Pulmonary Hypertension in Adults with Congenital Heart Disease: Real-World Data from the International COMPERA-CHD Registry

 ,
,  , , , , , , , , ,
, , , , , , , , ,  , , , , , , ,
, , , , , , , 
Abstract
:1. Introduction
2. Patients and Methods
2.1. Setting
2.2. Patient Selection
2.3. Data Documentation
2.4. Statistical Analysis
3. Results
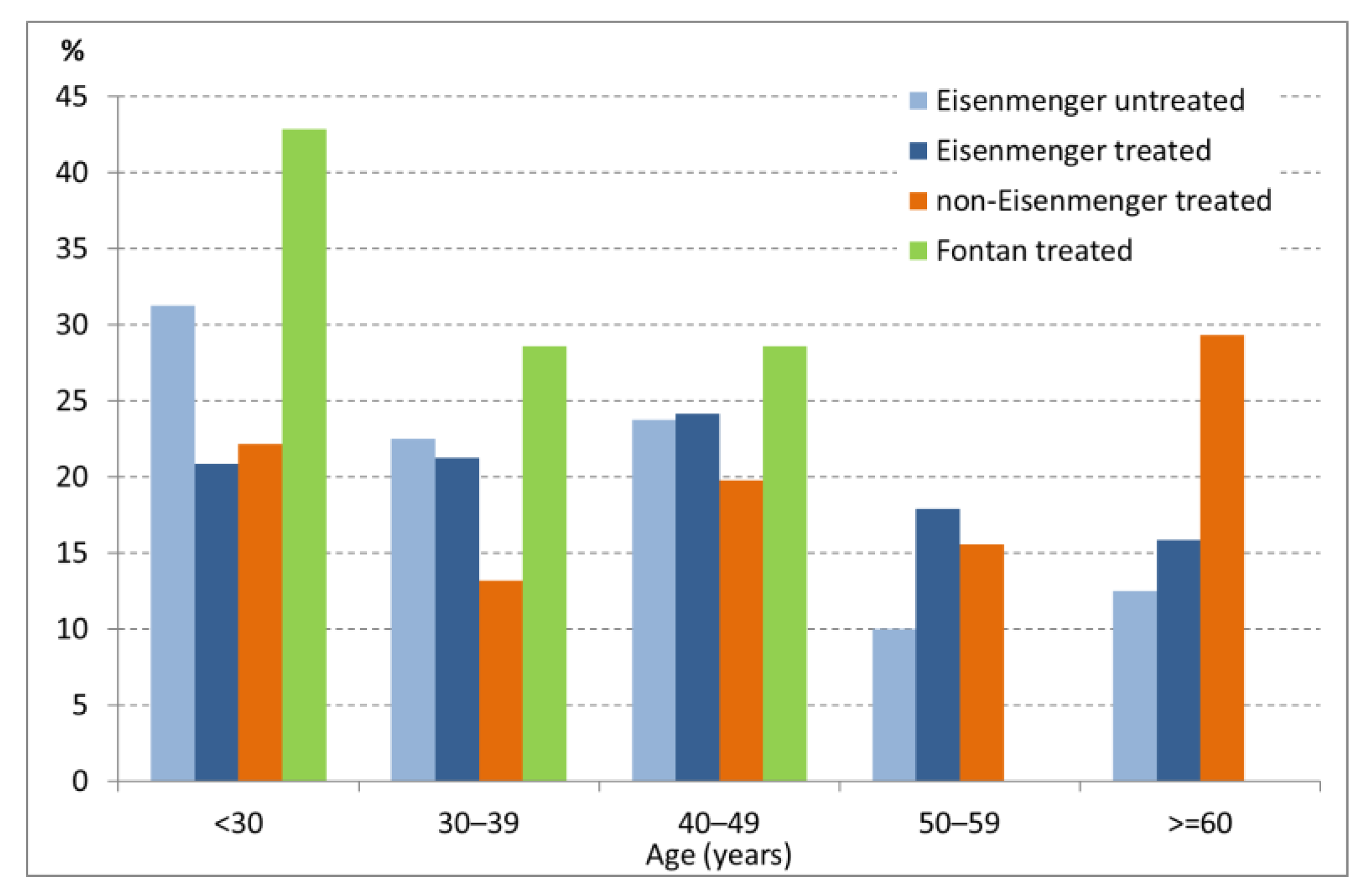
3.1. Patient Demographics and Baseline Characteristics
3.2. Type of Congenital Heart Defect
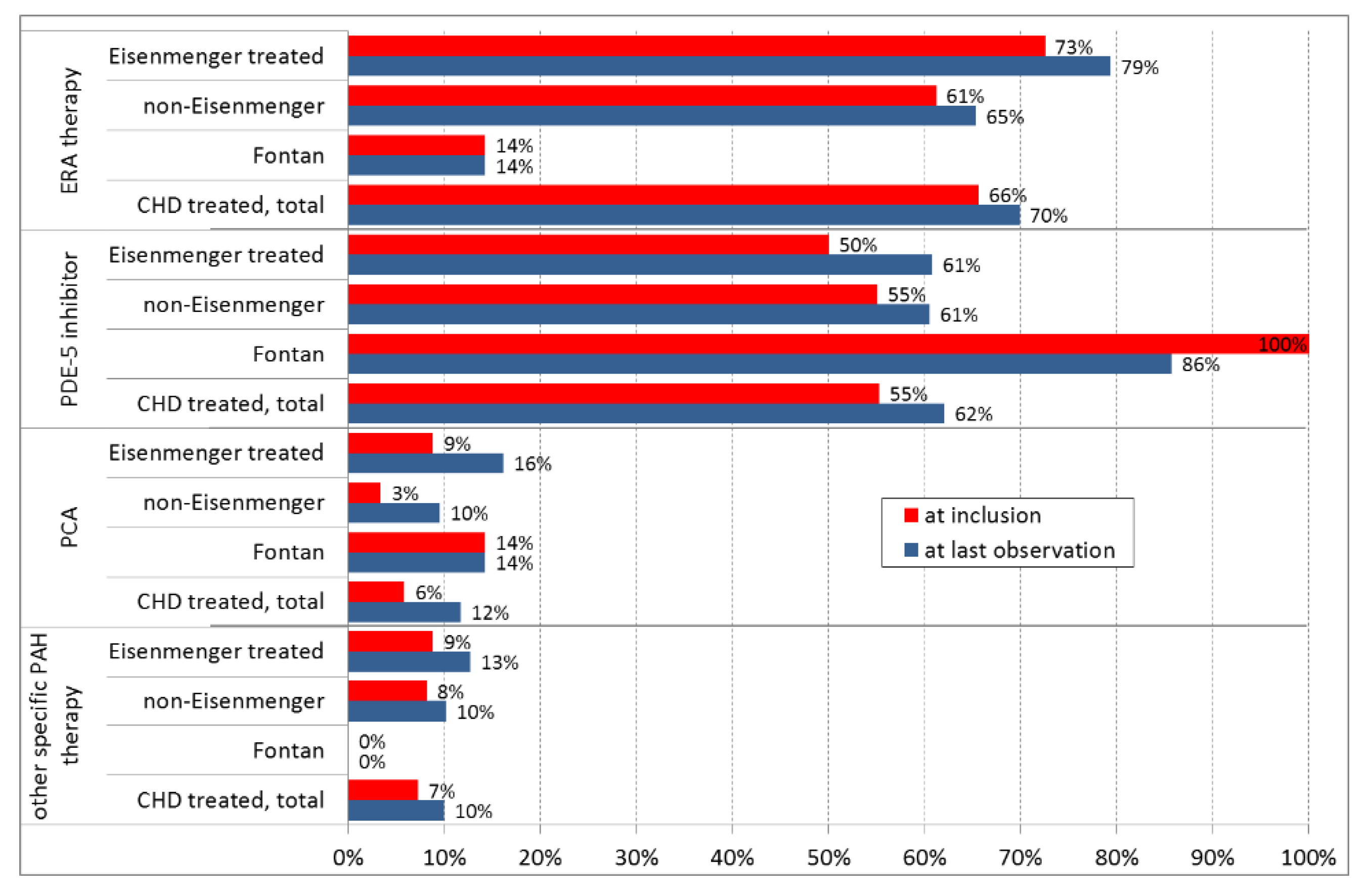
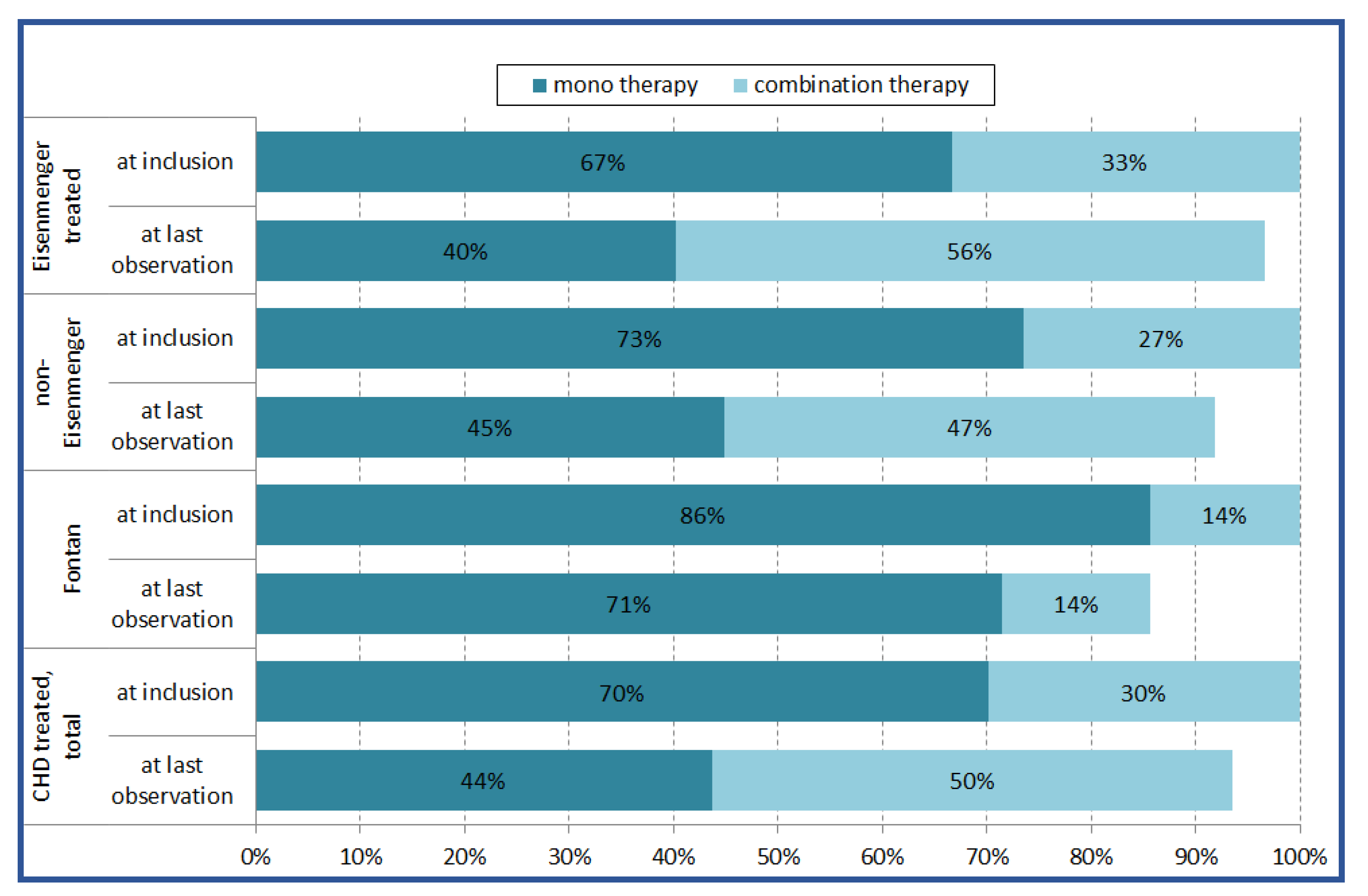
3.3. Medication and Treatment Strategy
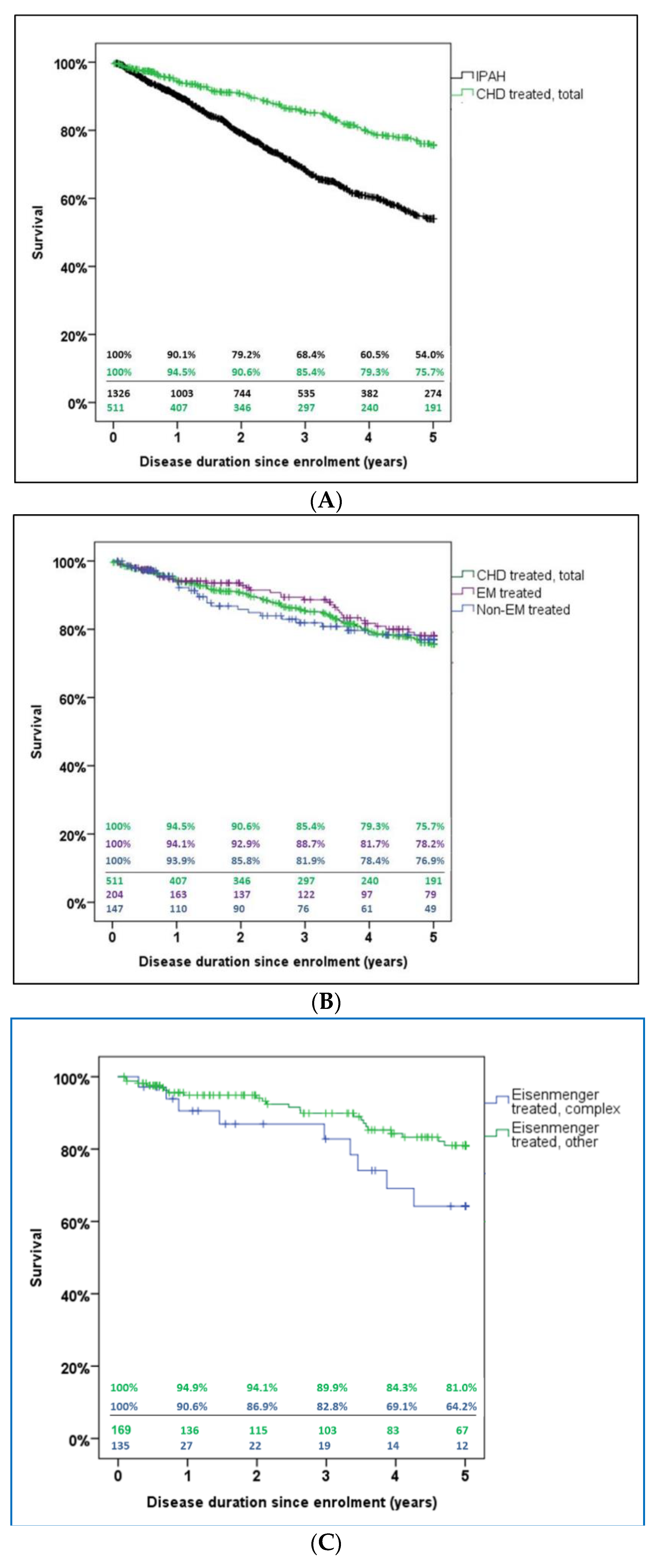
3.4. Survival
4. Discussion
4.1. Demographics, Hemodynamics, and Treatment
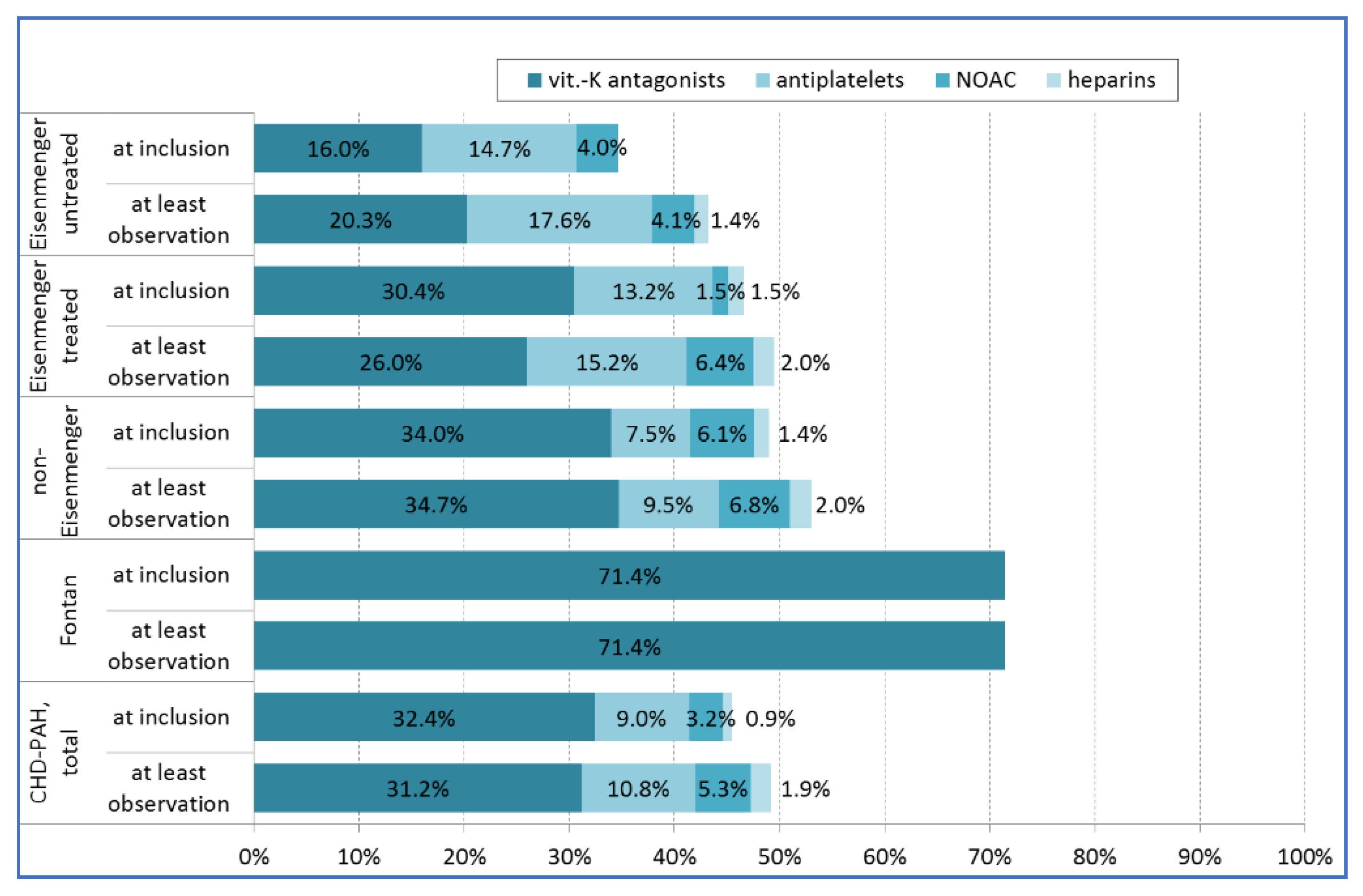
4.2. Co-Medication with Anticoagulants or Antiplatelets
4.3. Survival
4.4. Limitations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaemmerer, H.; Apitz, C.; Brockmeier, K.; Eicken, A.; Gorenflo, M.; Hager, A.; De Haan, F.; Huntgeburth, M.; Kozlik-Feldmann, R.G.; Miera, O.; et al. Pulmonary hypertension in adults with congenital heart disease: Updated recommendations from the Cologne Consensus Conference 2018. Int. J. Cardiol. 2018, 272, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galié, N.; Humbert, M.; Vachiéry, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Brida, M.; Gatzoulis, M.A. Pulmonary arterial hypertension in adult congenital heart disease. Heart 2018, 104, 1568–1574. [Google Scholar] [CrossRef]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.S.; Denton, C.; Ghofrani, H.A.; Sánchez, M.A.G.; Kumar, R.K.; Landzberg, M.; Machado, R.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [Green Version]
- Kaemmerer, H.; Apitz, C.; Brockmeier, K.; Eicken, A.; Gorenflo, M.; Hager, A.; De Haan, F.; Huntgeburth, M.; Kozlik-Feldmann, R.; Miera, O. Pulmonary hypertension in grown-ups with congenital heart disease: Recommendations of the Cologne Consensus Conference 2016. Dtsch. Med. Wochenschr. (1946) 2016, 141, S70–S79. [Google Scholar]
- Klinger, J.R.; Elliott, C.G.; Levine, D.J.; Bossone, E.; Duvall, L.; Fagan, K.; Frantsve-Hawley, J.; Kawut, S.M.; Ryan, J.J.; Rosenzweig, E.B.; et al. Therapy for pulmonary arterial hypertension in adults. Chest 2019, 155, 565–586. [Google Scholar] [CrossRef]
- Franklin, W.J.; Parekh, D.R.; Safdar, Z. Adult congenital heart disease and pulmonary arterial hypertension: The Texas adult congenital heart program experience. Postgrad. Med. 2011, 123, 32–45. [Google Scholar] [CrossRef]
