Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease

 ,
,  ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
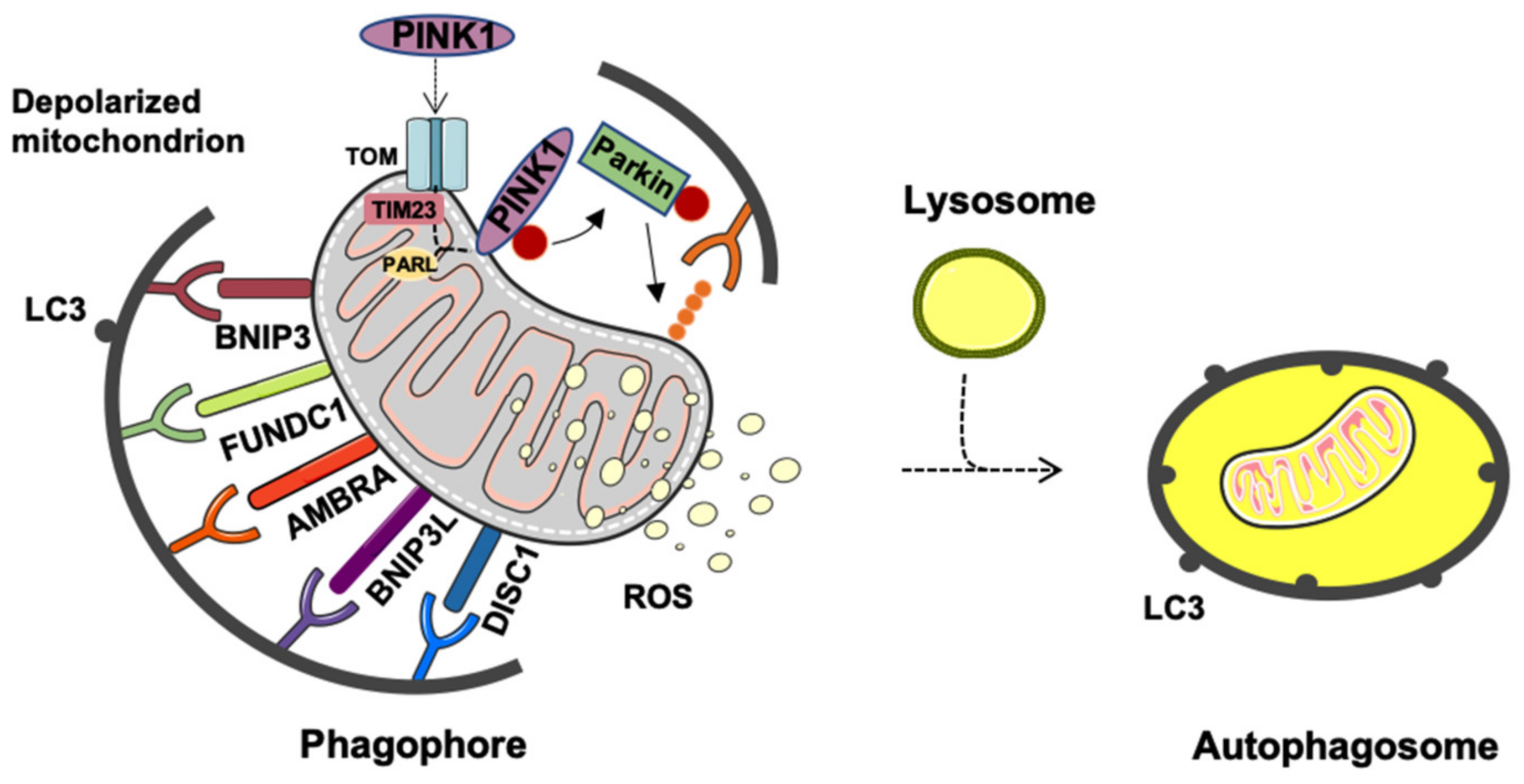
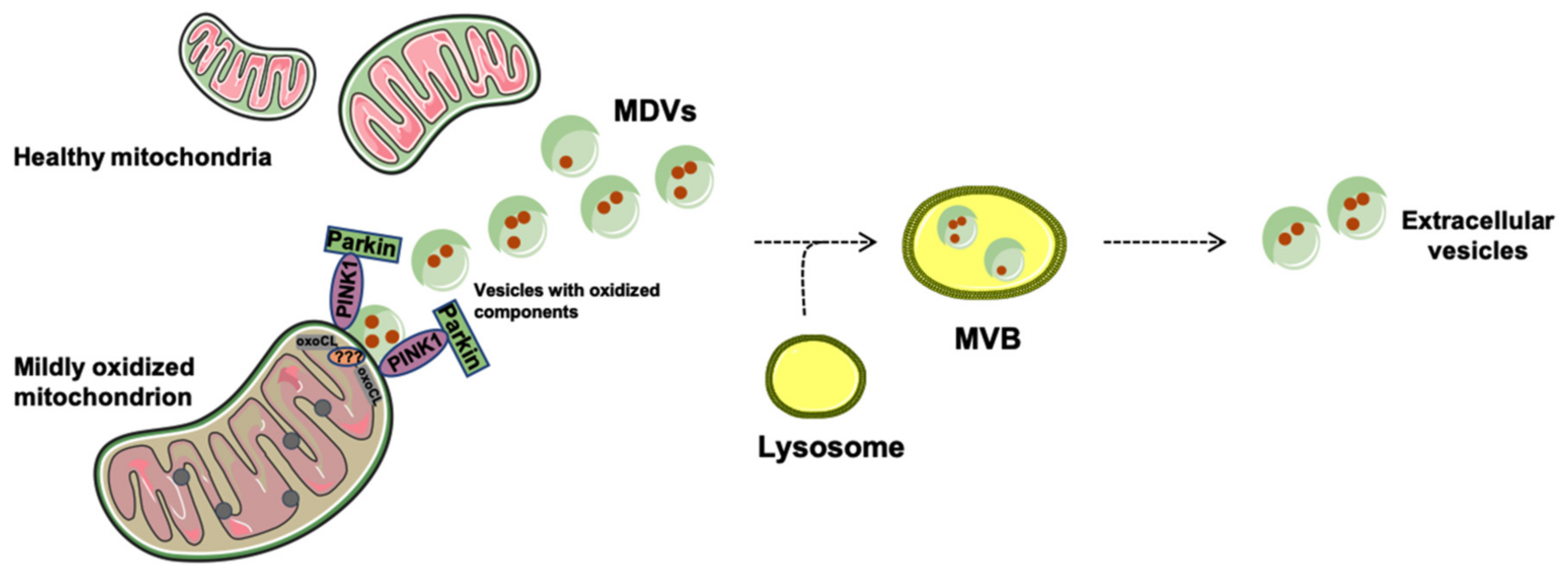
2. Mitophagy or Generation of Mitochondrial-Derived Vesicles: Easy Come Easy Go
Generation and Release of Mitochondria-Derived Vesicles
3. Failing Mitochondrial Quality Control and Inflammation during Aging: Partners in Crime
Human Immunodeficiency Virus Infection
4. Mitochondrial Quality Control in Neurodegeneration
4.1. Parkinson’s Disease
4.2. Alzheimer’s Disease
5. Mitochondrial Quality Control in Cardiovascular Disease
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossarizza, A.; Pinti, M.; Nasi, M.; Gibellini, L.; Manzini, S.; Roat, E.; Biasi, S.; De Bertoncelli, L.; Montagna, J.P.; Bisi, L.; et al. Increased plasma levels of extracellular mitochondrial DNA during HIV infection: A new role for mitochondrial damage-associated molecular patterns during inflammation. Mitochondrion 2011, 11, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.-S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.; et al. Mitochondrial signatures in circulating extracellular vesicles of older adults with Parkinson’s disease: Results from the EXosomes in PArkiNson’s Disease (EXPAND) Study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial dysfunction and aging: Insights from the analysis of extracellular vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef] [Green Version]
- Pradella, L.M.; Zuntini, R.; Magini, P.; Ceccarelli, C.; Neri, I.; Cerasoli, S.; Graziano, C.; Gasparre, G.; Turchetti, D. Two distinct thyroid tumours in a patient with Cowden syndrome carrying both a 10q23 and a mitochondrial DNA germline deletion. J. Med. Genet. 2011, 48, 779–782. [Google Scholar] [CrossRef]
- Daniele, T.; Schiaffino, M.V. Organelle biogenesis and interorganellar connections: Better in contact than in isolation. Commun. Integr. Biol. 2014, 7, 1–6. [Google Scholar] [CrossRef]
- Todkar, K.; Ilamathi, H.S.; Germain, M. Mitochondria and lysosomes: Discovering bonds. Front. Cell Dev. Biol. 2017, 5, 106. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Calvani, R.; Coelho-Junior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Inter-organelle membrane contact sites and mitochondrial quality control during aging: A geroscience view. Cells 2020, 9, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Nag, S.; Mason, A.B.; Barroso, M.M. Endosome-mitochondria interactions are modulated by iron release from transferrin. J. Cell Biol. 2016, 214, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Aston, D.; Capel, R.A.; Ford, K.L.; Christian, H.C.; Mirams, G.R.; Rog-Zielinska, E.A.; Kohl, P.; Galione, A.; Burton, R.A.B.; Terrar, D.A. High resolution structural evidence suggests the sarcoplasmic reticulum forms microdomains with scidic Stores (lysosomes) in the heart. Sci. Rep. 2017, 7, 40620. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; Baixauli, F.; Mittelbrunn, M. Interorganelle communication between mitochondria and the endolysosomal system. Front. Cell Dev. Biol. 2017, 5, 95. [Google Scholar] [CrossRef] [Green Version]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Stach, L.; Freemont, P.S. The AAA+ ATPase p97, a cellular multitool. Biochem. J. 2017, 474, 2953–2976. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Lezza, A.M.S. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions. Useful insights from aging and calorie restriction studies. Mitochondrion 2015, 25, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Kitamura, N.; Nakamura, Y.; Futamura, M.; Miyamoto, T.; Yoshida, M.; Ono, M.; Ichinose, S.; Arakawa, H. Possible existence of lysosome-like organella within mitochondria and its role in mitochondrial quality control. PLoS ONE 2011, 6, e16054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, A.; McLelland, G.-L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; Van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L.F. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-specific autophagy in inflammatory diseases: A potential therapeutic target underlying the quality control of multiple organelles. Autophagy 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazlauskaite, A.; Muqit, M.M.K. PINK1 and Parkin—mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J. 2015, 282, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Aerts, L.; Craessaerts, K.; De Strooper, B.; Morais, V.A. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J. Biol. Chem. 2015, 290, 2798–2811. [Google Scholar] [CrossRef] [Green Version]
- Koyano, F.; Matsuda, N. Molecular mechanisms underlying PINK1 and Parkin catalyzed ubiquitylation of substrates on damaged mitochondria. Biochim. Biophys. Acta 2015, 1853, 2791–2796. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba-Fukushima, K.; Imai, Y.; Yoshida, S.; Ishihama, Y.; Kanao, T.; Sato, S.; Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2012, 2, 1002. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, 4439–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Bowling, J.L.; Skolfield, M.C.; Riley, W.A.; Nolin, A.P.; Wolf, L.C.; Nelson, D.E. Temporal integration of mitochondrial stress signals by the PINK1:Parkin pathway. BMC Mol. Cell Biol. 2019, 20, 33. [Google Scholar] [CrossRef] [Green Version]
- Kolitsida, P.; Zhou, J.; Rackiewicz, M.; Nolic, V.; Dengjel, J.; Abeliovich, H. Phosphorylation of mitochondrial matrix proteins regulates their selective mitophagic degradation. Proc. Natl. Acad. Sci. USA 2019, 116, 20517–20527. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.; Tang, B. Rab7a and mitophagosome formation. Cells 2019, 8, 224. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Bucci, C. Multiple roles of the small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Role of the RAB7 protein in tumor progression and cisplatin chemoresistance. Cancers (Basel) 2019, 11, 1096. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife 2014, 3, e01612. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.-A.; Lamark, T.; Overvatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.