disease and Massive Gm1-Ganglioside Accumulation in Mice Defective in Lysosomal Acid Beta-Galactosidase. Hum. Mol. Genet. 1997, 6, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.; Suzuki, O.; Oshima, A.; Ogura, A.; Noguchi, Y.; Yamamoto, Y.; Asano, T.; Takimoto, K.; Sukegawa, K.; Suzuki, Y.; et al. Β-Galactosidase-Deficient Mouse as an Animal Model for Gm1-Gangliosidosis. Glycoconj J. 1997, 14, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, M.J.; Ou, L.; Tabaran, A.F.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; O’Sullivan, M.G.; Whitley, C.B. Comprehensive Behavioral and Biochemical Outcomes of Novel Murine Models of Gm1-Gangliosidosis and Morquio Syndrome Type B. Mol. Genet. Metab. 2019, 126, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Bidchol, A.M.; Dalal, A.; Trivedi, R.; Shukla, A.; Nampoothiri, S.; Sankar, V.H.; Danda, S.; Gupta, N.; Kabra, M.; Hebbar, S.A.; et al. Recurrent and Novel Glb1 Mutations in India. Gene 2015, 567, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Scaglia, F. Gm1 Gangliosidosis: Review of Clinical, Molecular, and Therapeutic Aspects. Mol. Genet. Metab. 2008, 94, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Caciotti, A.; Garman, S.C.; Rivera-Colon, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. Gm1 Gangliosidosis and Morquio B Disease: An Update on Genetic Alterations and Clinical Findings. Biochim. Biophys. Acta 2011, 1812, 782–790. [Google Scholar] [CrossRef]
- Celtikci, B.; Aydin, H.I.; Sivri, S.; Sönmez, M.; Topcu, M.; Özkara, H.A. Four Novel Mutations in the Β-Galactosidase Gene Identified in Infantile Type of Gm1 Gangliosidosis. Clin. Biochem. 2012, 45, 571–574. [Google Scholar] [CrossRef]
- Karimzadeh, P.; Naderi, S.; Modarresi, F.; Dastsooz, H.; Nemati, H.; Farokhashtiani, T.; Shamsian, B.S.; Inaloo, S.; Faghihi, M.A. Case Reports of Juvenile Gm1 Gangliosidosisis Type Ii Caused by Mutation in Glb1 Gene. BMC Med. Genet. 2017, 18, 73. [Google Scholar] [CrossRef] [Green Version]
- Tessitore, A.; del, P.M.M.; Sano, R.; Ma, Y.; Mann, L.; Ingrassia, A.; Laywell, E.D.; Steindler, D.A.; Hendershot, L.M.; d’Azzo, A. Gm1-Ganglioside-Mediated Activation of the Unfolded Protein Response Causes Neuronal Death in a Neurodegenerative Gangliosidosis. Mol. Cell 2004, 15, 753–766. [Google Scholar] [CrossRef]
- D’Azzo, A.; Tessitore, A.; Sano, R. Gangliosides as Apoptotic Signals in Er Stress Response. Cell Death Differ. 2006, 13, 404–414. [Google Scholar] [CrossRef] [Green Version]
- van der Voorn, J.P.; Kamphorst, W.; van der Knaap, M.S.; Powers, J.M. The Leukoencephalopathy of Infantile Gm1 Gangliosidosis: Oligodendrocytic Loss and Axonal Dysfunction. Acta Neuropathol. 2004, 107, 539–545. [Google Scholar] [CrossRef]
- Folkerth, R.D. Abnormalities of Developing White Matter in Lysosomal Storage Diseases. J. Neuropathol. Exp. Neurol. 1999, 58, 887–902. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Higaki, K.; Kajimaki, K.; Otsuka, S.; Ninomiya, H.; Matsuda, J.; Ohno, K.; Suzuki, Y.; Nanba, E. Enhanced Autophagy and Mitochondrial Aberrations in Murine Gm1-Gangliosidosis. Biochem. Biophys. Res. Commun. 2008, 367, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. Gm1-Ganglioside Accumulation at the Mitochondria-Associated Er Membranes Links Er Stress to Ca2+-Dependent Mitochondrial Apoptosis. Mol. Cell 2009, 36, 500–511. [Google Scholar] [CrossRef] [Green Version]
- Jeyakumar, M. Central Nervous System Inflammation Is a Hallmark of Pathogenesis in Mouse Models of Gm1 and Gm2 Gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef] [Green Version]