- Duffels, M.; Engelfriet, P.; Berger, R.; Van Loon, R.; Hoendermis, E.; Vriend, J.W.; Van der Velde, E.T.; Bresser, P.; Mulder, B. Pulmonary arterial hypertension in congenital heart disease: An epidemiologic perspective from a Dutch registry. Int. J. Cardiol. 2007, 120, 198–204. [Google Scholar] [CrossRef]
- Verheugt, C.L.; Uiterwaal, C.S.P.M.; Van der Velde, E.T.; Meijboom, F.J.; Pieper, P.G.; Vliegen, H.W.; Van Dijk, A.; Bouma, B.J.; Grobbee, D.E.; Mulder, B. Gender and outcome in adult congenital heart disease. Circulation 2008, 118, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Safdar, Z. Pulmonary hypertension: A woman’s disease. Tex. Heart Inst. J. 2013, 40, 302–303. [Google Scholar]
- Alonso-Gonzalez, R.; Lopez-Guarch, C.J.; Subirana-Domènech, M.T.; Ruiz, J.M.O.; González, I.O.; Cubero, J.S.; Del Cerro, M.J.; Salvador, M.L.; Dos Subira, L.; Gallego, P.; et al. Pulmonary hypertension and congenital heart disease: An insight from the REHAP National Registry. Int. J. Cardiol. 2015, 184, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Favilli, S.; Spaziani, G.; Ballo, P.; Fibbi, V.; Santoro, G.; Chiappa, E.; Arcangeli, C. Advanced therapies in patients with congenital heart disease-related pulmonary arterial hypertension: Results from a long-term, single center, real-world follow-up. Intern. Emerg. Med. 2015, 10, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Galié, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.-L.; Barberà, J.A.; Beghetti, M.; Corris, P.; Gaine, S.P.; Gibbs, J.S.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2009, 30, 2493–2537. [Google Scholar] [CrossRef] [PubMed]
- Derk, G.; Houser, L.; Miner, P.; Williams, R.; Moriarty, J.; Finn, P.; Alejos, J.; Aboulhosn, J. Efficacy of endothelin blockade in adults with f ontan physiology. Congenit. Heart Dis. 2015, 10, E11–E16. [Google Scholar] [CrossRef] [PubMed]
- Kempny, A.; Hjortshøj, C.S.; Gu, H.; Li, W.; Opotowsky, A.R.; Landzberg, M.J.; Jensen, A.S.; Søndergaard, L.; Estensen, M.-E.; Thilén, U.; et al. Predictors of death in contemporary adult patients with eisenmenger syndrome. Circulation 2017, 135, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Huscher, D.; Ghofrani, H.A.; Delcroix, M.; Distler, O.; Schweiger, C.; Gruenig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: Results from the COMPERA registry. Int. J. Cardiol. 2013, 168, 871–880. [Google Scholar] [CrossRef]