-L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, M.G.; Munafó, D.B.; Berón, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Huang, X.; Han, L.; Wang, X.; Cheng, H.; Zhao, Y.; Chen, Q.; Chen, J.; Cheng, H.; Xiao, R.; et al. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J. Biol. Chem. 2012, 287, 23615–23625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Orgaz, A.; Kvainickas, A.; Nägele, H.; Denner, J.; Eimer, S.; Dengjel, J.; Steinberg, F. Control of RAB 7 activity and localization through the retromer-TBC1D5 complex enables RAB7-dependent mitophagy. EMBO J. 2018, 37, 235–254. [Google Scholar] [CrossRef]
- Burd, C.; Cullen, P.J. Retromer: A master conductor of endosome sorting. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef]
- Jia, D.; Zhang, J.S.; Li, F.; Wang, J.; Deng, Z.; White, M.A.; Osborne, D.G.; Phillips-Krawczak, C.; Gomez, T.S.; Li, H.; et al. Structural and mechanistic insights into regulation of the retromer coat by TBC1d5. Nat. Commun. 2016, 7, 13305. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Zhang, J.; Chen, L.; Chen, Y.; Xu, X.; Hong, W.; Wang, T. Tyrosine phosphorylation of Rab7 by Src kinase. Cell. Signal. 2017, 35, 84–94. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Swarup, S.; Paulo, J.A.; Shen, K.; Sabatini, D.M.; Harper, J.W. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci. Adv. 2018, 4, eaav0443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baixauli, F.; Acín-Pérez, R.; Villarroya-Beltrí, C.; Mazzeo, C.; Nuñez-Andrade, N.; Gabandé-Rodriguez, E.; Ledesma, M.D.; Blázquez, A.; Martin, M.A.; Falcón-Pérez, J.M.; et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of mitochondrial function impairs lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assali, E.A.; Shlomo, D.; Zeng, J.; Taddeo, E.P.; Trudeau, K.M.; Erion, K.A.; Colby, A.H.; Grinstaff, M.W.; Liesa, M.; Las, G.; et al. Nanoparticle-mediated lysosomal reacidification restores mitochondrial turnover and function in β cells under lipotoxicity. FASEB J. 2018, 33, 4154–4165. [Google Scholar] [CrossRef] [PubMed]
- Soubannier, V.; McLelland, G.-L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Desdín-Micó, G.; Mittelbrunn, M. Role of exosomes in the protection of cellular homeostasis. Cell Adhes. Migr. 2017, 11, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 9066–9075. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.F.; Tang, M.Y.; Fon, E.A.; Durcan, T.M. Defending the mitochondria: The pathways of mitophagy and mitochondrial-derived vesicles. Int. J. Biochem. Cell Biol. 2016, 79, 427–436. [Google Scholar] [CrossRef]
- McLelland, G.-L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Bossola, M.; Manes-Gravina, E.; Landi, F.; Bernabei, R.; Marzetti, E. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res. 2018, 21, 350–359. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griessinger, E.; Moschoi, R.; Biondani, G.; Peyron, J.-F. Mitochondrial transfer in the leukemia microenvironment. Trends Cancer 2017, 3, 828–839. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F.; Bernabei, R.; Marzetti, E. Fueling inflamm-aging through mitochondrial dysfunction: Mechanisms and molecular targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Cesari, M.; Marzetti, E.; Canevelli, M.; Guaraldi, G. Geriatric syndromes: How to treat. Virulence 2017, 8, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Aswani, A.; Manson, J.; Itagaki, K.; Chiazza, F.; Collino, M.; Wupeng, W.L.; Chan, T.K.; Wong, W.S.F.; Hauser, C.J.; Thiemermann, C.; et al. Scavenging circulating mitochondrial DNA as a potential therapeutic option for multiple organ dysfunction in trauma hemorrhage. Front. Immunol. 2018, 9, 891. [Google Scholar] [CrossRef] [PubMed]
- Heil, M.; Brockmeyer, N.H. Self-DNA sensing fuels HIV-1-associated inflammation. Trends Mol. Med. 2019, 25, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef] [PubMed]
- D’Aquila, P.; Montesanto, A.; Rango, F.D.; Guarasci, F.; Passarino, G.; Bellizzi, D. Epigenetic signature: Implications for mitochondrial quality control in human aging. Aging 2019, 11, 1240–1251. [Google Scholar] [CrossRef]
- Blanch, M.; Mosquera, J.L.; Ansoleaga, B.; Ferrer, I.; Barrachina, M. Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am. J. Pathol. 2016, 186, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- Maass, D.L.; White, J.; Sanders, B.; Horton, J.W. Role of cytosolic vs. mitochondrial Ca2+ accumulation in burn injury-related myocardial inflammation and function. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Mankowski, R.T.; Kamenov, G.; Anton, S.D.; Manini, T.M.; Buford, T.W.; Saini, S.K.; Calvani, R.; Landi, F.; Bernabei, R.; et al. Advanced age is associated with iron dyshomeostasis and mitochondrial DNA damage in human skeletal muscle. Cells 2019, 8, 1525. [Google Scholar] [CrossRef] [Green Version]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Fiers, W.; Beyaert, R.; Declercq, W.; Vandenabeele, P. More than one way to die: Apoptosis, necrosis and reactive oxygen damage. Oncogene 1999, 18, 7719–7730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.-M.; Tarkowski, A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Mallette, F.A.; Mukhopadhyay, U.K.; Moores, A.; Ferbeyre, G. DNA damage signaling and p53-dependent senescence after prolonged β-interferon stimulation. Mol. Biol. Cell 2006, 17, 1583–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, U.; Chen, Q.; Chena, Z.J. CGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M.; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. The impact of SASP and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [Green Version]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17, e12734. [Google Scholar] [CrossRef]
- Belov, L.; Matic, K.J.; Hallal, S.; Best, O.G.; Mulligan, S.P.; Christopherson, R.I. Extensive surface protein profiles of extracellular vesicles from cancer cells may provide diagnostic signatures from blood samples. J. Extracell. Vesicles 2016, 5, 25355. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Greene, W.C. Dissecting how CD4 T cells are lost during HIV Infection. Cell Host Microbe 2016, 19, 280–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Muñoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feria, M.G.; Taborda, N.A.; Hernandez, J.C.; Rugeles, M.T. HIV replication is associated to inflammasomes activation, IL-1β, IL-18 and caspase-1 expression in GALT and peripheral blood. PLoS ONE 2018, 13, e0192845. [Google Scholar] [CrossRef]