- Sano, R.; Tessitore, A.; Ingrassia, A.; d’Azzo, A. Chemokine-Induced Recruitment of Genetically Modified Bone Marrow Cells into the Cns of Gm1-Gangliosidosis Mice Corrects Neuronal Pathology. Blood 2005, 106, 2259–2268. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Stryer Biochemie, 6th ed.; Elsevier GmbH: Munich, Germany, 2007. [Google Scholar]
- Futerman, A.H. Intracellular Trafficking of Sphingolipids: Relationship to Biosynthesis. Biochim. Biophys. Acta 2006, 1758, 1885–1892. [Google Scholar] [CrossRef] [Green Version]
- Kolter, T.; Sandhoff, K. Principles of Lysosomal Membrane Digestion: Stimulation of Sphingolipid Degradation by Sphingolipid Activator Proteins and Anionic Lysosomal Lipids. Annu. Rev. Cell Dev. Biol. 2005, 21, 81–103. [Google Scholar] [CrossRef] [Green Version]
- Skaper, S.D.; Leon, A.; Toffano, G. Ganglioside Function in the Development and Repair of the Nervous System. From Basic Science to Clinical Application. Mol. Neurobiol. 1989, 3, 173–199. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Milani, D.; Leon, A. Monosialoganglioside Gm1 Protects against Anoxia-Induced Neuronal Death in Vitro. Exp. Neurol. 1989, 106, 297–305. [Google Scholar] [CrossRef]
- Sandhoff, K.; Harzer, K. Gangliosides and Gangliosidoses: Principles of Molecular and Metabolic Pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [PubMed]
- Timur, Z.K.; Demir, S.A.; Marsching, C.; Sandhoff, R.; Seyrantepe, V. Neuraminidase-1 Contributes Significantly to the Degradation of Neuronal B-Series Gangliosides but Not to the Bypass of the Catabolic Block in Tay-Sachs Mouse Models. Mol. Genet. Metab. Rep. 2015, 4, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Phaneuf, D.; Wakamatsu, N.; Huang, J.Q.; Borowski, A.; Peterson, A.C.; Fortunato, S.R.; Ritter, G.; Igdoura, S.A.; Morales, C.R.; Benoit, G.; et al. Dramatically Different Phenotypes in Mouse Models of Human Tay-Sachs and Sandhoff Diseases. Hum. Mol. Genet. 1996, 5, 1–14. [Google Scholar] [CrossRef]
- Folkerth, R.D.; Alroy, J.; Bhan, I.; Kaye, E.M. Infantile Gm1 Gangliosidosis: Complete Morphology and Histochemistry of Two Autopsy Cases, with Particular Reference to Delayed Central Nervous System Myelination. Pediatr. Dev. Pathol. 2000, 3, 73–86. [Google Scholar] [CrossRef]
- Nada, R.; Gupta, K.; Lal, S.B.; Vasishta, R.K. An Autopsy Case of Infantile Gm1 Gangliosidosis with Adrenal Calcification. Metab. Brain Dis. 2011, 26, 307–310. [Google Scholar] [CrossRef]
- Müller, G.; Alldinger, S.; Moritz, A.; Zurbriggen, A.; Kirchhof, N.; Sewell, A.; Baumgärtner, W. Gm1-Gangliosidosis in Alaskan Huskies: Clinical and Pathologic Findings. Vet. Pathol. 2001, 38, 281–290. [Google Scholar] [CrossRef]
- Gray-Edwards, H.L.; Jiang, X.; Randle, A.N.; Taylor, A.R.; Voss, T.L.; Johnson, A.K.; McCurdy, V.J.; Sena-Esteves, M.; Ory, D.S.; Martin, D.R. Lipidomic Evaluation of Feline Neurologic Disease after Aav Gene Therapy. Mol. Ther. Methods Clin. Dev. 2017, 6, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Gray-Edwards, H.L.; Regier, D.S.; Shirley, J.L.; Randle, A.N.; Salibi, N.; Thomas, S.E.; Latour, Y.L.; Johnston, J.; Golas, G.; Maguire, A.S.; et al. Novel Biomarkers of Human Gm1 Gangliosidosis Reflect the Clinical Efficacy of Gene Therapy in a Feline Model. Mol. Ther. 2017, 25, 892–903. [Google Scholar] [CrossRef] [Green Version]
- Schalli, M.; Weber, P.; Tysoe, C.; Pabst, B.M.; Thonhofer, M.; Paschke, E.; Stutz, A.E.; Tschernutter, M.; Windischhofer, W.; Withers, S.G. A New Type of Pharmacological Chaperone for Gm1-Gangliosidosis Related Human Lysosomal Beta-Galactosidase: N-Substituted 5-Amino-1-Hydroxymethyl-Cyclopentanetriols. Bioorg. Med. Chem. Lett. 2017, 27, 3431–3435. [Google Scholar] [CrossRef]
