Novel Insights into the Role of UBE3A in Regulating Apoptosis and Proliferation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. MEFs Generation
2.2. BrdU Incorporation
2.3. Cell Proliferation Assay
2.4. Apoptosis
2.5. Caspase 3/7 Activity
2.6. Western Blot Analysis
2.7. Live Cell Imaging
2.8. CytoPainter Cell Proliferation Assay
2.9. SILAC
2.10. Mass Spectrometry Analysis
2.11. RNA-Seq Library Preparation of MEFs
2.12. Bioinformatics Analysis of MEFs Gene Expression
2.13. Bioinformatics Analysis of Mouse Hippocampi Dataset
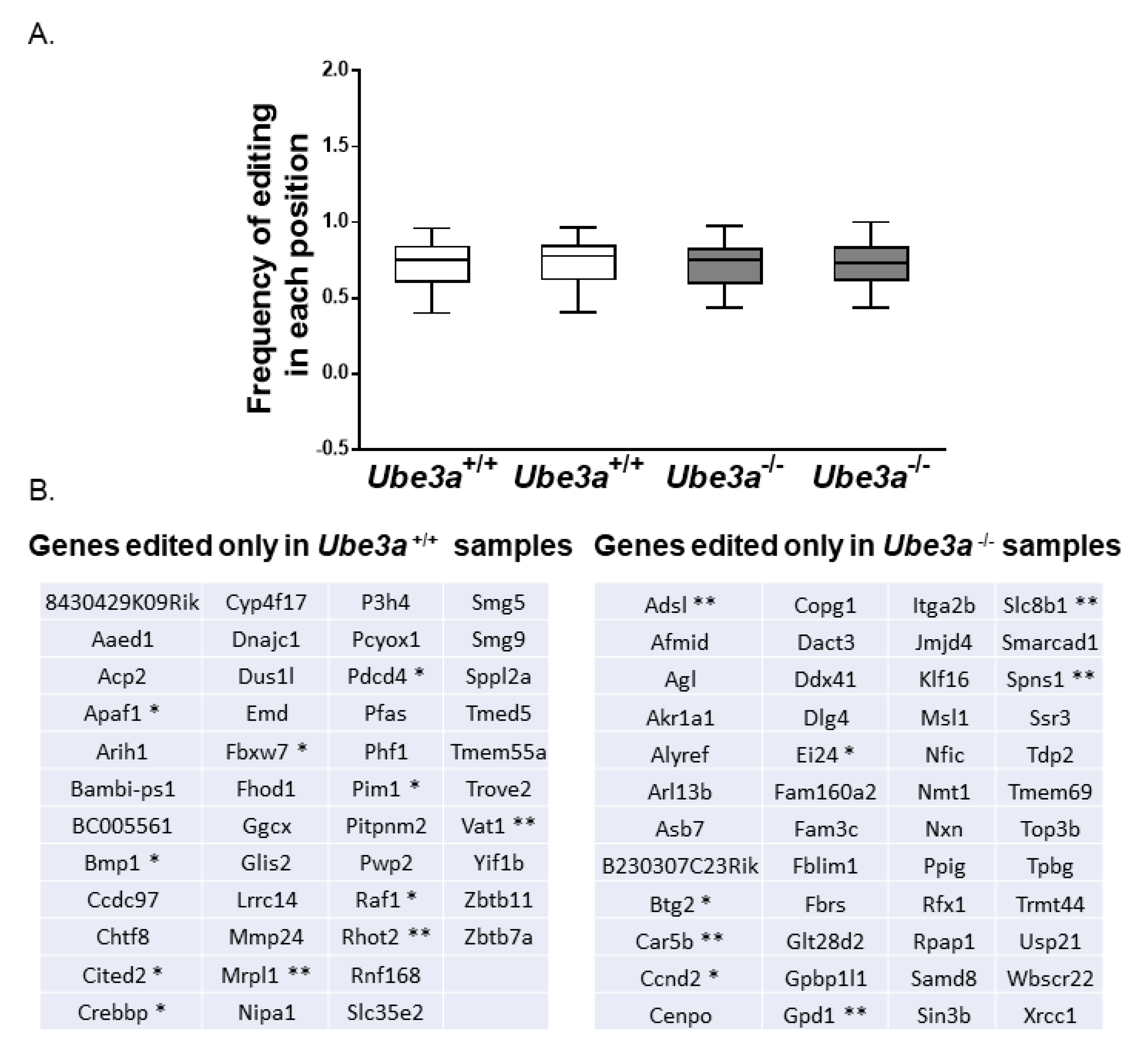
2.14. Bioinformatics Analysis of RNA-Editing Sites in MEFs RNA-Seq Data