- Perfettini, J.L.; Castedo, M.; Roumier, T.; Andreau, K.; Nardacci, R.; Piacentini, M.; Kroemer, G. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005, 12, 916–923. [Google Scholar] [CrossRef] [Green Version]
- Muthumani, K.; Choo, A.Y.; Hwang, D.S.; Premkumar, A.; Dayes, N.S.; Harris, C.; Green, D.R.; Wadsworth, S.A.; Siekierka, J.J.; Weiner, D.B. HIV-1 Nef-induced FasL induction and bystander killing requires p38 MAPK activation. Blood 2005, 106, 2059–2068. [Google Scholar] [CrossRef] [Green Version]
- Varbanov, M.; Espert, L.; Biard-Piechaczyk, M. Mechanisms of CD4 T-cell depletion triggered by HIV-1 viral proteins. AIDS Rev. 2006, 8, 221–236. [Google Scholar]
- Nasi, M.; De Biasi, S.; Gibellini, L.; Bianchini, E.; Pecorini, S.; Bacca, V.; Guaraldi, G.; Mussini, C.; Pinti, M.; Cossarizza, A. Ageing and inflammation in patients with HIV infection. Clin. Exp. Immunol. 2017, 187, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Nasi, M.; Pinti, M.; Mussini, C.; Cossarizza, A. Persistent inflammation in HIV infection: Established concepts, new perspectives. Immunol. Lett. 2014, 161, 184–188. [Google Scholar] [CrossRef]
- Dinkins, C.; Arko-Mensah, J.; Deretic, V. Autophagy and HIV. Semin. Cell Dev. Biol. 2010, 21, 712–718. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumptner-Cuvelette, P.; Jouve, M.; Helft, J.; Dugast, M.; Glouzman, A.-S.; Jooss, K.; Raposo, G.; Benaroch, P. Human immunodeficiency virus-1 Nef expression induces intracellular accumulation of multivesicular bodies and major histocompatibility complex class II complexes: Potential role of phosphatidylinositol 3-kinase. Mol. Biol. Cell 2003, 14, 4857–4870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandrin, V.; Cosset, F.L. Intracellular versus cell surface assembly of retroviral pseudotypes is determined by the cellular localization of the viral glycoprotein, its capacity to interact with Gag, and the expression of the Nef protein. J. Biol. Chem. 2006, 281, 528–542. [Google Scholar] [CrossRef] [Green Version]
- Sanfridson, A.; Hester, S.; Doyle, C. Nef proteins encoded by human and simian immunodeficiency viruses induce the accumulation of endosomes and lysosomes in human T cells. Proc. Natl. Acad. Sci. USA 1997, 94, 873–878. [Google Scholar] [CrossRef] [Green Version]
- Arshad, O.; Gadawska, I.; Sattha, B.; Côté, H.C.F.; Hsieh, A.Y.Y. Elevated cell-free mitochondrial DNA in filtered plasma is associated with HIV infection and inflammation. J. Acquir. Immune Defic. Syndr. 2018, 78, 111–118. [Google Scholar] [CrossRef]
- Dai, Z.; Cai, W.; Hu, F.; Lan, Y.; Li, L.; Chung, C.; Caughey, B.; Zhang, K.; Tang, X. Plasma mitochondrial DNA levels as a biomarker of lipodystrophy among HIV-infected patients treated with highly active antiretroviral therapy (HAART). Curr. Mol. Med. 2015, 15, 975–979. [Google Scholar] [CrossRef] [Green Version]