- Schalli, M.; Tysoe, C.; Fischer, R.; Pabst, B.M.; Thonhofer, M.; Paschke, E.; Rappitsch, T.; Stutz, A.E.; Tschernutter, M.; Windischhofer, W.; et al. N-Substituted 5-Amino-1-Hydroxymethyl-Cyclopentanetriols: A New Family of Activity Promotors for a Gm1-Gangliosidosis Related Human Lysosomal Beta-Galactosidase Mutant. Carbohydr. Res. 2017, 443, 15–22. [Google Scholar] [CrossRef]
- Suzuki, Y. Chaperone Therapy Update: Fabry Disease, Gm1-Gangliosidosis and Gaucher Disease. Brain Dev. 2013, 35, 515–523. [Google Scholar] [CrossRef]
- Deodato, F.; Procopio, E.; Rampazzo, A.; Taurisano, R.; Donati, M.A.; Dionisi-Vici, C.; Caciotti, A.; Morrone, A.; Scarpa, M. The Treatment of Juvenile/Adult Gm1-Gangliosidosis with Miglustat May Reverse Disease Progression. Metab. Brain Dis. 2017, 32, 1529–1536. [Google Scholar] [CrossRef]
- Shield, J.P.; Stone, J.; Steward, C.G. Bone Marrow Transplantation Correcting Β-Galactosidase Activity Does Not Influence Neurological Outcome in Juvenile Gm1-Gangliosidosis. J. Inherit. Metab. Dis. 2005, 28, 797–798. [Google Scholar] [CrossRef]
- Condori, J.; Acosta, W.; Ayala, J.; Katta, V.; Flory, A.; Martin, R.; Radin, J.; Cramer, C.L.; Radin, D.N. Enzyme Replacement for Gm1-Gangliosidosis: Uptake, Lysosomal Activation, and Cellular Disease Correction Using a Novel Β-Galactosidase:Rtb Lectin Fusion. Mol. Genet. Metab. 2016, 117, 199–209. [Google Scholar] [CrossRef]
- Beck, M. Treatment Strategies for Lysosomal Storage Disorders. Dev. Med. Child. Neurol. 2018, 60, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Broekman, M.L.; Baek, R.C.; Comer, L.A.; Fernandez, J.L.; Seyfried, T.N.; Sena-Esteves, M. Complete Correction of Enzymatic Deficiency and Neurochemistry in the Gm1-Gangliosidosis Mouse Brain by Neonatal Adeno-Associated Virus-Mediated Gene Delivery. Mol. Ther. 2007, 15, 30–37. [Google Scholar] [CrossRef]
- Baek, R.C.; Broekman, M.L.; Leroy, S.G.; Tierney, L.A.; Sandberg, M.A.; d’Azzo, A.; Seyfried, T.N.; Sena-Esteves, M. Aav-Mediated Gene Delivery in Adult Gm1-Gangliosidosis Mice Corrects Lysosomal Storage in Cns and Improves Survival. PLoS ONE 2010, 5, e13468. [Google Scholar] [CrossRef] [Green Version]
- Hayward, C.; Patel, H.C.; Manohar, S.G.; Lyon, A.R. Gene Therapy for Gm1 Gangliosidosis: Challenges of Translational Medicine. Ann. Transl. Med. 2015, 3 (Suppl. 1), S28. [Google Scholar]
- Takaura, N.; Yagi, T.; Maeda, M.; Nanba, E.; Oshima, A.; Suzuki, Y.; Yamano, T.; Tanaka, A. Attenuation of Ganglioside Gm1 Accumulation in the Brain of Gm1 Gangliosidosis Mice by Neonatal Intravenous Gene Transfer. Gene Ther. 2003, 10, 1487–1493. [Google Scholar] [CrossRef] [Green Version]
- Weismann, C.M.; Ferreira, J.; Keeler, A.M.; Su, Q.; Qui, L.; Shaffer, S.A.; Xu, Z.; Gao, G.; Sena-Esteves, M. Systemic Aav9 Gene Transfer in Adult Gm1 Gangliosidosis Mice Reduces Lysosomal Storage in Cns and Extends Lifespan. Hum. Mol. Genet. 2015, 24, 4353–4364. [Google Scholar] [CrossRef] [Green Version]
- Gröne, A.; Alldinger, S.; Baumgärtner, W. Interleukin-1beta, -6, -12 and Tumor Necrosis Factor-Alpha Expression in Brains of Dogs with Canine Distemper Virus Infection. J. Neuroimmunol. 2000, 110, 20–30. [Google Scholar] [CrossRef]
- Vangipuram, M.; Ting, D.; Kim, S.; Diaz, R.; Schule, B. Skin Punch Biopsy Explant Culture for Derivation of Primary Human Fibroblasts. J. Vis. Exp. 2013, 77, e3779. [Google Scholar] [CrossRef]
- Kreutzer, R.; Kreutzer, M.; Sewell, A.C.; Techangamsuwan, S.; Leeb, T.; Baumgärtner, W. Impact of Beta-Galactosidase Mutations on the Expression of the Canine Lysosomal Multienzyme Complex. Biochim. Biophys. Acta 2009, 1792, 982–987. [Google Scholar] [CrossRef] [Green Version]