2.15. Statistical Analysis
2.16. Data Availability
3. Results
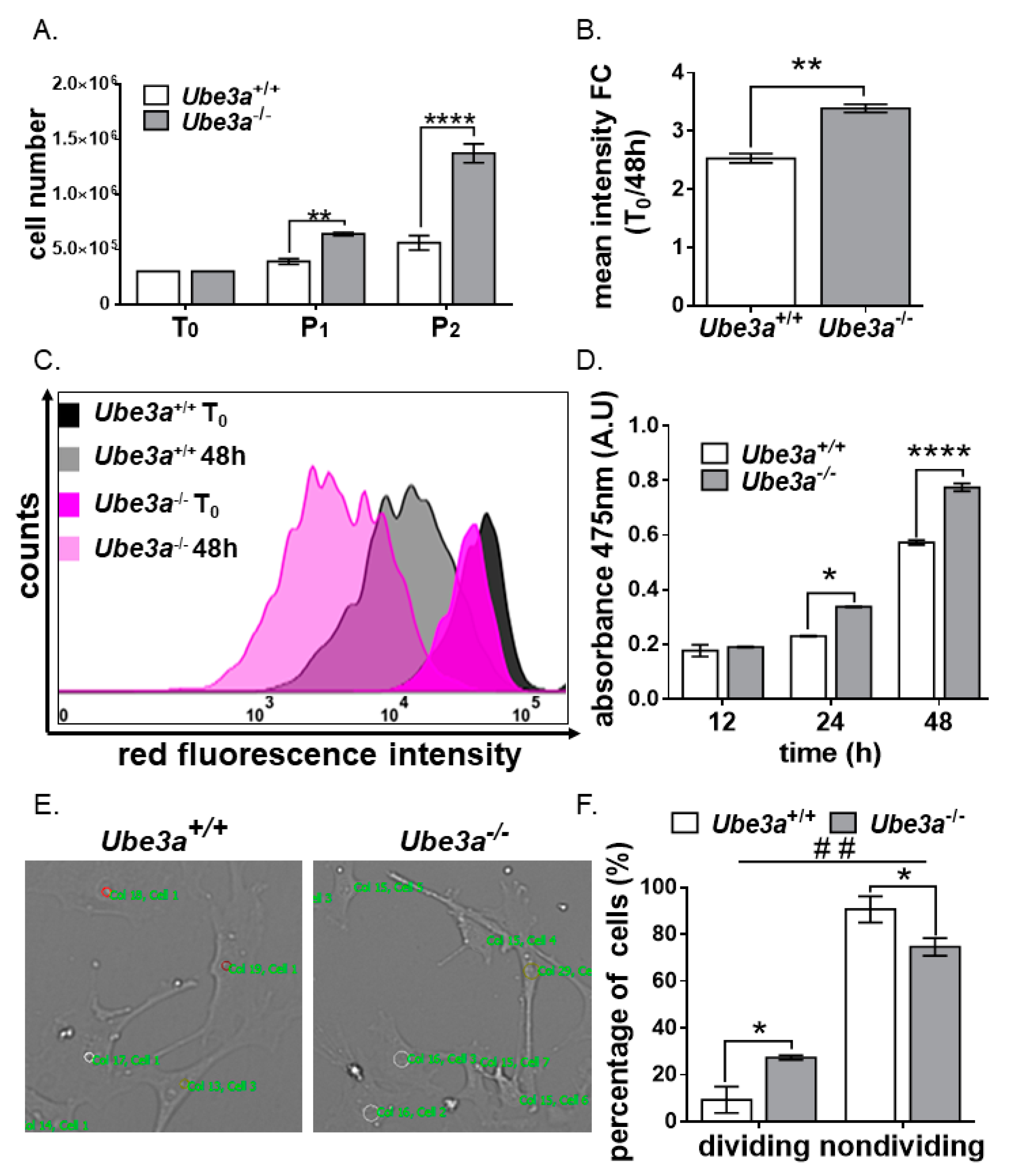
3.1. Ube3a Deletion Enhances the Growth Capacity of MEFs
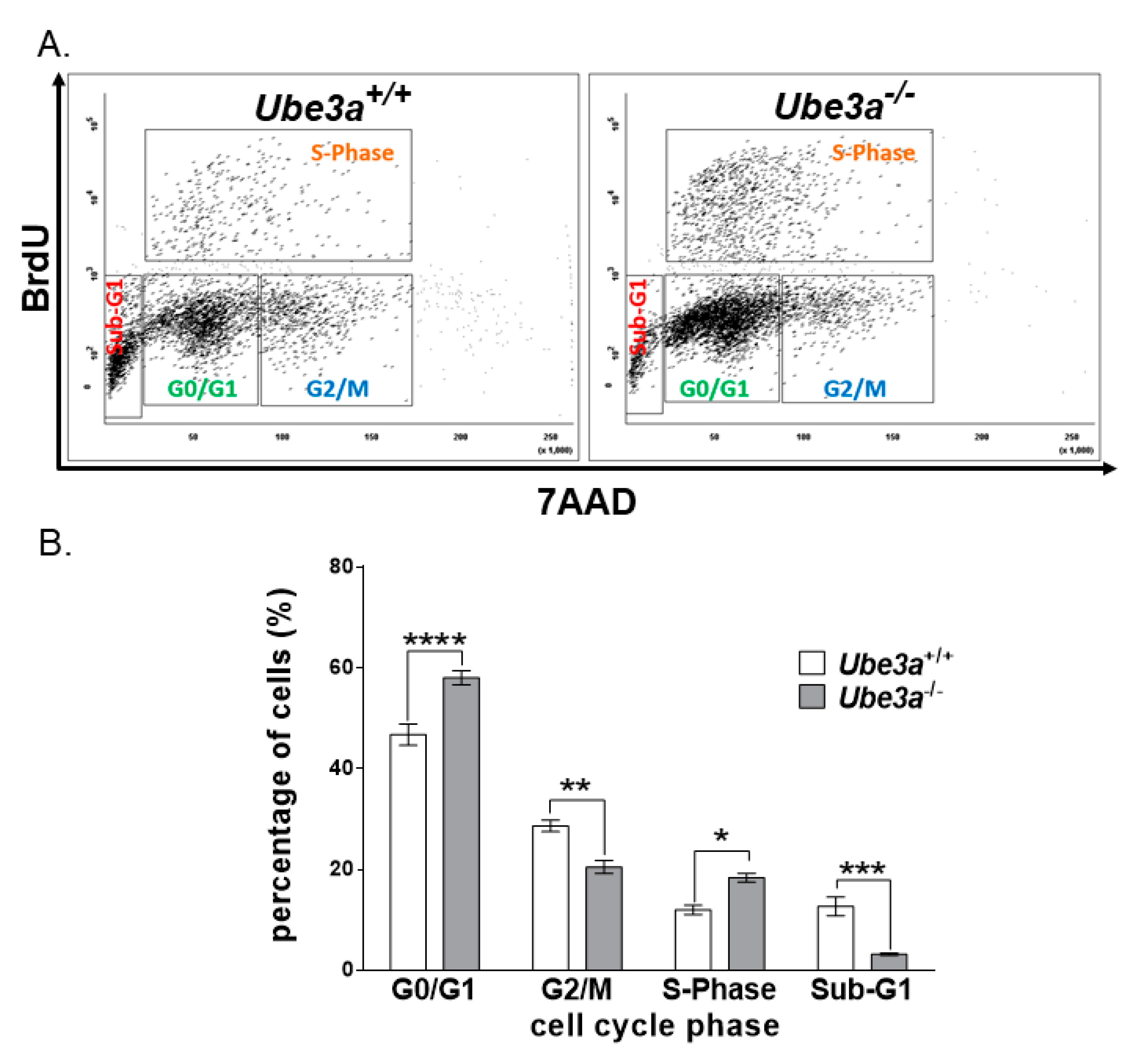
