Detection of Platelet-Activating Antibodies Associated with Heparin-Induced Thrombocytopenia

 ,
,
Abstract
:1. Introduction
2. From Pathogenesis of HIT to Functional Tests for the Detection of Clinically Relevant Antibodies
3. Shared Aspects of Functional Assays
3.1. Test Platelet Preparation
3.2. Platelet Donor Selection
3.3. Preparation of Samples from Patients with Suspected HIT
3.4. Heparin Concentrations
3.5. Platelets, Patient Samples, and Heparin: Volumes to Be Used
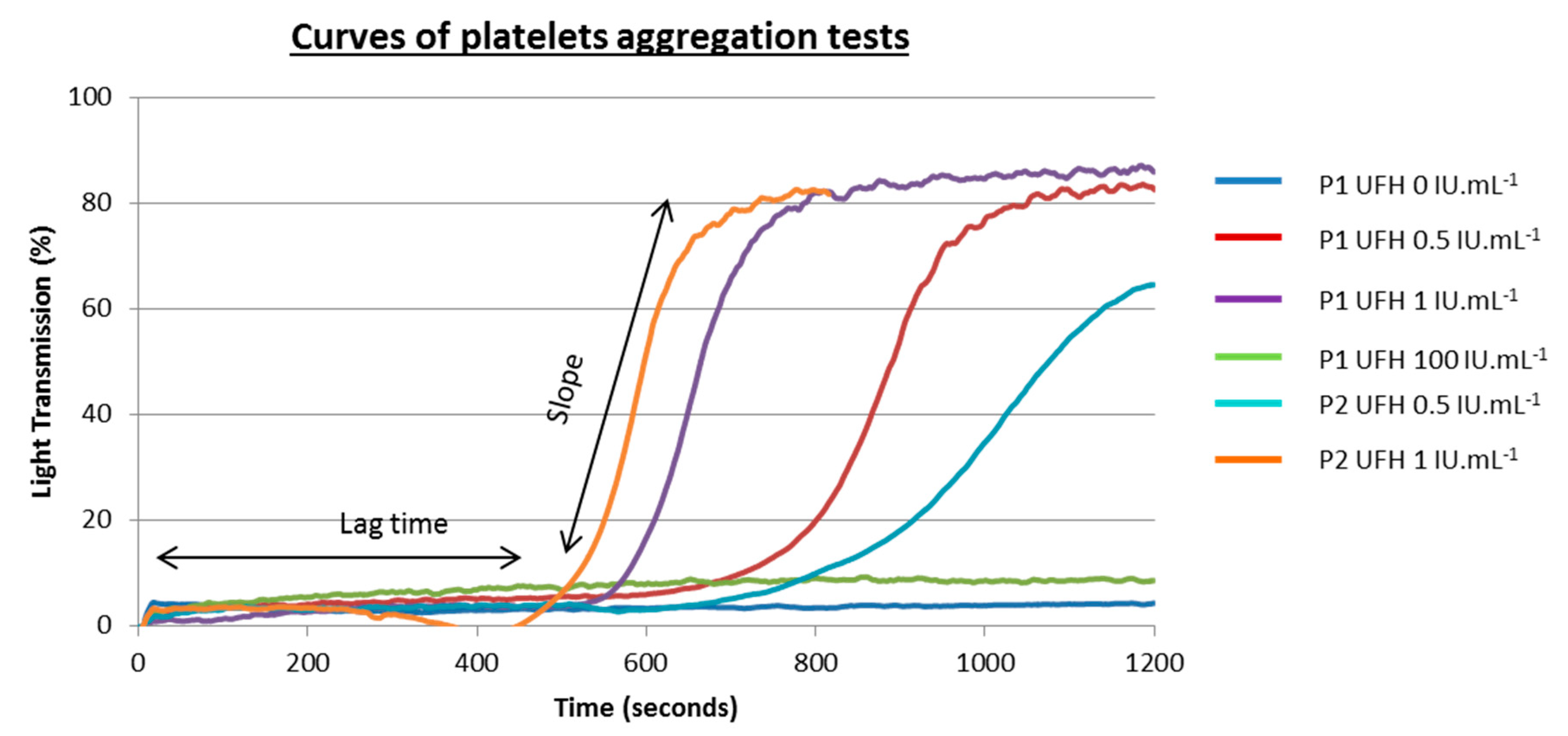
4. Optical Aggregometry
5. Flow Cytometric Assays
6. Heparin-Induced Platelet Activation (HIPA)
7. Whole Blood Aggregometry
Potential Pitfalls in Interpretation of the HIMEA
8. Serotonin Release Assay
9. Other Functional Assays
10. Shared Difficulties when Interpreting Functional Tests
10.1. Persistent Platelet Activation with Supratherapeutic Heparin Concentration
10.2. Interpretation of Platelet Activation by HIT Antibodies in the Absence of Heparin
11. Quality Controls
12. Comparison of Washed Platelet and PRP Activation Assays
13. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Greinacher, A.; Kohlmann, T.; Strobel, U.; Sheppard, J.A.; Warkentin, T.E. The temporal profile of the anti-PF4/heparin immune response. Blood 2009, 113, 4970–4976. [Google Scholar] [CrossRef]
- Cines, D.B.; Tomaski, A.; Tannenbaum, S. Immune endothelial-cell injury in heparin-associated thrombocytopenia. N. Engl. J. Med. 1987, 316, 581–589. [Google Scholar] [CrossRef]
- Pouplard, C.; Iochmann, S.; Renard, B.; Herault, O.; Colombat, P.; Amiral, J.; Gruel, Y. Induction of monocyte tissue factor expression by antibodies to heparin-platelet factor 4 complexes developed in heparin-induced thrombocytopenia. Blood 2001, 97, 3300–3302. [Google Scholar] [CrossRef] [Green Version]
- Arepally, G.M. Heparin-induced thrombocytopenia. Blood 2017, 129, 2864–2872. [Google Scholar] [CrossRef]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greinacher, A.; Selleng, S. How I evaluate and treat thrombocytopenia in the intensive care unit patient. Blood 2016, 128, 3032–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuker, A. Clinical and laboratory diagnosis of heparin-induced thrombocytopenia: An integrated approach. Semin. Thromb. Hemost. 2014, 40, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Kelton, J.G.; Warkentin, T.E. Heparin-induced thrombocytopenia: A historical perspective. Blood 2008, 112, 2607–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaloro, E.J.; McCaughan, G.; Mohammed, S.; Lau, K.K.E.; Gemmell, R.; Cavanaugh, L.; Donikian, D.; Kondo, M.; Brighton, T.; Pasalic, L. HIT or miss? A comprehensive contemporary investigation of laboratory tests for heparin induced thrombocytopenia. Pathology 2018, 50, 426–436. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Greinacher, A.; Gruel, Y.; Aster, R.H.; Chong, B.H. Laboratory testing for heparin-induced thrombocytopenia: A conceptual framework and implications for diagnosis. Semin. Thromb. Hemost. JTH 2011, 9, 2498–2500. [Google Scholar] [CrossRef]
- Amiral, J.; Marfaing-Koka, A.; Wolf, M.; Alessi, M.C.; Tardy, B.; Boyer-Neumann, C.; Vissac, A.M.; Fressinaud, E.; Poncz, M.; Meyer, D. Presence of autoantibodies to interleukin-8 or neutrophil-activating peptide-2 in patients with heparin-associated thrombocytopenia. Blood 1996, 88, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regnault, V.; de Maistre, E.; Carteaux, J.P.; Gruel, Y.; Nguyen, P.; Tardy, B.; Lecompte, T. Platelet activation induced by human antibodies to interleukin-8. Blood 2003, 101, 1419–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauova, L.; Poncz, M.; McKenzie, S.E.; Reilly, M.P.; Arepally, G.; Weisel, J.W.; Nagaswami, C.; Cines, D.B.; Sachais, B.S. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005, 105, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Rauova, L.; Zhai, L.; Kowalska, M.A.; Arepally, G.M.; Cines, D.B.; Poncz, M. Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis: Diagnostic and therapeutic implications. Blood 2006, 107, 2346–2353. [Google Scholar] [CrossRef] [PubMed]
- Pouplard, C.; May, M.A.; Iochmann, S.; Amiral, J.; Vissac, A.M.; Marchand, M.; Gruel, Y. Antibodies to platelet factor 4-heparin after cardiopulmonary bypass in patients anticoagulated with unfractionated heparin or a low-molecular-weight heparin: Clinical implications for heparin-induced thrombocytopenia. Circulation 1999, 99, 2530–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolovic, M.; Pratt, A.K.; Vukicevic, V.; Sarumi, M.; Johnson, L.S.; Shah, N.S. Platelet Count Trends and Prevalence of Heparin-Induced Thrombocytopenia in a Cohort of Extracorporeal Membrane Oxygenator Patients. Crit. Care Med. 2016, 44, e1031–e1037. [Google Scholar] [CrossRef]