- Herzog, A.; Hartung, R.; Reuser, A.J.; Hermanns, P.; Runz, H.; Karabul, N.; Gökce, S.; Pohlenz, J.; Kampmann, C.; Lampe, C.; et al. A Cross-Sectional Single-Centre Study on the Spectrum of Pompe Disease, German Patients: Molecular Analysis of the Gaa Gene, Manifestation and Genotype-Phenotype Correlations. Orphanet J. Rare Dis. 2012, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Ichinomiya, S.; Watanabe, H.; Maruyama, K.; Toda, H.; Iwasaki, H.; Kurosawa, M.; Matsuda, J.; Suzuki, Y. Motor and Reflex Testing in Gm1-Gangliosidosis Model Mice. Brain Dev. 2007, 29, 210–216. [Google Scholar] [CrossRef]
- Crawley, J.N. Behavioral Phenotyping of Transgenic and Knockout Mice: Experimental Design and Evaluation of General Health, Sensory Functions, Motor Abilities, and Specific Behavioral Tests. Brain Res. 1999, 835, 18–26. [Google Scholar] [CrossRef]
- Matsuda, J.; Suzuki, O.; Oshima, A.; Ogura, A.; Naiki, M.; Suzuki, Y. Neurological Manifestations of Knockout Mice with Β-Galactosidase Deficiency. Brain Dev. 1997, 19, 19–20. [Google Scholar] [CrossRef]
- Gerhauser, I.; Alldinger, S.; Baumgärtner, W. Ets-1 Represents a Pivotal Transcription Factor for Viral Clearance, Inflammation, and Demyelination in a Mouse Model of Multiple Sclerosis. J. Neuroimmunol. 2007, 188, 86–94. [Google Scholar] [CrossRef]
- Kummerfeld, M.; Meens, J.; Haas, L.; Baumgärtner, W.; Beineke, A. Generation and Characterization of a Polyclonal Antibody for the Detection of Theiler’s Murine Encephalomyelitis Virus by Light and Electron Microscopy. J. Virol. Methods 2009, 160, 185–188. [Google Scholar] [CrossRef]
- Gerhauser, I.; Li, L.; Li, D.; Klein, S.; Elmarabet, S.A.; Deschl, U.; Kalkuhl, A.; Baumgärtner, W.; Ulrich, R.; Beineke, A. Dynamic Changes and Molecular Analysis of Cell Death in the Spinal Cord of Sjl Mice Infected with the Bean Strain of Theiler’s Murine Encephalomyelitis Virus. Apoptosis 2018, 23, 170–186. [Google Scholar] [CrossRef]
- Itoh, M.; Matsuda, J.; Suzuki, O.; Ogura, A.; Oshima, A.; Tai, T.; Suzuki, Y.; Takashima, S. Development of Lysosomal Storage in Mice with Targeted Disruption of the Β-Galactosidase Gene: A Model of Human Gm1-Gangliosidosis. Brain Dev. 2001, 23, 379–384. [Google Scholar] [CrossRef]
- Kreutzer, M.; Seehusen, F.; Kreutzer, R.; Pringproa, K.; Kummerfeld, M.; Claus, P.; Deschl, U.; Kalkul, A.; Beineke, A.; Baumgärtner, W.; et al. Axonopathy Is Associated with Complex Axonal Transport Defects in a Model of Multiple Sclerosis. Brain Pathol. 2012, 22, 454–471. [Google Scholar] [CrossRef]
- Nadeem, M.; Spitzbarth, I.; Haist, V.; Rohn, K.; Tauscher, K.; Rohn, K.; Bossers, A.; Langeveld, J.; Papasavva-Stylianou, P.; Groschup, M.H.; et al. Immunolabelling of Non-Phosphorylated Neurofilament Indicates Damage of Spinal Cord Axons in Tse-Infected Goats. Vet. Rec. 2016, 178, 141. [Google Scholar] [CrossRef]
- Herder, V.; Gerhauser, I.; Klein, S.K.; Almeida, P.; Kummerfeld, M.; Ulrich, R.; Seehusen, F.; Rohn, K.; Schaudien, D.; Baumgärtner, W.; et al. Interleukin-10 Expression During the Acute Phase Is a Putative Prerequisite for Delayed Viral Elimination in a Murine Model for Multiple Sclerosis. J. Neuroimmunol. 2012, 249, 27–39. [Google Scholar] [CrossRef]
- Tongtako, W.; Lehmbecker, A.; Wang, Y.; Hahn, K.; Baumgärtner, W.; Gerhauser, I. Canine Dorsal Root Ganglia Satellite Glial Cells Represent an Exceptional Cell Population with Astrocytic and Oligodendrocytic Properties. Sci Rep. 2017, 7, 13915. [Google Scholar] [CrossRef] [Green Version]
- Hortobagyi, T.; Wise, S.; Hunt, N.; Cary, N.; Djurovic, V.; Fegan-Earl, A.; Shorrock, K.; Rouse, D.; Al-Sarraj, S. Traumatic Axonal Damage in the Brain Can Be Detected Using Beta-App Immunohistochemistry within 35 Min after Head Injury to Human Adults. Neuropathol. Appl. Neurobiol. 2007, 33, 226–237. [Google Scholar] [CrossRef]