- Younes, S.-A.; Talla, A.; Pereira Ribeiro, S.; Saidakova, E.V.; Korolevskaya, L.B.; Shmagel, K.V.; Shive, C.L.; Freeman, M.L.; Panigrahi, S.; Zweig, S.; et al. Cycling CD4+ T cells in HIV-infected immune nonresponders have mitochondrial dysfunction. J. Clin. Investig. 2018, 128, 5083–5094. [Google Scholar] [CrossRef]
- Brunet-Ratnasingham, E.; Dubé, M.; Kaufmann, D.E. Targeting mitochondria to revive dysfunctional regulatory T cells. Trends Mol. Med. 2019, 25, 1–3. [Google Scholar] [CrossRef]
- Heil, M.; Vega-Muñoz, I. Nucleic acid sensing in mammals and plants: Facts and caveats. Int. Rev. Cell Mol. Biol. 2019, 345, 225–285. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.; Kim, T.; Huh, Y.J.; Lee, J.; Lee, Y. Il Amelioration of mitochondrial quality control and proteostasis by natural compounds in Parkinson’s disease models. Int. J. Mol. Sci. 2019, 20, 5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, A.J.; Wijeyekoon, R.S.; Scott, K.M.; Gunawardana, N.P.; Hayat, S.; Solim, I.H.; McMahon, H.T.; Barker, R.A.; Williams-Gray, C.H. The peripheral inflammatory response to alpha-synuclein and endotoxin in Parkinson’s disease. Front. Neurol. 2018, 9, 946. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Guerra, F.; Girolimetti, G.; Beli, R.; Mitruccio, M.; Pacelli, C.; Ferretta, A.; Gasparre, G.; Cocco, T.; Bucci, C. Synergistic effect of mitochondrial and lysosomal dysfunction in Parkinson’s disease. Cells 2019, 8, 452. [Google Scholar] [CrossRef] [Green Version]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H.M. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 2010, 20, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Trajkovic, K.; Tsunemi, T.; Krainc, D. Parkin modulates endosomal organization and function of the endo-lysosomal pathway. J. Neurosci. 2016, 36, 2425–2437. [Google Scholar] [CrossRef] [Green Version]
- Small, S.A.; Petsko, G.A. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat. Rev. Neurosci. 2015, 16, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Perrett, R.M.; Alexopoulou, Z.; Tofaris, G.K. The endosomal pathway in Parkinson’s disease. Mol. Cell. Neurosci. 2015, 66, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Restelli, L.M.; Oettinghaus, B.; Halliday, M.; Agca, C.; Licci, M.; Sironi, L.; Savoia, C.; Hench, J.; Tolnay, M.; Neutzner, A.; et al. Neuronal mitochondrial dysfunction activates the integrated stress response to induce fibroblast growth factor 21. Cell Rep. 2018, 24, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, M.R.; Nagele, R.G.; Wang, H.Y.; Peterson, P.A.; Lee, D.H.S. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 2001, 38, 120–134. [Google Scholar] [CrossRef]
- Willén, K.; Edgar, J.R.; Hasegawa, T.; Tanaka, N.; Futter, C.E.; Gouras, G.K. Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol. Neurodegener. 2017, 12, 61. [Google Scholar] [CrossRef]