- Vayne, C.; May, M.A.; Bourguignon, T.; Lemoine, E.; Guery, E.A.; Rollin, J.; Gruel, Y.; Pouplard, C. Frequency and Clinical Impact of Platelet Factor 4-Specific Antibodies in Patients Undergoing Extracorporeal Membrane Oxygenation. Thromb. Haemost. 2019, 119, 1138–1146. [Google Scholar] [CrossRef]
- Chen, J.; Dong, J.F.; Sun, C.; Bergeron, A.; McBride, L.; Pillai, M.; Barnard, M.R.; Salmon, J.; Michelson, A.D.; Bray, P.F. Platelet FcgammaRIIA His131Arg polymorphism and platelet function: Antibodies to platelet-bound fibrinogen induce platelet activation. J. Thromb. Haemost. 2003, 1, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Rollin, J.; Pouplard, C.; Gratacap, M.P.; Leroux, D.; May, M.A.; Aupart, M.; Gouilleux-Gruart, V.; Payrastre, B.; Gruel, Y. Polymorphisms of protein tyrosine phosphatase CD148 influence FcgammaRIIA-dependent platelet activation and the risk of heparin-induced thrombocytopenia. Blood 2012, 120, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Rollin, J.; Pouplard, C.; Sung, H.C.; Leroux, D.; Saada, A.; Gouilleux-Gruart, V.; Thibault, G.; Gruel, Y. Increased risk of thrombosis in FcgammaRIIA 131RR patients with HIT due to defective control of platelet activation by plasma IgG2. Blood 2015, 125, 2397–2404. [Google Scholar] [CrossRef]
- Rollin, J.; Pouplard, C.; Gruel, Y. Risk factors for heparin-induced thrombocytopenia: Focus on Fcgamma receptors. Thromb. Haemost. 2016, 116, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Amiral, J.; Wolf, M.; Fischer, A.; Boyer-Neumann, C.; Vissac, A.; Meyer, D. Pathogenicity of IgA and/or IgM antibodies to heparin-PF4 complexes in patients with heparin-induced thrombocytopenia. Br. J. Haematol. 1996, 92, 954–959. [Google Scholar] [CrossRef]
- Cattaneo, M.; Cerletti, C.; Harrison, P.; Hayward, C.P.; Kenny, D.; Nugent, D.; Nurden, P.; Rao, A.K.; Schmaier, A.H.; Watson, S.P.; et al. Recommendations for the Standardization of Light Transmission Aggregometry: A Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J. Thromb. Haemost. 2013, 11, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Alessi, M.C.; Sié, P.; Payrastre, B. Strengths and Weaknesses of Light Transmission Aggregometry in Diagnosing Hereditary Platelet Function Disorders. J. Clin. Med. 2020, 9, 763. [Google Scholar] [CrossRef] [Green Version]
- Menitove, J.E.; Frenzke, M.; Aster, R.H. Use of PGE1 for preparation of platelet concentrates. Transfusion 1986, 26, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Arnold, D.M.; Nazi, I.; Kelton, J.G. The platelet serotonin-release assay. Am. J. Hematol. 2015, 90, 564–572. [Google Scholar] [CrossRef]
- Greinacher, A.; Michels, I.; Kiefel, V.; Mueller-Eckhardt, C. A rapid and sensitive test for diagnosing heparin-associated thrombocytopenia. Thromb. Haemost. 1991, 66, 734–736. [Google Scholar] [CrossRef]
- Feinstein, M.B.; Fraser, C. Human platelet secretion and aggregation induced by calcium ionophores. Inhibition by PGE1 and dibutyryl cyclic AMP. J. Gen. Physiol. 1975, 66, 561–581. [Google Scholar] [CrossRef] [Green Version]
- Zahavi, M.; Zahavi, J.; Kakkar, V.V. The effect of pyridoxal-5-phosphate on the inhibition of platelet aggregation and adenosine-3’-5’-cyclic monophosphate accumulation in human platelets. Life Sci. 1984, 35, 1497–1503. [Google Scholar] [CrossRef]
- Kinlough-Rathbone, R.L.; Mustard, J.F.; Perry, D.W.; Dejana, E.; Cazenave, J.P.; Packham, M.A.; Harfenist, E.J. Factors influencing the deaggregation of human and rabbit platelets. Thromb. Haemost. 1983, 49, 162–167. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Hayward, C.P.; Smith, C.A.; Kelly, P.M.; Kelton, J.G. Determinants of donor platelet variability when testing for heparin-induced thrombocytopenia. J. Lab. Clin. Med. 1992, 120, 371–379. [Google Scholar]
- Harrison, P.; Mackie, I.; Mumford, A.; Briggs, C.; Liesner, R.; Winter, M.; Machin, S. Guidelines for the laboratory investigation of heritable disorders of platelet function. Br. J. Haematol. 2011, 155, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Chong, B.H.; Burgess, J.; Ismail, F. The clinical usefulness of the platelet aggregation test for the diagnosis of heparin-induced thrombocytopenia. Thromb. Haemost. 1993, 69, 344–350. [Google Scholar] [CrossRef]
- Morel-Kopp, M.C.; Mullier, F.; Gkalea, V.; Bakchoul, T.; Minet, V.; Elalamy, I.; Ward, C.M. Heparin-induced multi-electrode aggregometry method for heparin-induced thrombocytopenia testing: Communication from the SSC of the ISTH. Semin. Thromb. Hemost. JTH 2016, 14, 2548–2552. [Google Scholar] [CrossRef]