- Olsson, K.M.; Delcroix, M.; Ghofrani, H.A.; Tiede, H.; Huscher, D.; Speich, R.; Grünig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Anticoagulation and survival in pulmonary arterial hypertension: Results from the comparative, prospective registry of newly initiated therapies for pulmonary hypertension (COMPERA). Circulation 2014, 129, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.; Hoeper, M.M.; Gibbs, J.S.R.; Kaemmerer, H.; Pepke-Zaba, J.; Coghlan, J.G.; Scelsi, L.; D’Alto, M.; Olsson, K.; Ulrich, S.; et al. Pre-capillary, combined, and post-capillary pulmonary hypertension. J. Am. Coll. Cardiol. 2016, 68, 368–378. [Google Scholar] [CrossRef] [Green Version]
- National Institutes of Health, US Library of Medicine. COMPERA/COMPERA-KIDS. Available online: https://clinicaltrials.gov/ct2/show/NCT01347216. (accessed on 11 May 2020).
- Stout, K.K.; Daniels, C.J.; Aboulhosn, J.A.; Bozkurt, B.; Broberg, C.S.; Colman, J.M.; Crumb, S.R.; Dearani, J.A.; Fuller, S.; Gurvitz, M.; et al. 2018 AHA/ACC guideline for the management of adults with congenital heart disease: Executive summary: A report of the american college of cardiology/american heart association task force on clinical practice guidelines. J. Am. Coll. Cardiol. 2019, 73, 1494–1563. [Google Scholar] [CrossRef]
- Coats, A.; Shewan, L. Statement on authorship and publishing ethics in the international journal of cardiology. Int. J. Cardiol. 2011, 153, 239–240. [Google Scholar] [CrossRef] [PubMed]
- Tulloh, R.M.R.; Dimopoulos, K.; Condliffe, R.; Clift, P.; CHAMPION Steering Committee. Management of adults with congenital heart disease and pulmonary arterial hypertension in the UK: Survey of current practice, unmet needs and expert commentary. Heart Lung Circ. 2017, 27, 1018–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diller, G.P.; Körten, M.-A.; Bauer, U.M.; Miera, O.; Tutarel, O.; Kaemmerer, H.; Berger, F.; Baumgartner, H. Current therapy and outcome of Eisenmenger syndrome: Data of the German National Register for congenital heart defects. Eur. Heart J. 2016, 37, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Vijarnsorn, C.; Durongpisitkul, K.; Chungsomprasong, P.; Bositthipichet, D.; Ketsara, S.; Titaram, Y.; Chanthong, P.; Kanjanauthai, S.; Soongswang, J. Contemporary survival of patients with pulmonary arterial hypertension and congenital systemic to pulmonary shunts. PLoS ONE 2018, 13, e0195092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condliffe, R.C.; Clift, P.; Dimopoulos, K.; Tulloh, R.M.R. Management dilemmas in pulmonary arterial hypertension associated with congenital heart disease. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hascoët, S.; Fournier, E.; Jaïs, X.; Le Gloan, L.; Dauphin, C.; Houeijeh, A.; Godart, F.; Iriart, X.; Richard, A.; Radojevic, J.; et al. Outcome of adults with Eisenmenger syndrome treated with drugs specific to pulmonary arterial hypertension: A French multicentre study. Arch. Cardiovasc. Dis. 2017, 110, 303–316. [Google Scholar] [CrossRef]