3.2. Ube3a Deletion Alters Cell Cycle Progression
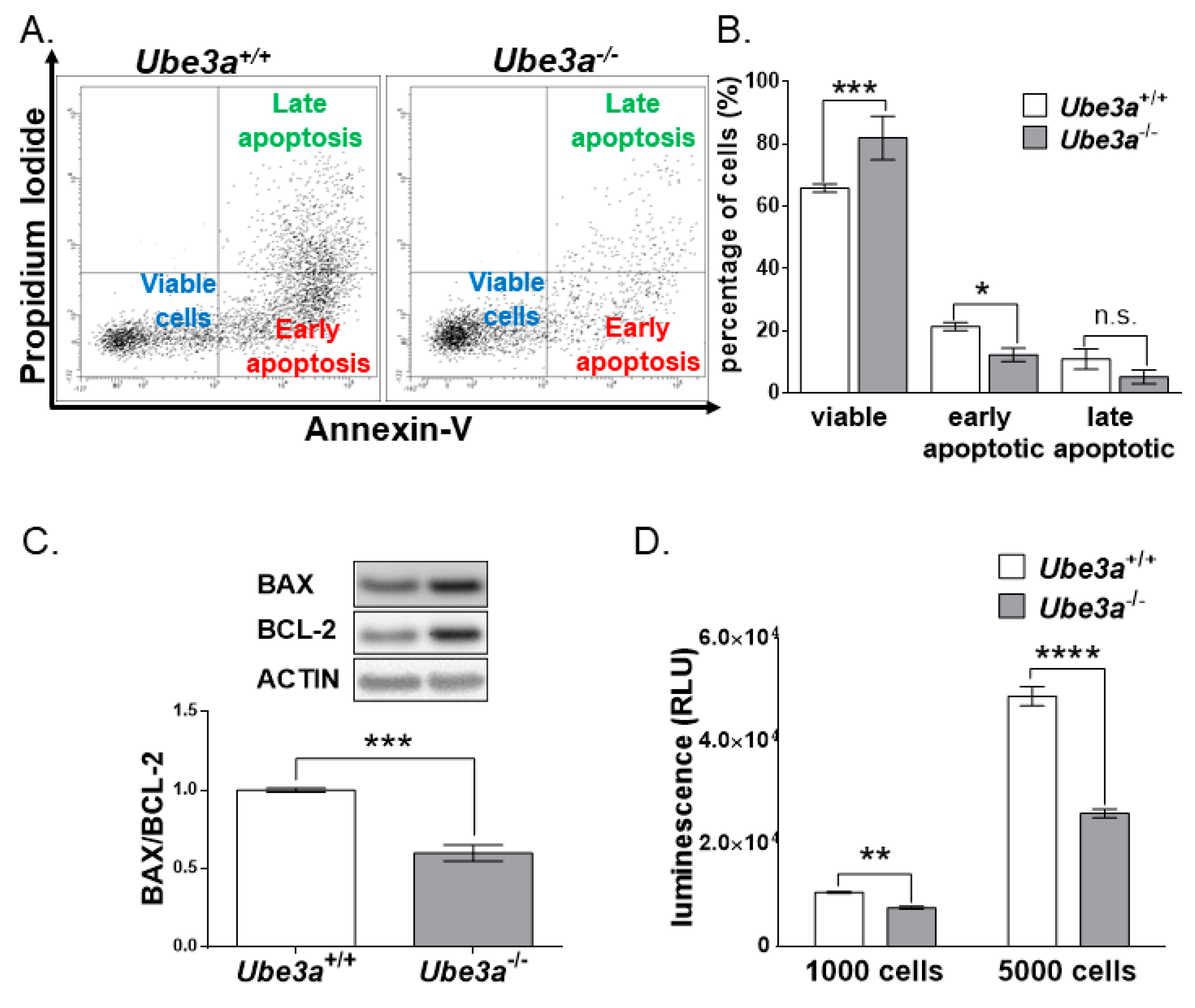
3.3. Ube3a Deletion Reduces the Apoptotic Capacity of MEFs
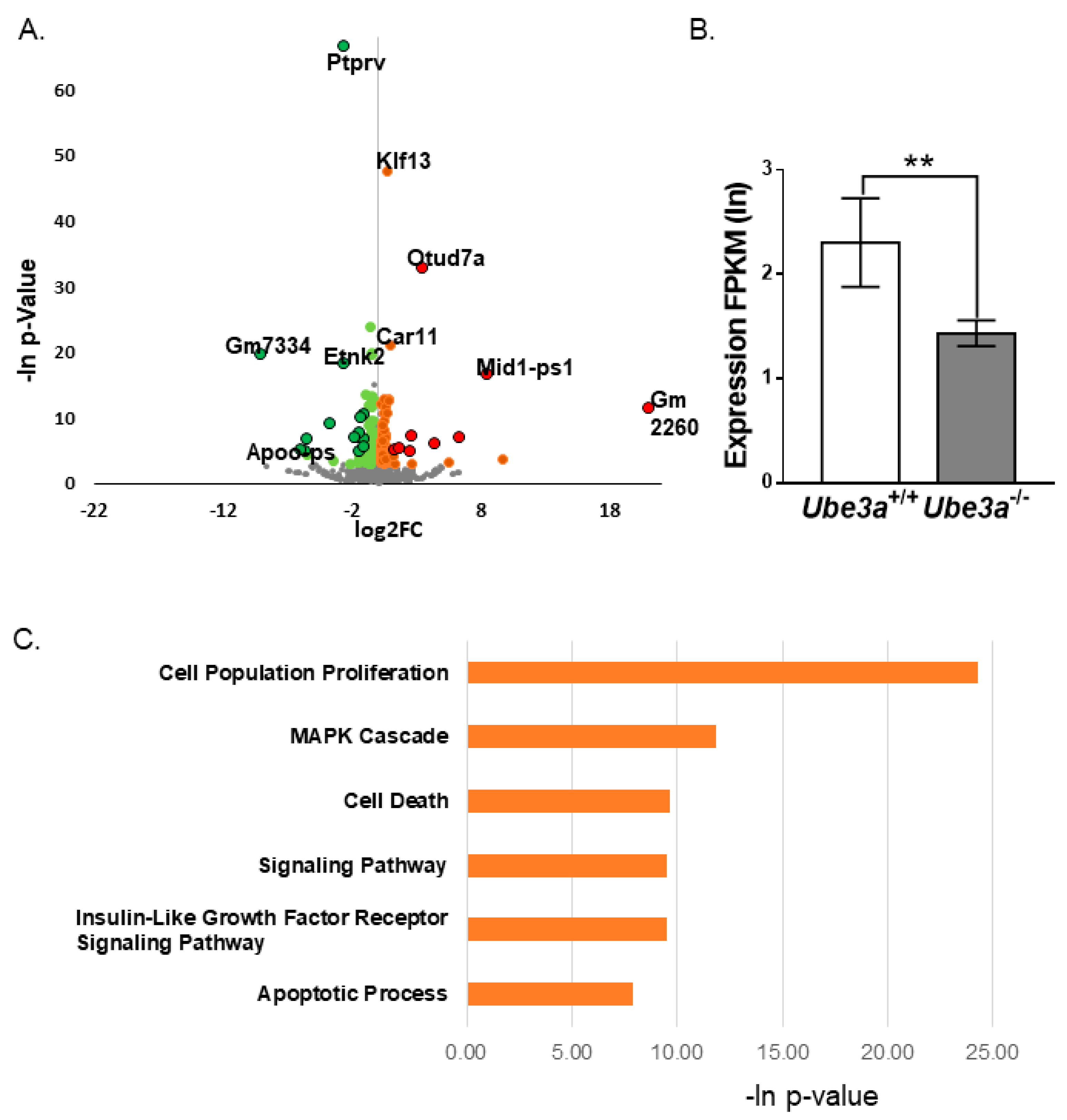