- Bruhns, P.; Jonsson, F. Mouse and human FcR effector functions. Immunol. Rev. 2015, 268, 25–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vayne, C.; Guery, E.A.; Charuel, N.; Besombes, J.; Lambert, W.C.; Rollin, J.; Gruel, Y.; Pouplard, C. Evaluation of functional assays for the diagnosis of heparin induced thrombocytopenia using 5B9, a monoclonal IgG that mimics human antibodies. J. Thromb. Haemost. 2020, 18, 968–975. [Google Scholar] [CrossRef]
- Greinacher, A.; Warkentin, T.E.; Chong, B.H. Heparin-Induced Thrombocytopenia. In Platelets, 4th ed.; Michelson, A.D., Cattaneo, M., Frelinger, A.L., Newman, P.J., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 741–767. [Google Scholar]
- Isenhart, C.E.; Brandt, J.T. Platelet aggregation studies for the diagnosis of heparin-induced thrombocytopenia. Am. J. Clin. Pathol. 1993, 99, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Michaut, L.; Laurent, N.; Kentsch, K.; Spindeldreher, S.; Deckert-Salva, F. Stability of anti-immunotherapeutic antibodies in frozen human serum samples. Bioanalysis 2014, 6, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S. Inhibition of in vitro platelet aggregation and release and fibrinolysis. Ann. Clin. Lab. Sci. 1989, 19, 260–265. [Google Scholar] [PubMed]
- Favaloro, E.J. Laboratory tests for identification or exclusion of heparin induced thrombocytopenia: HIT or miss? Am. J. Hematol. 2018, 93, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Minet, V.; Dogne, J.M.; Mullier, F. Functional Assays in the Diagnosis of Heparin-Induced Thrombocytopenia: A Review. Molecules 2017, 22, 617. [Google Scholar] [CrossRef] [Green Version]
- Brandt, E.; Ludwig, A.; Petersen, F.; Flad, H.D. Platelet-derived CXC chemokines: Old players in new games. Immunol. Rev. 2000, 177, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Newman, P.M.; Chong, B.H. Heparin-induced thrombocytopenia: New evidence for the dynamic binding of purified anti-PF4-heparin antibodies to platelets and the resultant platelet activation. Blood 2000, 96, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E.; Kelton, J.G. Temporal aspects of heparin-induced thrombocytopenia. N. Engl. J. Med. 2001, 344, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Chong, B.H.; Ismail, F.; Cade, J.; Gallus, A.S.; Gordon, S.; Chesterman, C.N. Heparin-induced thrombocytopenia: Studies with a new low molecular weight heparinoid, Org 10172. Blood 1989, 73, 1592–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauel, K.; Furll, B.; Warkentin, T.E.; Weitschies, W.; Kohlmann, T.; Sheppard, J.I.; Greinacher, A. Heparin-induced thrombocytopenia--therapeutic concentrations of danaparoid, unlike fondaparinux and direct thrombin inhibitors, inhibit formation of platelet factor 4-heparin complexes. J. Thromb. Haemost. 2008, 6, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Eekels, J.J.; Pachler, C.; Krause, N.; Muhr, T.; Waltl, G.; Greinacher, A. Ticagrelor causes false negative functional tests for heparin-induced thrombocytopenia. Blood 2020, 135, 875–878. [Google Scholar] [CrossRef]
- White, M.M.; Siders, L.; Jennings, L.K.; White, F.L. The effect of residual heparin on the interpretation of heparin-induced platelet aggregation in the diagnosis of heparin-associated thrombocytopenia. Thromb. Haemost. 1992, 68, 88. [Google Scholar] [CrossRef]
- Potzsch, B.; Keller, M.; Madlener, K.; Muller-Berghaus, G. The use of heparinase improves the specificity of crossreactivity testing in heparin-induced thrombocytopenia. Thromb. Haemost. 1996, 76, 1121. [Google Scholar] [CrossRef]
- Amiral, J.; Seghatchian, J. An update on evidence based diagnostic and confirmatory testing strategies for heparin induced thrombocytopenia using combined immunological and functional assays. Transfus. Apher. Sci. Off. J. World Apher. Assoc. Off. J. Eur. Soc. Haemapher. 2018, 57, 804–811. [Google Scholar] [CrossRef]
- Sheridan, D.; Carter, C.; Kelton, J.G. A diagnostic test for heparin-induced thrombocytopenia. Blood 1986, 67, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Morel-Kopp, M.C.; Tan, C.W.; Brighton, T.A.; McRae, S.; Baker, R.; Tran, H.; Mollee, P.; Kershaw, G.; Joseph, J.; Ward, C.; et al. Validation of whole blood impedance aggregometry as a new diagnostic tool for HIT: Results of a large Australian study. Thromb. Haemost. 2012, 107, 575–583. [Google Scholar]