- Varela, D.; Teleb, M.; El-Mallah, W. Advanced therapies for the management of adults with pulmonary arterial hypertension due to congenital heart disease: A systematic review. Open Heart 2018, 5, e000744. [Google Scholar] [CrossRef] [Green Version]
- Rosenkranz, S. Pulmonary hypertension 2015: Current definitions, terminology, and novel treatment options. Clin. Res. Cardiol. 2015, 104, 197–207. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galié, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Hoeper, M.M.; Apitz, C.; Grünig, E.; Halank, M.; Ewert, R.; Kaemmerer, H.; Kabitz, H.-J.; Kähler, C.; Klose, H.; Leuchte, H.; et al. Targeted therapy of pulmonary arterial hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int. J. Cardiol. 2018, 272, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Ghofrani, H.A. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax 2016, 71, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGoon, M.D.; Benza, R.L.; Escribano-Subías, P.; Jiang, X.; Miller, D.P.; Peacock, A.J.; Pepke-Zaba, J.; Pulido, T.; Rich, S.; Rosenkranz, S.; et al. Pulmonary arterial hypertension: Epidemiology and registries. J. Am. Coll. Cardiol. 2013, 62, D51–D59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, M.L.; Strange, G.; King, I.; Arnup, S.; Vidmar, S.; O’Donnell, C.; Kermeen, F.; Grigg, L.; Weintraub, R.G.; Celermajer, D.S. Congenital heart disease-associated pulmonary arterial hypertension: Preliminary results from a novel registry. Intern. Med. J. 2012, 42, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Jansa, P.; Jarkovsky, J.; Al-Hiti, H.; Popelová, J.R.; Ambrož, D.; Zatočil, T.; Votavova, R.; Polacek, P.; Marešová, J.; Aschermann, M.; et al. Epidemiology and long-term survival of pulmonary arterial hypertension in the Czech Republic: A retrospective analysis of a nationwide registry. BMC Pulm. Med. 2014, 14, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Bruaene, A.; Delcroix, M.; Pasquet, A.; De Backer, J.; De Pauw, M.; Naeije, R.; Vachiery, J.-L.; Paelinck, B.; Morissens, M.; Budts, W. The Belgian Eisenmenger syndrome registry: Implications for treatment strategies? Acta Cardiol. 2009, 64, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Idrees, M.; Al-Najashi, K.; Khan, A.; Al-Dammas, S.; Al-Awwad, H.; Batubara, E.; Al Otai, A.; AbdulHameed, J.; Fayed, A.; Kashour, T. Pulmonary arterial hypertension in Saudi Arabia: Patients’ clinical and physiological characteristics and hemodynamic parameters. A single center experience. Ann. Thorac. Med. 2014, 9, 209–215. [Google Scholar] [CrossRef]