- Wilquet, V.; Strooper, B. De Amyloid-beta precursor protein processing in neurodegeneration. Curr. Opin. Neurobiol. 2004, 14, 582–588. [Google Scholar] [CrossRef]
- Tang, B.L. Neuronal protein trafficking associated with Alzheimer disease: From APP and BACE1 to glutamate receptors. Cell Adhes. Migr. 2009, 3, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.Y.; Tan, M.S.; Yu, J.T.; Tan, L. The role of retromer in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 4201–4209. [Google Scholar] [CrossRef]
- Zafar, S.; Younas, N.; Correia, S.; Shafiq, M.; Tahir, W.; Schmitz, M.; Ferrer, I.; Andréoletti, O.; Zerr, I. Strain-specific altered regulatory response of Rab7a and Tau in Creutzfeldt-Jakob disease and Alzheimer’s disease. Mol. Neurobiol. 2017, 54, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Mohamed, N.V.; Desjardins, A.; Lippé, R.; Fon, E.A.; Leclerc, N. Rab7A regulates tau secretion. J. Neurochem. 2017, 141, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Melber, A.; Haynes, C.M. UPRmt regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef]
- Wang, Z.T.; Lu, M.H.; Zhang, Y.; Ji, W.L.; Lei, L.; Wang, W.; Fang, L.P.; Wang, L.W.; Yu, F.; Wang, J.; et al. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell 2019, 18, e12860. [Google Scholar] [CrossRef]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Fang, E.F. Mitophagy and NAD+ inhibit Alzheimer disease. Autophagy 2019, 15, 1112–1114. [Google Scholar] [CrossRef] [Green Version]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Butchart, J. Systemic inflammation and Alzheimer’s disease. Biochem. Soc. Trans. 2011, 39, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C.; Marzetti, E. Contribution of impaired mitochondrial autophagy to cardiac aging: Mechanisms and therapeutic opportunities. Circ. Res. 2012, 110, 1125–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzetti, E.; Wohlgemuth, S.E.; Anton, S.D.; Bernabei, R.; Carter, C.S.; Leeuwenburgh, C. Cellular mechanisms of cardioprotection by calorie restriction: State of the science and future perspectives. Clin. Geriatr. Med. 2009, 25, 715–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring in vivo mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.-L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [Green Version]
- Maejima, Y.; Isobe, M.; Sadoshima, J. Regulation of autophagy by Beclin 1 in the heart. J. Mol. Cell. Cardiol. 2016, 95, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tan, V.P.; Miyamoto, S. Nutrient-sensing mTORC1: Integration of metabolic and autophagic signals. J. Mol. Cell. Cardiol. 2016, 95, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Ock, S.; Lee, W.S.; Ahn, J.; Kim, H.M.; Kang, H.; Kim, H.S.; Jo, D.; Abel, E.D.; Lee, T.J.; Kim, J. Deletion of IGF-1 receptors in cardiomyocytes attenuates cardiac aging in male mice. Endocrinology 2016, 157, 336–345. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125, 1134–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.; He, Z.; Huang, H.; Zhuang, H.; Liu, H.; Liu, X.; Yang, S.; He, P.; Yang, H.; Feng, D. Mitochondrial quality control in cardiomyocytes: A critical role in the progression of cardiovascular diseases. Front. Physiol. 