- Baumgartel, M.W.; Eichler, P.; Glockner, W.M.; Ranze, O.; Greinacher, A. Heparin-induced thrombocytopenia (HIT): In vitro and in vivo cross-reactivity to danaparoid sodium and successful treatment with recombinant hirudin (lepirudin). Eur. J. Haematol. 2000, 65, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Horlait, G.; Minet, V.; Mullier, F.; Michaux, I. Persistent heparin-induced thrombocytopenia: Danaparoid cross-reactivity or delayed-onset heparin-induced thrombocytopenia? A case report. Blood Coagul. Fibrinolysis 2017, 28, 193–197. [Google Scholar] [CrossRef]
- Rijkers, M.; Saris, A.; Heidt, S.; Mulder, A.; Porcelijn, L.; Claas, F.H.J.; Bierings, R.; Leebeek, F.W.G.; Jansen, A.J.G.; Vidarsson, G.; et al. A subset of anti-HLA antibodies induces FcgammaRIIa-dependent platelet activation. Haematologica 2018, 103, 1741–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratantoni, J.C.; Pollet, R.; Gralnick, H.R. Heparin-induced thrombocytopenia: Confirmation of diagnosis with in vitro methods. Blood 1975, 45, 395–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaloro, E.J.; Bernal-Hoyos, E.; Exner, T.; Koutts, J. Heparin-induced thrombocytopenia: Laboratory investigation and confirmation of diagnosis. Pathology 1992, 24, 177–183. [Google Scholar] [CrossRef]
- Greinacher, A.; Amiral, J.; Dummel, V.; Vissac, A.; Kiefel, V.; Mueller-Eckhardt, C. Laboratory diagnosis of heparin-associated thrombocytopenia and comparison of platelet aggregation test, heparin-induced platelet activation test, and platelet factor 4/heparin enzyme-linked immunosorbent assay. Transfusion 1994, 34, 381–385. [Google Scholar] [CrossRef]
- Pouplard, C.; Amiral, J.; Borg, J.Y.; Laporte-Simitsidis, S.; Delahousse, B.; Gruel, Y. Decision analysis for use of platelet aggregation test, carbon 14-serotonin release assay, and heparin-platelet factor 4 enzyme-linked immunosorbent assay for diagnosis of heparin-induced thrombocytopenia. Am. J. Clin. Pathol. 1999, 111, 700–706. [Google Scholar] [CrossRef] [Green Version]
- Brodard, J.; Alberio, L.; Angelillo-Scherrer, A.; Nagler, M. Accuracy of heparin-induced platelet aggregation test for the diagnosis of heparin-induced thrombocytopenia. Thromb. Res. 2020, 185, 27–30. [Google Scholar] [CrossRef]
- Galea, V.; Khaterchi, A.; Robert, F.; Gerotziafas, G.; Hatmi, M.; Elalamy, I. Heparin-induced multiple electrode aggregometry is a promising and useful functional tool for heparin-induced thrombocytopenia diagnosis: Confirmation in a prospective study. Platelets 2013, 24, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Tomer, A. A sensitive and specific functional flow cytometric assay for the diagnosis of heparin-induced thrombocytopenia. Br. J. Haematol. 1997, 98, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poley, S.; Mempel, W. Laboratory diagnosis of heparin-induced thrombocytopenia: Advantages of a functional flow cytometric test in comparison to the heparin-induced platelet-activation test. Eur. J. Haematol. 2001, 66, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Garritsen, H.S.; Probst-Kepper, M.; Legath, N.; Eberl, W.; Samaniego, S.; Woudenberg, J.; Schuitemaker, J.H.; Kroll, H.; Gurney, D.A.; Moore, G.W.; et al. High sensitivity and specificity of a new functional flow cytometry assay for clinically significant heparin-induced thrombocytopenia antibodies. Int. J. Lab. Hematol. 2014, 36, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Mullier, F.; Minet, V.; Bailly, N.; Devalet, B.; Douxfils, J.; Chatelain, C.; Elalamy, I.; Dogne, J.M.; Chatelain, B. Platelet microparticle generation assay: A valuable test for immune heparin-induced thrombocytopenia diagnosis. Thromb. Res. 2014, 133, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Malicev, E.; Kozak, M.; Rozman, P. Evaluation of a flow cytometric assay for the confirmation of heparin-induced thrombocytopenia. Int. J. Lab. Hematol. 2016, 38, 240–245. [Google Scholar] [CrossRef]
- Tardy-Poncet, B.; Montmartin, A.; Piot, M.; Alkhalfioui, F.; Maes, H.; Tardy, B.; Group, G.-H.S. A standardized Functional Assay for Routine Reliable HIT diagnosis: A potential Alternative to the Serotonin Release Assay. Res. Pract. Thromb. Haemost. 2017, 1 (Suppl. 1), 1–1451. [Google Scholar]
- Cipok, M.; Tomer, A.; Elalamy, I.; Kirgner, I.; Dror, N.; Kay, S.; Deutsch, V.R. Pathogenic heparin-induced thrombocytopenia and thrombosis (HIT) antibodies determined by rapid functional flow cytometry. Eur. J. Haematol. 2019, 103, 225–233. [Google Scholar] [CrossRef]
- Althaus, K.; Pelzl, L.; Hidiatov, O.; Amiral, J.; Marini, I.; Bakchoul, T. Evaluation of a flow cytometer-based functional assay using platelet-rich plasma in the diagnosis of heparin-induced thrombocytopenia. Thromb. Res. 2019, 180, 55–61. [Google Scholar] [CrossRef]