- Ammer, J.J.; Grothe, B.; Felmy, F. Late Postnatal Development of Intrinsic and Synaptic Properties Promotes Fast and Precise Signaling in the Dorsal Nucleus of the Lateral Lemniscus. J. Neurophysiol. 2012, 107, 1172–1185. [Google Scholar] [CrossRef] [Green Version]
- Franzen, D.L.; Gleiss, S.A.; Berger, C.; Kümpfbeck, F.S.; Ammer, J.J.; Felmy, F. Development and Modulation of Intrinsic Membrane Properties Control the Temporal Precision of Auditory Brain Stem Neurons. J. Neurophysiol. 2015, 113, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Rautenberg, P.L.; Grothe, B.; Felmy, F. Quantification of the Three-Dimensional Morphology of Coincidence Detector Neurons in the Medial Superior Olive of Gerbils During Late Postnatal Development. J. Comp. Neurol. 2009, 517, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Brogden, G.; Husein, D.M.; Steinberg, P.; Naim, H.Y. Isolation and Quantification of Sphingosine and Sphinganine from Rat Serum Revealed Gender Differences. Biomolecules 2019, 9, 459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogden, G.; Shammas, H.; Maalouf, K.; Naim, S.L.; Wetzel, G.; Amiri, M.; von Köckritz-Blickwede, M.; Das, A.M.; Naim, H.Y. Case Study on the Pathophysiology of Fabry Disease: Abnormalities of Cellular Membranes Can Be Reversed by Substrate Reduction in Vitro. Biosci. Rep. 2017, 37, BSR20160402. [Google Scholar] [CrossRef]
- Loos, B.; Toit, A.d.; Hofmeyr, J.H. Defining and Measuring Autophagosome Flux-Concept and Reality. Autophagy 2014, 10, 2087–2096. [Google Scholar] [CrossRef]
- Kandel, E.; Schwartz, H.; Jessell, T. Principles of Neural Science, 4th ed.; Butler, J., Lebowitz, H., Eds.; McGraw-Hill Education Ltd.: New York, NY, USA, 2000. [Google Scholar]
- Seehusen, F.; Baumgärtner, W. Axonal Pathology and Loss Precede Demyelination and Accompany Chronic Lesions in a Spontaneously Occurring Animal Model of Multiple Sclerosis. Brain Pathol. 2010, 20, 551–559. [Google Scholar] [CrossRef]
- Lempp, C.; Spitzbarth, I.; Puff, C.; Cana, A.; Kegler, K.; Techangamsuwan, S.; Baumgärtner, W.; Seehusen, F. New Aspects of the Pathogenesis of Canine Distemper Leukoencephalitis. Viruses 2014, 6, 2571–2601. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Wang, Z.; Li, H.; Wiese, M.; Zheng, H. App Physiological and Pathophysiological Functions: Insights from Animal Models. Cell Res. 2012, 22, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Coleman, M.P.; Adalbert, R.; Beirowski, B. Neuroprotective Strategies in Ms: Lessons from C57bl/Wlds Mice. J. Neurol. Sci. 2005, 233, 133–138. [Google Scholar] [CrossRef]
- Gudi, V.; Gai, L.; Herder, V.; Tejedor, L.S.; Kipp, M.; Amor, S.; Suhs, K.W.; Hansmann, F.; Beineke, A.; Baumgärtner, W.; et al. Synaptophysin Is a Reliable Marker for Axonal Damage. J. Neuropathol. Exp. Neurol. 2017, 76, 109–125. [Google Scholar] [CrossRef]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, N.; Noda, Y. Intracellular Transport and Kinesin Superfamily Proteins, Kifs: Structure, Function, and Dynamics. Physiol. Rev. 2008, 88, 1089–1118. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin Superfamily Motor Proteins and Intracellular Transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- Bock, P.; Spitzbarth, I.; Haist, V.; Stein, V.M.; Tipold, A.; Puff, C.; Beineke, A.; Baumgärtner, W. Spatio-Temporal Development of Axonopathy in Canine Intervertebral Disc Disease as a Translational Large Animal Model for Nonexperimental Spinal Cord Injury. Brain Pathol. 2013, 23, 82–99. [Google Scholar] [CrossRef]
- Dale, J.M.; Garcia, M.L. Neurofilament Phosphorylation During Development and Disease: Which Came First, the Phosphorylation or the Accumulation? J. Amino Acids 2012, 2012, e382107. [Google Scholar] [CrossRef] [Green Version]