- Benza, R.; Miller, D.P.; Barst, R.J.; Badesch, D.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Pascall, E.; Tulloh, R.M.R. Pulmonary hypertension in congenital heart disease. Future Cardiol. 2018, 14, 343–353. [Google Scholar] [CrossRef] [Green Version]
- D’Alto, M.; Romeo, E.; Argiento, P.; Sarubbi, B.; Santoro, G.; Grimaldi, N.; Correra, A.; Scognamiglio, G.; Russo, V.; Calabrò, R. Bosentan–sildenafil association in patients with congenital heart disease-related pulmonary arterial hypertension and Eisenmenger physiology. Int. J. Cardiol. 2012, 155, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Iversen, K.; Jensen, A.S.; Jensen, T.V.; Vejlstrup, N.G.; Søndergaard, L. Combination therapy with bosentan and sildenafil in Eisenmenger syndrome: A randomized, placebo-controlled, double-blinded trial. Eur. Heart J. 2010, 31, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manes, A.; Palazzini, M.; Leci, E.; Reggiani, M.L.B.; Branzi, A.; Galié, N. Current era survival of patients with pulmonary arterial hypertension associated with congenital heart disease: A comparison between clinical subgroups. Eur. Heart J. 2014, 35, 716–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frogoudaki, A.A.; Gatzoulis, M.A. Pulmonary arterial hypertension in congenital heart disease. Contin. Cardiol. Educ. 2018, 4, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, H.; Bonhoeffer, P.; De Groot, N.M.; De Haan, F.; Deanfield, J.E.; Galié, N.; Gatzoulis, M.A.; Gohlke-Baerwolf, C.; Kaemmerer, H.; Kilner, P.; et al. ESC Guidelines for the management of grown-up congenital heart disease (new version 2010): The task force on the management of grown-up congenital heart disease of the european society of cardiology (ESC). Eur. Heart J. 2010, 31, 2915–2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, M.C.; Perloff, J.K.; Child, J.S. Pathogenesis of thrombocytopenia in cyanotic congenital heart disease. Am. J. Cardiol. 2006, 98, 254–258. [Google Scholar] [CrossRef]
- Viswanathan, S. Thromboembolism and anticoagulation after Fontan surgery. Ann. Pediatr. Cardiol. 2016, 9, 236–240. [Google Scholar] [CrossRef]
- Deutsche Herzstiftung. Deutscher Herzbericht 2018; Frankfurt am Main; Deutsche Herzstiftung e.V.: Frankfurt am Main, Germany, 2018. [Google Scholar]