- Eichler, P.; Budde, U.; Haas, S.; Kroll, H.; Loreth, R.M.; Meyer, O.; Pachmann, U.; Potzsch, B.; Schabel, A.; Albrecht, D.; et al. First workshop for detection of heparin-induced antibodies: Validation of the heparin-induced platelet-activation test (HIPA) in comparison with a PF4/heparin ELISA. Thromb. Haemost. 1999, 81, 625–629. [Google Scholar] [CrossRef]
- Minet, V.; Baudar, J.; Bailly, N.; Douxfils, J.; Laloy, J.; Lessire, S.; Gourdin, M.; Devalet, B.; Chatelain, B.; Dogne, J.M.; et al. Rapid exclusion of the diagnosis of immune HIT by AcuStar HIT and heparin-induced multiple electrode aggregometry. Thromb. Res. 2014, 133, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Baker, S.A.; Hall, E.T.; Gombar, S.; Bao, A.; Zehnder, J.L. Implementation of Whole-Blood Impedance Aggregometry for Heparin-Induced Thrombocytopenia Functional Assay and Case Discussion. Am. J. Clin. Pathol. 2019, 152, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Gkalea, V.; Khaterchi, A.; Levy, P.; Jourdi, G.; Elalamy, I. Prospective Evaluation of a Rapid Functional Assay for Heparin-Induced Thrombocytopenia Diagnosis in Critically Ill Patients. Crit. Care Med. 2019, 47, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Tomer, A.; Masalunga, C.; Abshire, T.C. Determination of heparin-induced thrombocytopenia: A rapid flow cytometric assay for direct demonstration of antibody-mediated platelet activation. Am. J. Hematol. 1999, 61, 53–61. [Google Scholar] [CrossRef]
- Jy, W.; Mao, W.W.; Horstman, L.L.; Valant, P.A.; Ahn, Y.S. A flow cytometric assay of platelet activation marker P-selectin (CD62P) distinguishes heparin-induced thrombocytopenia (HIT) from HIT with thrombosis (HITT). Thromb. Haemost. 1999, 82, 1255–1259. [Google Scholar] [CrossRef] [Green Version]
- Solano, C.; Mutsando, H.; Self, M.; Morel-Kopp, M.C.; Mollee, P. Using HitAlert flow cytometry to detect heparin-induced thrombocytopenia antibodies in a tertiary care hospital. Blood Coagul. 2013, 24, 365–370. [Google Scholar] [CrossRef]
- Eekels, J.J.M.; Althaus, K.; Bakchoul, T.; Kroll, H.; Kiefel, V.; Nazy, I.; Lee, L.S.; Sachs, U.; Warkentin, T.E.; Greinacher, A. An international external quality assessment for laboratory diagnosis of heparin-induced thrombocytopenia. Semin. Thromb. Hemost. JTH 2019, 17, 525–531. [Google Scholar] [CrossRef]
- Selleng, S.; Selleng, K.; Friesecke, S.; Grundling, M.; Kuhn, S.O.; Raschke, R.; Heidecke, O.J.; Hinz, C.; Hron, G.; Warkentin, T.E.; et al. Prevalence and clinical implications of anti-PF4/heparin antibodies in intensive care patients: A prospective observational study. J. Thromb. Thrombolysis 2015, 39, 60–67. [Google Scholar] [CrossRef]
- Elalamy, I.; Galea, V.; Hatmi, M.; Gerotziafas, G.T. Heparin-induced multiple electrode aggregometry: A potential tool for improvement of heparin-induced thrombocytopenia diagnosis. Semin. Thromb. Hemost. JTH 2009, 7, 1932–1934. [Google Scholar] [CrossRef]
- Greinacher, A.; Selleng, K.; Warkentin, T.E. Autoimmune heparin-induced thrombocytopenia. Semin. Thromb. Hemost. JTH 2017, 15, 2099–2114. [Google Scholar] [CrossRef] [Green Version]
- Pfueller, S.L.; Luscher, E.F. The effects of aggregated immunoglobulins on human blood platelets in relation to their complement-fixing abilities. II. Structural requirements of the immunoglobulin. J. Immunol. (Baltim. Md. 1950) 1972, 109, 526–533. [Google Scholar]
- Fouassier, M.; Bourgerette, E.; Libert, F.; Pouplard, C.; Marques-Verdier, A. Determination of serotonin release from platelets by HPLC and ELISA in the diagnosis of heparin-induced thrombocytopenia: Comparison with reference method by [C]-serotonin release assay. Semin. Thromb. Hemost. JTH 2006, 4, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Nazi, I.; Arnold, D.M.; Warkentin, T.E.; Smith, J.W.; Staibano, P.; Kelton, J.G. Distinguishing between anti-platelet factor 4/heparin antibodies that can and cannot cause heparin-induced thrombocytopenia. Semin. Thromb. Hemost. JTH 2015, 13, 1900–1907. [Google Scholar] [CrossRef] [PubMed]
- Vayne, C.; Guery, E.A.; Kizlik-Masson, C.; Rollin, J.; Bauters, A.; Gruel, Y.; Pouplard, C. Beneficial effect of exogenous platelet factor 4 for detecting pathogenic heparin-induced thrombocytopenia antibodies. Br. J. Haematol. 2017, 179, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W.; Etches, W.S.; Boshkov, L.K.; Gordon, P.A. Heparin-induced thrombocytopenia: An improved method of detection based on lumi-aggregometry. Br. J. Haematol. 1995, 91, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Nazi, I.; Arnold, D.M.; Smith, J.W.; Horsewood, P.; Moore, J.C.; Warkentin, T.E.; Crowther, M.A.; Kelton, J.G. FcgammaRIIa proteolysis as a diagnostic biomarker for heparin-induced thrombocytopenia. Semin. Thromb. Hemost. JTH 2013, 11, 1146–1153. [Google Scholar] [CrossRef]