- Petzold, A. Neurofilament Phosphoforms: Surrogate Markers for Axonal Injury, Degeneration and Loss. J. Neurol. Sci. 2005, 233, 183–198. [Google Scholar] [CrossRef] [Green Version]
- Grant, P.; Pant, H.C. Neurofilament Protein Synthesis and Phosphorylation. J. Neurocytol. 2000, 29, 843–872. [Google Scholar] [CrossRef]
- Yabe, J.T.; Jung, C.; Chan, W.K.; Shea, T.B. Phospho-Dependent Association of Neurofilament Proteins with Kinesin in Situ. Cell Motil. Cytoskelet. 2000, 45, 249–262. [Google Scholar] [CrossRef]
- Motil, J.; Chan, W.K.; Dubey, M.; Chaudhury, P.; Pimenta, A.; Chylinski, T.M.; Ortiz, D.T.; Shea, T.B. Dynein Mediates Retrograde Neurofilament Transport within Axons and Anterograde Delivery of Nfs from Perikarya into Axons: Regulation by Multiple Phosphorylation Events. Cell Motil. Cytoskelet. 2006, 63, 266–286. [Google Scholar] [CrossRef]
- Kummerfeld, M.; Seehusen, F.; Klein, S.; Ulrich, R.; Kreutzer, R.; Gerhauser, I.; Herder, V.; Baumgartner, W.; Beineke, A. Periventricular Demyelination and Axonal Pathology Is Associated with Subependymal Virus Spread in a Murine Model for Multiple Sclerosis. Intervirology 2012, 55, 401–416. [Google Scholar] [CrossRef]
- Spitzbarth, I.; Lempp, C.; Kegler, K.; Ulrich, R.; Kalkuhl, A.; Deschl, U.; Baumgartner, W.; Seehusen, F. Immunohistochemical and Transcriptome Analyses Indicate Complex Breakdown of Axonal Transport Mechanisms in Canine Distemper Leukoencephalitis. Brain Behav. 2016, 6, e00472. [Google Scholar] [CrossRef]
- Twelvetrees, A.E.; Pernigo, S.; Sanger, A.; Guedes-Dias, P.; Schiavo, G.; Steiner, R.A.; Dodding, M.P.; Holzbaur, E.L. The Dynamic Localization of Cytoplasmic Dynein in Neurons Is Driven by Kinesin-1. Neuron 2016, 90, 1000–1015. [Google Scholar] [CrossRef] [Green Version]
- Walkley, S.U.; Baker, H.J.; Rattazzi, M.C. Initiation and Growth of Ectopic Neurites and Meganeurites During Postnatal Cortical Development in Ganglioside Storage Disease. Dev. Brain Res. 1990, 51, 167–178. [Google Scholar] [CrossRef]
- Di Rocco, M.; Rossi, A.; Parenti, G.; Allegri, A.E.; Filocamo, M.; Pessagno, A.; Tortori-Donati, P.; Minetti, C.; Biancheri, R. Different Molecular Mechanisms Leading to White Matter Hypomyelination in Infantile Onset Lysosomal Disorders. Neuropediatrics 2005, 36, 265–269. [Google Scholar] [CrossRef]
- Erol, I.; Alehan, F.; Pourbagher, M.A.; Canan, O.; Yildirim, S.V. Neuroimaging Findings in Infantile Gm1 Gangliosidosis. Eur. J. Paediatr. Neurol. 2006, 10, 245–248. [Google Scholar] [CrossRef]
- Cathala, L.; Brickley, S.; Cull-Candy, S.; Farrant, M. Maturation of Epscs and Intrinsic Membrane Properties Enhances Precision at a Cerebellar Synapse. J. Neurosci. 2003, 23, 6074–6085. [Google Scholar] [CrossRef]
- Rodriguez-Molina, V.M.; Aertsen, A.; Heck, D.H. Spike Timing and Reliability in Cortical Pyramidal Neurons: Effects of Epsc Kinetics, Input Synchronization and Background Noise on Spike Timing. PLoS ONE 2007, 2, e319. [Google Scholar] [CrossRef]
- Zsiros, V.; Hestrin, S. Background Synaptic Conductance and Precision of Epsp-Spike Coupling at Pyramidal Cells. J. Neurophysiol. 2005, 93, 3248–3256. [Google Scholar] [CrossRef] [Green Version]
- van Meer, G.; Lisman, Q. Sphingolipid Transport: Rafts and Translocators. J. Biol. Chem. 2002, 277, 25855–25858. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Norton, W.T.; Poduslo, S.E. Myelination in Rat Brain: Method of Myelin Isolation. J. Neurochem. 1973, 21, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Jackman, N.; Ishii, A.; Bansal, R. Oligodendrocyte Development and Myelin Biogenesis: Parsing out the Roles of Glycosphingolipids. Physiology 2009, 24, 290–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, K.; Ando, S.; Yu, R.K. Gangliosides of Human, Cat, and Rabbit Spinal Cords and Cord Myelin. J. Lipid Res. 1978, 19, 863–871. [Google Scholar] [PubMed]