- Lowe, B.S.; Therrien, J.; Ionescu-Ittu, R.; Pilote, L.; Martucci, G.; Marelli, A.J. Diagnosis of pulmonary hypertension in the congenital heart disease adult population impact on outcomes. J. Am. Coll. Cardiol. 2011, 58, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Diller, G.P.; Kempny, A.; Inuzuka, R.; Radke, R.; Wort, S.J.; Baumgartner, H.; Gatzoulis, M.A.; Dimopoulos, K. Survival prospects of treatment naïve patients with Eisenmenger: A systematic review of the literature and report of own experience. Heart 2014, 100, 1366–1372. [Google Scholar] [CrossRef]
- Diller, G.P.; Dimopoulos, K.; Broberg, C.S.; Kaya, M.G.; Naghotra, U.S.; Uebing, A.; Harries, C.; Goktekin, O.; Gibbs, S.; Gatzoulis, M.A. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: A combined retrospective and case-control study. Eur. Heart J. 2006, 27, 1737–1742. [Google Scholar] [CrossRef] [Green Version]
- Nunan, D.; Aronson, J.; Bankhead, C. Catalogue of bias: Attrition bias. BMJ Evid.-Based Med. 2018, 23, 21–22. [Google Scholar] [CrossRef]
- Miller, D.P.; Gomberg-Maitland, M.; Humbert, M. Survivor bias and risk assessment. Eur. Respir. J. 2012, 40, 530–532. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Rodriguez, M.; Llorca, J. Bias. J. Epidemiol. Community Health 2004, 58, 635–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, Y.; Johnson, M.K.; Kiely, D.G.; Condliffe, R.; Elliot, C.A.; Gibbs, S.; Howard, L.S.; Pepke-Zaba, J.; Sheares, K.K.; Corris, P.A.; et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Farber, H.W.; Miller, D.P.; Poms, A.; Badesch, D.; Frost, A.E.; Rouzic, E.M.-L.; Romero, A.J.; Benton, W.W.; Elliott, C.G.; McGoon, M.D.; et al. Five-year outcomes of patients enrolled in the REVEAL registry. Chest 2015, 148, 1043–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IPAH (n = 1481) | PAH-CHD Total (n = 680) | ES Untreated (n = 80) | ES Treated (n = 240) | Non-ES-PAH Treated (n = 167) | Fontan Treated (n = 7) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 66.3 ± 15.5 | 45.5 ± 16.8 | 40.1 ± 14.6 | 43.8 ± 15.1 | 47.6 ± 18.3 | 31.9 ± 9.4 | |||||||
| Sex | male | 604 | 40.8% | 227 | 33.4% | 26 | 32.5% | 76 | 31.7% | 55 | 32.9% | 3 | 42.9% |
| female | 877 | 59.2% | 453 | 66.6% | 54 | 67.5% | 164 | 68.3% | 112 | 67.1% | 4 | 57.1% | |
| FC | unknown | 78 | 5.3% | 80 | 11.8% | 30 | 37.5% | 19 | 7.9% | 14 | 8.4% | 4 | 57.1% |
| I | 3 | 0.2% | 22 | 3.2% | 1 | 1.3% | 8 | 3.3% | 5 | 3.0% | 0 | 0% | |
| II | 178 | 12.0% | 159 | 23.4% | 11 | 13.8% | 65 | 27.1% | 44 | 26.3% | 3 | 42.9% | |
| III | 997 | 67.3% | 392 | 57.6% | 36 | 45.0% | 136 | 56.7% | 101 | 60.5% | 0 | 0% | |
| IV | 225 | 15.2% | 27 | 4.0% | 2 | 2.5% | 12 | 5.0% | 3 | 1.8% | 0 | 0% | |
| 6-min-walk distance (m) | 294 ± 123 | 367 ± 120 | 392 ± 118 | 354 ± 121 | 381 ± 120 | 475 ± 50 | |||||||
| Treatment Characteristics | Number of Patients Included |