- Mullier, F.; Bailly, N.; Cornet, Y.; Dubuc, E.; Robert, S.; Osselaer, J.C.; Chatelain, C.; Dogne, J.M.; Chatelain, B. Contribution of platelet microparticles generation assay to the diagnosis of type II heparin-induced thrombocytopenia. Thromb. Haemost. 2010, 103, 1277–1281. [Google Scholar]
- Tardy-Poncet, B.; Piot, M.; Chapelle, C.; France, G.; Campos, L.; Garraud, O.; Decousus, H.; Mismetti, P.; Tardy, B. Thrombin generation and heparin-induced thrombocytopenia. Semin. Thromb. Hemost. JTH 2009, 7, 1474–1481. [Google Scholar] [CrossRef]
- Cuker, A.; Rux, A.H.; Hinds, J.L.; Dela Cruz, M.; Yarovoi, S.V.; Brown, I.A.; Yang, W.; Konkle, B.A.; Arepally, G.M.; Watson, S.P.; et al. Novel diagnostic assays for heparin-induced thrombocytopenia. Blood 2013, 121, 3727–3732. [Google Scholar] [CrossRef] [Green Version]
- Prechel, M.M.; McDonald, M.K.; Jeske, W.P.; Messmore, H.L.; Walenga, J.M. Activation of platelets by heparin-induced thrombocytopenia antibodies in the serotonin release assay is not dependent on the presence of heparin. Semin. Thromb. Hemost. JTH 2005, 3, 2168–2175. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Medvedev, N.; Delcea, M.; Greinacher, A. Anti-platelet factor 4/polyanion antibodies mediate a new mechanism of autoimmunity. Nat. Commun. 2017, 8, 14945. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E. Laboratory diagnosis of heparin-induced thrombocytopenia. Int. J. Lab. Hematol. 2019, 41 (Suppl. 1), 15–25. [Google Scholar] [CrossRef] [Green Version]
- Kizlik-Masson, C.; Vayne, C.; McKenzie, S.E.; Poupon, A.; Zhou, Y.; Champier, G.; Pouplard, C.; Gruel, Y.; Rollin, J. 5B9, a monoclonal anti-platelet factor 4/heparin IgG with a human Fc fragment that mimics heparin-induced thrombocytopenia antibodies. Semin. Thromb. Hemost. JTH 2017, 15, 2065–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warkentin, T.E.; Nazy, I.; Sheppard, J.I.; Smith, J.W.; Kelton, J.G.; Arnold, D.M. Serotonin-release assay-negative heparin-induced thrombocytopenia. Am. J. Hematol. 2020, 95, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Cuker, A.; Arepally, G.M.; Chong, B.H.; Cines, D.B.; Greinacher, A.; Gruel, Y.; Linkins, L.A.; Rodner, S.B.; Selleng, S.; Warkentin, T.E.; et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: Heparin-induced thrombocytopenia. Blood Adv. 2018, 2, 3360–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruel, Y.; De Maistre, E.; Pouplard, C.; Mullier, F.; Susen, S.; Roullet, S.; Blais, N.; Le Gal, G.; Vincentelli, A.; Lasne, D.; et al. Diagnosis and management of heparin-induced thrombocytopenia. Anaesth. Crit. Care Pain Med. 2020. [Google Scholar] [CrossRef]
- Pouplard, C.; Gueret, P.; Fouassier, M.; Ternisien, C.; Trossaert, M.; Regina, S.; Gruel, Y. Prospective evaluation of the ‘4Ts’ score and particle gel immunoassay specific to heparin/PF4 for the diagnosis of heparin-induced thrombocytopenia. J. Thromb. Haemost. 2007, 5, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Barelli, S.; Zermatten, M.G.; Monnin-Respen, F.; Matthey-Guirao, E.; Nicolas, N.; Gomez, F.; Goodyer, M.; Gerschheimer, C.; Alberio, L. Rapid and Accurate Bayesian Diagnosis of Heparin-induced thrombocytopenia. Blood 2020, 135, 875–878. [Google Scholar] [CrossRef]
- Liederman, Z.; Van Cott, E.M.; Smock, K.; Meijer, P.; Selby, R. Heparin-induced thrombocytopenia: An international assessment of the quality of laboratory testing. J. Thromb. Haemost. 2019, 17, 2123–2130. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Greinacher, A. Platelet factor 4/heparin complexes present epitopes differently on solid-phase vs. platelet surfaces. Blood 2017, 129, 3498–3501. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Environment | Test | Instrument | Principle of the Assay: Endpoint | Advantages | Drawbacks |
|---|---|---|---|---|---|
| Whole Blood | Heparin-Induced Multiple Aggregometry (HIMEA) | Multiplate® analyzer | measurement of impedance increase due to platelet aggregation and deposition onto the electrodes | - no need for platelet isolation (whole blood) - easy to perform and rapid assay (one hour) - very good sensitivity: potential alternative to SRA | - need for each laboratory to determine its own cut-off - need for collection of donors’ blood into tubes containing hirudin |
| Platelet-Rich Plasma | Platelet Aggregation Test (PAT)/Light Transmission Aggregometry (LTA) | aggregometer (light transmission) | measurement of increase in light transmission due to platelet aggregation | - easily available in many laboratories | - performance highly dependent on the donor′s platelet (role of plasma environment) - time-consuming - large volumes needed |