- Ikonen, E. Roles of Lipid Rafts in Membrane Transport. Curr. Opin. Cell Biol. 2001, 13, 470–477. [Google Scholar] [CrossRef]
- Kuech, E.M.; Brogden, G.; Naim, H.Y. Alterations in Membrane Trafficking and Pathophysiological Implications in Lysosomal Storage Disorders. Biochimie 2016, 130, 152–162. [Google Scholar] [CrossRef]
- Hering, H.; Lin, C.C.; Sheng, M. Lipid Rafts in the Maintenance of Synapses, Dendritic Spines, and Surface Ampa Receptor Stability. J. Neurosci. 2003, 23, 3262–3271. [Google Scholar] [CrossRef] [Green Version]
- Kreutzer, R.; Kreutzer, M.; Propsting, M.J.; Sewell, A.C.; Leeb, T.; Naim, H.Y.; Baumgärtner, W. Insights into Post-Translational Processing of Beta-Galactosidase in an Animal Model Resembling Late Infantile Human G-Gangliosidosis. J. Cell Mol. Med. 2008, 12, 1661–1671. [Google Scholar] [CrossRef] [Green Version]
- Lei, H.L.; Ye, J.; Qiu, W.J.; Zhang, H.W.; Han, L.S.; Wang, Y.; Gu, X.F. Beta-Galactosidase Deficiencies and Novel Glb1 Mutations in Three Chinese Patients with Morquio B Disease or Gm1 Gangliosidosis. World J. Pediatrics 2012, 8, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.M.; Adams, D.A.; Markello, T.; Golas, G.; Yang, S.; Sincan, M.; Simeonov, D.R.; Fajardo, K.F.; Hansen, N.F.; Cherukuri, P.F.; et al. Exome Sequencing as a Diagnostic Tool in a Case of Undiagnosed Juvenile-Onset Gm1-Gangliosidosis. Neurology 2012, 79, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, M.F.; Lacerda, L.; Macedo-Ribeiro, S.; Baptista, E.; Ribeiro, H.; Prata, M.J.; Alves, S. Lysosomal Multienzymatic Complex-Related Diseases: A Genetic Study among Portuguese Patients. Clin. Genet. 2012, 81, 379–393. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to Silence and Understanding Nonsense: Exonic Mutations That Affect Splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Kwak, J.E.; Son, M.Y.; Son, Y.S.; Son, M.J.; Cho, Y.S. Biochemical and Molecular Characterization of Novel Mutations in Glb1 and Neu1 in Patient Cells with Lysosomal Storage Disorders. Biochem. Biophys. Res. Commun. 2015, 457, 554–560. [Google Scholar] [CrossRef]
- Denny, C.A.; Alroy, J.; Pawlyk, B.S.; Sandberg, M.A.; d’Azzo, A.; Seyfried, T.N. Neurochemical, Morphological, and Neurophysiological Abnormalities in Retinas of Sandhoff and Gm1 Gangliosidosis Mice. J. Neurochem. 2007, 101, 1294–1302. [Google Scholar] [CrossRef]
- Heinecke, K.A.; Luoma, A.; d’Azzo, A.; Kirschner, D.A.; Seyfried, T.N. Myelin Abnormalities in the Optic and Sciatic Nerves in Mice with Gm1-Gangliosidosis. ASN Neuro 2015, 7, e1759091415568913. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1st Antibody | Clonality | Dilution | Manufacturer | 2nd Antibody |
|---|---|---|---|---|
| Amyloid precursor protein (APP), MAB348; 22C11 | Mouse monoclonal | 1:2000 | Millipore, Burlington, USA | Goat-anti- mouse (GAM, BioLogo, BA-9200) |
| 2′,3′-Cyclic-nucleotide-3′-phosphodiesterase (CNPase), 11-5B | 1:100 | |||
| Dynein, MMS-400P | 1:25 | Covance Inc., Princeton, USA | None | |
| Non-phosphorylated neurofilament (nNF), SMI-311 | 1:8000 | Calbiochem, Merck KGaA, Darmstadt, Germany | Goat-anti- mouse (GAM, BioLogo, BA-9200) | |
| Phosphorylated neurofilament (pNF), SMI-312 | 1:8000 | Sternberger Monoclonals Incorporated, MD, USA | ||
| Glial fibrillary acidic protein (GFAP), 6F2 | Rabbit polyclonal | 1:1000 | Dako/Agilent Technologies, Santa Clara, CA, USA | Goat-anti- rabbit (GAR, 1:200, BioLogo, BA-1000) |
| Iba1, PA5-27436 | 1:1000 | Thermo Electron LED GmbH, Langenselbold, Germany | ||
| Kinesin, K0889-100UG | 1:400 | Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany | ||