|---|---|
| CHD-PAH total | 680 |
| Eisenmenger - untreated | 80 (11.8%) |
| Eisenmenger - treated | 240 (35.3%) |
| Non-Eisenmenger-PAH - treated | 167 (24.6%) |
| Fontan - treated | 7 (1.0%) |
| Not categorized | 186 (27.4%) |
| Targeted PAH medication | |
| Endothelin receptor antagonists (ERA) | 389 (57.2%) |
| Phosphodiesterase type-5 inhibitors (PDE5i) | 353 (51.9%) |
| Prostanoids | 35 (5.1%) |
| Soluble guanylate cyclase (sGC) stimulator | 17 (2.5%) |
| Tyrosine kinase inhibitor | 1 (0.1%) |
| Treatment strategy and supportive treatment | |
| Monotherapy with targeted PAH medication | 408 (60.0%) |
| Combination therapy with targeted PAH medication | 192 (28.3%) |
| Oral anticoagulation: Vitamin K antagonists | 217 (31.9%) |
| Oral anticoagulation: Non-vitamin K antagonists (NOAC) | 30 (4.4%) |
| Antiplatelet therapy (Aspirin, Clopidogrel) | 68 (10.0%) |
| Treatment Group | Number of Patients Included (Total) | Targeted PAH Medication | n (%) | p-Value | Mono-/Combination-Therapy | p-Value |
|---|---|---|---|---|---|---|
| CHD-associated PAH - total | 680 | ERA PDE5-I Prostanoid sGC-stimulator Tyrosine kinase inhibitor | 389 (57.2%) 353 (51.9%) 35 (5.1%) 17 (2.5%) 1 (0.1%) | 408 (60.0%)/ 192 (28.3%) | ||
| Eisenmenger, treated | 240 | ERA PDE5-I Prostanoid sGC-stimulator Tyrosine kinase inhibitor | 172 (71.7%) 134 (55.8%) 20 (8.3%) 7 (2.9%) 0 | 152 (63.3%)/ 88 (36.7%) | ||
| Non-Eisenmenger-PAH, treated | 167 | ERA PDE5-I Prostanoid sGC-stimulator Tyrosine kinase inhibitor | 100 (59.9%) 97 (58.1%) 7 (4.2%) 6 (3.6%) 1 (0.6%) | 0.013 0.652 0.099 0.703 0.410 | 121 (72.5%)/ 46 (27.5%) | 0.054 |
| Fontan, treated | 7 | ERA PDE5-I Prostanoid sGC-stimulator Tyrosine kinase inhibitor | 1 (14.3%) 7 (100 %) 1 (14.3%) 0 0 | 6 (85.7%)/ 1 (14.3%) | ||
| Not categorized, treated | 187 | ERA PDE5-I Prostanoid sGC-stimulator Tyrosine kinase inhibitor | 116 (62.4%) 115 (61.8%) 7 (3.8%) 4 (2.2%) 0 | 129 (69.4%)/ 57 (30.6%) |
| n (%) | ||
|---|---|---|
| 1. Pre-tricuspid shunts (n = 213) | Persisting foramen ovale | 5 (0.7) |
| Atrial septal defect | 186 (27.4) | |
| Partial atrioventricular septal defect | 4 (0.6) | |
| Partial anomalous pulmonary venous return | 16 (2.4) | |
| Total anomalous pulmonary venous return | 1 (0.1) | |
| details not stated | 1 (0.1) | |
| 2. Post-tricuspid shunts (n = 325) | Complete atrioventricular septal defect | 79 (11.6) |
| Ventricular septal defect | 199 (29.3) | |
| Patent ductus arteriosus Botalli | 40 (5.9) | |
| Aortopulmonary window | 6 (0.9) | |
| details not stated | 1 (0.1) | |