| Flow Cytometry (FCM) | flow cytometer | measurement of increase in P-selectin or phosphatidylserine expression on an activated platelet (HIT Confirm® assay and HIT Alert®) | - commercialized marked- in vitro diagnostic assay - performed in 2–3 h | - lower sensitivity than the reference test (i.e., SRA) - performance highly dependent on the donor’s platelet (role of plasma environment) | |
| Washed Platelets | Serotonin Release Assay (SRA) | scintillation counter (β-radioactivity) | measurement of 14C-serotonin release from dense granules upon platelet activation | - rid of the inhibitory effect of plasma on platelet activation - considered “gold standard” | - platelet washing steps - use of radioelements (costly, specific agreement and premises) - time-consuming method: delayed results |
| Heparin-Induced Platelet Aggregation (HIPA) | 96-well plate with visual reading | visible macro-aggregates | - rid of the inhibitory effect of plasma on platelet activation | - platelet washing steps - lack of data on the performance - subjectivity of visual reading |
| Platelets | Patient’s Sample | Heparin | Other | Final Volume | Final Sample Dilution | |
|---|---|---|---|---|---|---|
| PAT | 135 (PRP) | 90 | 25 | 0 | 250 | 1/2.7 |
| SRA | 75 (WP) | 20 | 5 | 0 | 100 | 1/5 |
| HIPA | 75 (WP) | 20 | 10 | 0 | 100 | 1/5 |
| FCM (HIT Confirm®) | 10 (PRP) | 10 | 5 | 20 (labelled antibodies + buffer) | 50 | 1/5 |
| HIMEA | 300 (WB) | 200 | 20 | 100 (saline) | 620 | 1/3 |
| Functional Assay | Study | Positivity Criteria of the Test | Patients Studied | Criteria for the Diagnosis of HIT | Diagnostic Performance (*) | Comparison |
|---|---|---|---|---|---|---|
| PAT | A. Greinacher et al. [59] | Platelet aggregation > 25% with LCH and no aggregation in the presence of the buffer (4 random donors) | 209 patients with suspicion of HIT | A positive reaction with platelets of 2 or more donors | Not applicable (NA) | HIPA performed with platelets from same donors. Poor agreement between HIPA and PAT. |
| B. Chong et al. [33] | Platelet aggregation > 25% with LCH | Thrombocytopenia due to other causes (n = 20) Non-thrombocytopenic patients who received heparin (n = 17) Patients with HIT (n = 17) Healthy donors (n = 23) | Clinical diagnosis; n = 17 | Ss 39% with the least reactive donor Ss 81% with the most reactive donor Sp: 90% | with SRA | |
| C. Pouplard et al. [60] | Platelet aggregation > 20% with LCH and with a sharp slope (5 random donors) | 100 patients with clinical suspicion of HIT | Clinical diagnosis; n = 40 | Ss: 91% Sp: 77% | NA | |
| V. Galea et al. [62] | Maximal aggregation > 25% with LCH, no response in the presence of saline, and platelet aggregation inhibited with HCH | 200 consecutive patients with clinical suspicion of HIT | Clinical context and positive SRA; n = 21 | Ss: 76% Sp: 96% PPV: 80% NPV: 97% | NA | |
| J. Brodard et al. [61] | Platelet aggregation > 50% with LCH and with two out of four selected platelet donors | 122 patients with clinical suspicion of HIT and positive anti-PF4/H ELISA | Clinical context and positive HIPA; n = 39 | Ss: 69% Sp: 100% | NA | |
| Flow Cytometry Assay | A. Tomer et al. [63] | Annexin V binding. ≥6.6% platelet activation with LCH and inhibition with HCH Grey zone 6–6.6%. The number of platelet donors is not mentioned | 25 patients with clinical suspicion of HIT | Clinical diagnosis + and positive SRA; n = 19 | Ss: 95% Sp: 100% | SRA |
| S. Poley et al. [64] | Annexin V binding. >13% platelet activation with LCH and inhibition of platelet activation with HCH. Pooled platelets of selected donors | 248 patients with clinical suspicion of HIT | Clinical diagnosis and positive HIPA; n = 17 | Ss: 95% Sp: 96% | HIPA (4 donors) | |
| HS. Garritsen et al. [65] | Annexin V binding (HIT Alert®). ≥7.6% platelet activation in the presence of LCH and platelet activation reduced by ≥50% in the presence of HCH. One selected platelet donor | 346 patients with clinical suspicion of HIT | Clinical diagnosis; n = 17 | Ss: 88.2% Sp: 99.1% | For IgG ELISA negative sera: 98% agreement with HIT Alert®. For IgG ELSA positive sera: 52.7% agreement with HIT Alert® | |
| F. Mullier et al. [66] | Ratio PMP annexin V expression (LDH/HDH). One platelet donor only | 53 patients with clinical suspicion of HIT | Clinical diagnosis; n = 9 | Ss: 88.9% Sp: 100% | NA | |
| E. Malicev et al. [67] | >10% CD62P-positive platelets at LCH and ≥50% and inhibition of platelet activation at HDH. Two platelet donors | 41 patients with clinical suspicion of HIT and positive ELISA IgG | Clinical context and positive HIPA; n = 14 | Ss: 82% Sp: 83% | NA | |