2020, 11, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Lin, K.; Zhang, Y.; Li, M.; Xu, J.; Chen, K.; Zhu, P.; Yu, R. Mst1 deletion reduces septic cardiomyopathy via activating Parkin-related mitophagy. J. Cell. Physiol. 2020, 235, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Wang, W.; Wada, S.; McDermott-Roe, C.; Evans, C.S.; Gosis, B.; Morley, M.P.; Rathi, K.S.; Li, J.; Li, K.; et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019, 575, 375–379. [Google Scholar] [CrossRef]
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Binding of FUN14 Domain Containing 1 with Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 2017, 136, 2248–2266. [Google Scholar] [CrossRef]
- Yu, W.; Xu, M.; Zhang, T.; Zhang, Q.; Zou, C. Mst1 promotes cardiac ischemia–reperfusion injury by inhibiting the ERK-CREB pathway and repressing FUNDC1-mediated mitophagy. J. Physiol. Sci. 2019, 69, 113–127. [Google Scholar] [CrossRef]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, Å.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012, 3, 265. [Google Scholar] [CrossRef] [Green Version]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; López, B.; González, A.; Ravassa, S.; Díez, J.; et al. Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Zhang, H.; Hu, S. Mitochondrial aldehyde dehydrogenase 2 activation and cardioprotection. J. Mol. Cell. Cardiol. 2013, 55, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Yu, L.; Wang, Y.; Wang, H.; Li, C.; Yin, Y.; Yang, J.; Wang, Z.; Zheng, Q.; Ma, H. Aldehyde dehydrogenase 2 activation in aged heart improves the autophagy by reducing the carbonyl modification on SIRT1. Oncotarget 2016, 7, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Cadete, V.J.J.; Deschênes, S.; Cuillerier, A.; Brisebois, F.; Sugiura, A.; Vincent, A.; Turnbull, D.; Picard, M.; McBride, H.M.; Burelle, Y. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. 2016, 594, 5343–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Servier Medical Art. Available online: http://www.servier.com/Powerpoint-image-bank.


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. https://doi.org/10.3390/jcm9051440
Picca A, Guerra F, Calvani R, Coelho-Junior HJ, Bossola M, Landi F, Bernabei R, Bucci C, Marzetti E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. Journal of Clinical Medicine. 2020; 9(5):1440. https://doi.org/10.3390/jcm9051440
Chicago/Turabian StylePicca, Anna, Flora Guerra, Riccardo Calvani, Hélio José Coelho-Junior, Maurizio Bossola, Francesco Landi, Roberto Bernabei, Cecilia Bucci, and Emanuele Marzetti. 2020. "Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease" Journal of Clinical Medicine 9, no. 5: 1440. https://doi.org/10.3390/jcm9051440
APA StylePicca, A., Guerra, F., Calvani, R., Coelho-Junior, H. J., Bossola, M., Landi, F., Bernabei, R., Bucci, C., & Marzetti, E. (2020). Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. Journal of Clinical Medicine, 9(5), 1440. https://doi.org/10.3390/jcm9051440