| 3. Complex anomalies (n = 121) | Complete transposition of great arteries | 19 (2.8) |
| Congenitally corrected transposition of great arteries | 12 (1.8) | |
| Double-outlet right ventricle with transposition of great arteries | 5 (0.7) | |
| Truncus arteriosus | 4 (0.6) | |
| Tricuspid atresia | 12 (1.8) | |
| Double-inlet ventricle | 13 (1.9) | |
| Pulmonary atresia with intact ventricular septum | 1 (0.1) | |
| Fallot´s Tetralogy | 13 (1.9) | |
| Double-outlet right ventricle—Fallot type | 9 (1.3) | |
| Pulmonary atresia with ventricular septal defect | 30 (4.4) | |
| Ebstein’s anomaly | 2 (0.3) | |
| details not stated | 1 (0.1) | |
| 4. Left heart disease/aortic valve, and aortic anomalies (n = 9) | Aortic coarctation | 2 (0.3) |
| Aortic valve stenosis | 5 (0.7) | |
| Subaortic stenosis | 1 (0.1) | |
| Aortic valve regurgitation | 1 (0.1) | |
| 5. Other congenital cardiac anomalies (n = 12) | Atrioventricular valve anomalies | 2 (0.3) |
| other | 5 (0.7) | |
| Pulmonary artery stenosis | 3 (0.4) | |
| details not stated | 2 (0.3) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaemmerer, H.; Gorenflo, M.; Huscher, D.; Pittrow, D.; Apitz, C.; Baumgartner, H.; Berger, F.; Bruch, L.; Brunnemer, E.; Budts, W.; et al. Pulmonary Hypertension in Adults with Congenital Heart Disease: Real-World Data from the International COMPERA-CHD Registry. J. Clin. Med. 2020, 9, 1456. https://doi.org/10.3390/jcm9051456
Kaemmerer H, Gorenflo M, Huscher D, Pittrow D, Apitz C, Baumgartner H, Berger F, Bruch L, Brunnemer E, Budts W, et al. Pulmonary Hypertension in Adults with Congenital Heart Disease: Real-World Data from the International COMPERA-CHD Registry. Journal of Clinical Medicine. 2020; 9(5):1456. https://doi.org/10.3390/jcm9051456
Chicago/Turabian StyleKaemmerer, Harald, Matthias Gorenflo, Dörte Huscher, David Pittrow, Christian Apitz, Helmut Baumgartner, Felix Berger, Leonhard Bruch, Eva Brunnemer, Werner Budts, and et al. 2020. "Pulmonary Hypertension in Adults with Congenital Heart Disease: Real-World Data from the International COMPERA-CHD Registry" Journal of Clinical Medicine 9, no. 5: 1456. https://doi.org/10.3390/jcm9051456
APA StyleKaemmerer, H., Gorenflo, M., Huscher, D., Pittrow, D., Apitz, C., Baumgartner, H., Berger, F., Bruch, L., Brunnemer, E., Budts, W., Claussen, M., Coghlan, G., Dähnert, I., D’Alto, M., Delcroix, M., Distler, O., Dittrich, S., Dumitrescu, D., Ewert, R., ... Rosenkranz, S. (2020). Pulmonary Hypertension in Adults with Congenital Heart Disease: Real-World Data from the International COMPERA-CHD Registry. Journal of Clinical Medicine, 9(5), 1456. https://doi.org/10.3390/jcm9051456