| B. Tardy et al. [68] | P-selectin expression. >16.5% platelet activation with LCH and inhibition with HCH Two selected platelet donors | 228 patients with clinical suspicion | Expert opinion adjudication (clinical diagnosis + local laboratory results); n = 106 | Ss: 83% Sp: 97% | NA | |
| M. Cipok et al. [69] | ≥2-fold greater P-selectin expression than that of the normal control. One platelet donor only | 63 patients with clinical suspicion | Positive SRA; n = 21 | Ss: 90.5% Sp: 95% | NA | |
| K. Althaus et al. [70] | P-selectin expression (Emo-test HIT®). %HEPLA: >13.0%. Grey zone 9.6–13%. One unselected platelet donor only | 164 surgical or medical patients with clinical suspicion of HIT and positive EIA IgG | Positive HIPA; n = 33 | Ss: 69.7% Sp: 75.4% | NA | |
| HIPA | A. Greinacher et al. [27] | HIPA was positive if the suspension became transparent with LCH, but not with heparin HCH (4 random donors) | 34 patients with suspicion of HIT | Not applicable | Not applicable | Excellent agreement with SRA: Kappa = 0.85 Moderate agreement with PAT: Kappa = 0.46 |
| P. Eichler et al. [71] | HIPA was positive if the suspension became transparent with LCH, but not with HDH. A sample was judged positive if positive results were obtained with test platelets of at least 2 of the 4 donors | Workshop involving 9 laboratories with 8 samples: 2 from healthy blood donors, 5 from HIT patients (with HIT antibodies), 1 from a patient with sepsis | Not applicable | Not applicable | Expected results in 82% of cases | |
| HIMEA | M.C. Morel-Kopp et al. [34] | ISTH criteria | 181 patients with suspicion of HIT and positive EIA | Clinical context and positive SRA; n = 72 | Ss: 90.3% Sp: 89% | HIMEA and SRA were performed with the same good responder donors |
| V. Galea et al. [62] | AUC with LCH > 267 AU with a representative shape of a platelet aggregation curve and a decrease in the AUC value with HCH > 50% | 200 consecutive patients with suspicion of HIT | Clinical context and positive SRA; n = 21 | Ss: 81% Sp: 99% NPV: 98% PPV: 89% | NA | |
| V. Minet et al. [72] | Platelet aggregation occurred in the presence of LCH with a reduction of >80% with HCH | 116 patients with suspicion of HIT | 4Ts score and Accustar HIT; n = 2 | Ss: 100% Sp: 90% | NA | |
| J. Jin et al. [73] | AUC > 50 with LCH and AUC = 0 or inhibition of at least 50% of the AUC obtained with HCH | 70 patients with suspicion of HIT | 4Ts score > 4 and positive EIA IgG and positive SRA; n = 7 | Ss: 85% Sp: 98% | NA | |
| V. Galea et al. [74] | Aggregation curve at LCH was typical and AUC decreased by 50% or more with HDH | 87 patients with suspicion of HIT | Clinical context, positive SRA, and positive IgG ELISA; n = 12 | Ss: 91% Sp: 100% | NA | |
| SRA | D. Sheridan et al. [52] | Release > 20% with LCH and < 20% with HCH. One donor | 28 patients with suspicion of HIT 573 non-HIT patients | Clinical diagnosis: n = 6 | Ss: 100% Sp 99% | NA |
| C. Pouplard et al. [60] | Release > 20% with LCH and < 20% with HCH. One donor | 100 patients with suspicion of HIT | Clinical diagnosis: n = 40 | Ss: 88% Sp: 100% | NA | |
| B. Chong et al. [33] | Release > 20% with LCH and < 20% with HCH. One donor | Thrombocytopenia due to other causes (n = 20) | Clinical diagnosis: n = 17 | Ss: 65% with the least reactive donor Ss: 94% with the most reactive donor Sp: 90% | Comparison with PAT, Kappa = 0.60 | |
| F. Mullier et al. [66] | Release > 20% with LCH and < 20% with HCH or less than 50% of that observed with LCH. | 53 patients with suspicion of HIT | Clinical diagnosis: n = 9 | Ss: 88.9% Sp: 95.5% | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tardy, B.; Lecompte, T.; Mullier, F.; Vayne, C.; Pouplard, C. Detection of Platelet-Activating Antibodies Associated with Heparin-Induced Thrombocytopenia. J. Clin. Med. 2020, 9, 1226. https://doi.org/10.3390/jcm9041226
Tardy B, Lecompte T, Mullier F, Vayne C, Pouplard C. Detection of Platelet-Activating Antibodies Associated with Heparin-Induced Thrombocytopenia. Journal of Clinical Medicine. 2020; 9(4):1226. https://doi.org/10.3390/jcm9041226
Chicago/Turabian StyleTardy, Brigitte, Thomas Lecompte, François Mullier, Caroline Vayne, and Claire Pouplard. 2020. "Detection of Platelet-Activating Antibodies Associated with Heparin-Induced Thrombocytopenia" Journal of Clinical Medicine 9, no. 4: 1226. https://doi.org/10.3390/jcm9041226
APA StyleTardy, B., Lecompte, T., Mullier, F., Vayne, C., & Pouplard, C. (2020). Detection of Platelet-Activating Antibodies Associated with Heparin-Induced Thrombocytopenia. Journal of Clinical Medicine, 9(4), 1226. https://doi.org/10.3390/jcm9041226