| Myelin basic protein (MBP), AB980 | 1:500 | Millipore, Burlington, USA | ||
| Periaxin (PRX), HPA001868 | 1:5000 | Sigma-Aldrich, Taufkirchen, Germany |
| Tests for General Health Condition | ||
| Test | Grade | Points |
| General appearance | Normal posture, smooth and shiny hair | 0 |
| Normal posture, dull and shaggy hair | 1 | |
| Mildly crooked back, dull and shaggy hair | 2 | |
| Severely crooked back, dull, shaggy and dirty hair, incontinence | 3 | |
| Behavior and activity | Attentive and curious | 0 |
| Very calm, mildly reduced spontaneous activity, unreduced induced activity | 1 | |
| Apathy, moderately reduced spontaneous activity, mildly reduced, induced activity | 2 | |
| Stupor, no spontaneous activity, little induced activity | 3 | |
| Gait | Normal gait | 0 |
| Mild ataxia, occasionally mild unsteady gait | 1 | |
| Moderate ataxia, frequently mild to moderate unsteady gait, mild staggering and stumbling | 2 | |
| Severe ataxia, frequently moderate to severe unsteady gait | 3 | |
| Neurologic Tests for the Characterization of GM1-Gangliosidosis | ||
| Test | Grade | Points |
| “Parachute” reflex | Extension and abduction of the hindlimbs, extension of the knee | 0 |
| Mildly delayed reaction, intermitting extension of the knee | 1 | |
| Moderately delayed reaction, flexion and adduction of the hind limbs, slow movement | 2 | |
| No reaction, continuous flexing and adducting of the hind limbs | 3 | |
| Grid walking | Animal does not step into mesh circuit | 0 |
| 21–30 s until stepping into mesh circuit | 1 | |
| 11–20 s until stepping into mesh circuit | 2 | |
| 0–10 s until stepping into mesh circuit | 3 | |
| Hang test | Mouse is able to hang horizontally upside down at a grid for more than 30 s | 0 |
| Mouse adheres for 21–30 s | 1 | |
| Mouse adheres for 11–20 s | 2 | |
| Mouse adheres for 10–0 s | 3 | |
| Avoidance behavior after pinching the base of the tail | Strong reaction, squeaking | 0 |
| Mildly delayed reaction | 1 | |
| Turning of the trunk, extension of the hindlimbs | 2 | |
| No reaction | 3 | |
| Correction of the body position after turning onto the back | Immediate correction | 0 |
| Mildly delayed correction | 1 | |
| Moderate to severely delayed correction | 2 | |
| No correction | 3 | |
| Total | 24 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eikelberg, D.; Lehmbecker, A.; Brogden, G.; Tongtako, W.; Hahn, K.; Habierski, A.; Hennermann, J.B.; Naim, H.Y.; Felmy, F.; Baumgärtner, W.; et al. Axonopathy and Reduction of Membrane Resistance: Key Features in a New Murine Model of Human GM1-Gangliosidosis. J. Clin. Med. 2020, 9, 1004. https://doi.org/10.3390/jcm9041004
Eikelberg D, Lehmbecker A, Brogden G, Tongtako W, Hahn K, Habierski A, Hennermann JB, Naim HY, Felmy F, Baumgärtner W, et al. Axonopathy and Reduction of Membrane Resistance: Key Features in a New Murine Model of Human GM1-Gangliosidosis. Journal of Clinical Medicine. 2020; 9(4):1004. https://doi.org/10.3390/jcm9041004
Chicago/Turabian StyleEikelberg, Deborah, Annika Lehmbecker, Graham Brogden, Witchaya Tongtako, Kerstin Hahn, Andre Habierski, Julia B. Hennermann, Hassan Y. Naim, Felix Felmy, Wolfgang Baumgärtner, and et al. 2020. "Axonopathy and Reduction of Membrane Resistance: Key Features in a New Murine Model of Human GM1-Gangliosidosis" Journal of Clinical Medicine 9, no. 4: 1004. https://doi.org/10.3390/jcm9041004
APA StyleEikelberg, D., Lehmbecker, A., Brogden, G., Tongtako, W., Hahn, K., Habierski, A., Hennermann, J. B., Naim, H. Y., Felmy, F., Baumgärtner, W., & Gerhauser, I. (2020). Axonopathy and Reduction of Membrane Resistance: Key Features in a New Murine Model of Human GM1-Gangliosidosis. Journal of Clinical Medicine, 9(4), 1004. https://doi.org/10.3390/jcm9041004