3.4. Transcriptomic Analyses Support Altered Apoptosis Processes Triggered by Ube3a Deletion
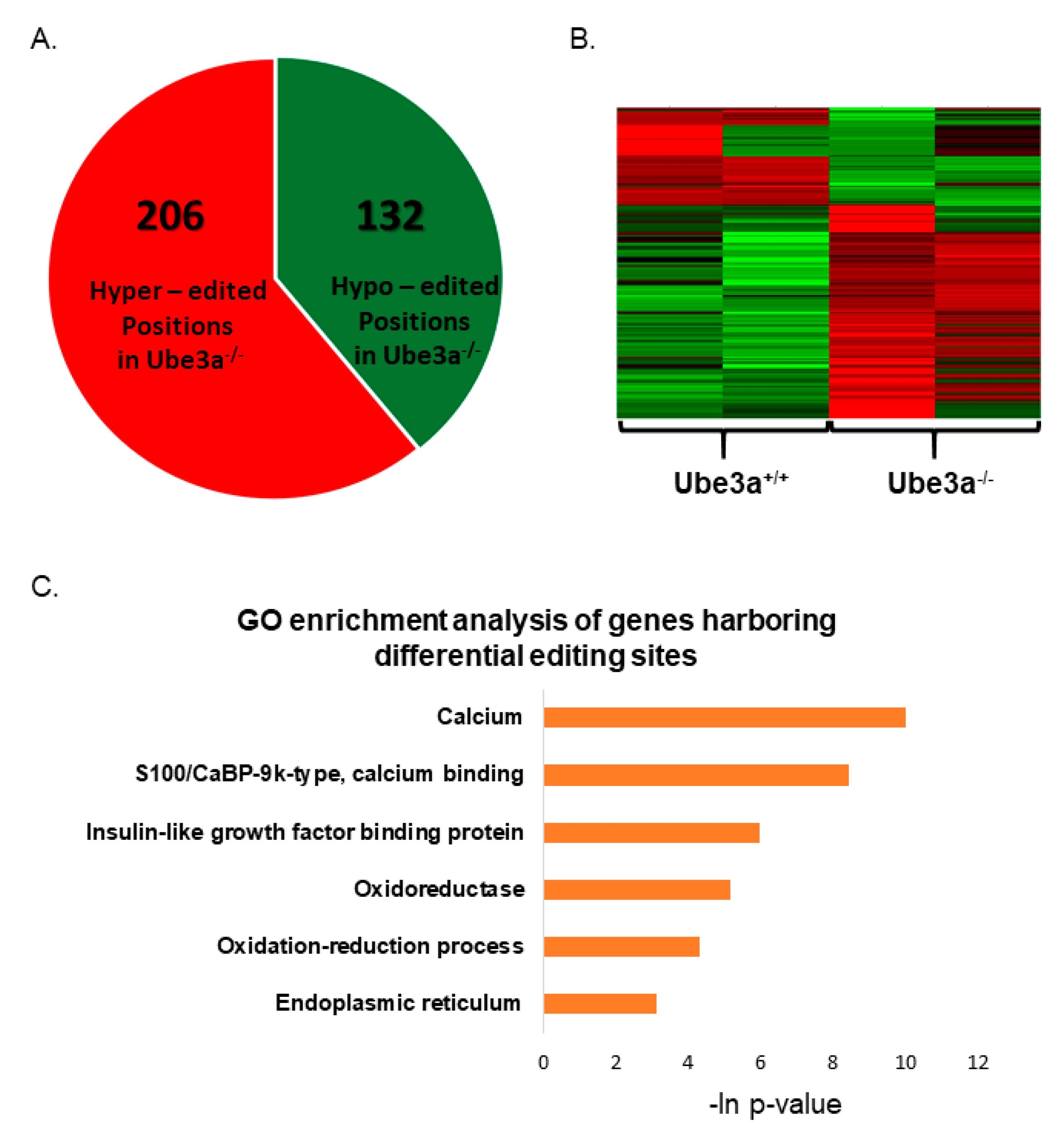
3.5. Dysregulation of RNA Editing Affects Apoptosis-Related Pathways
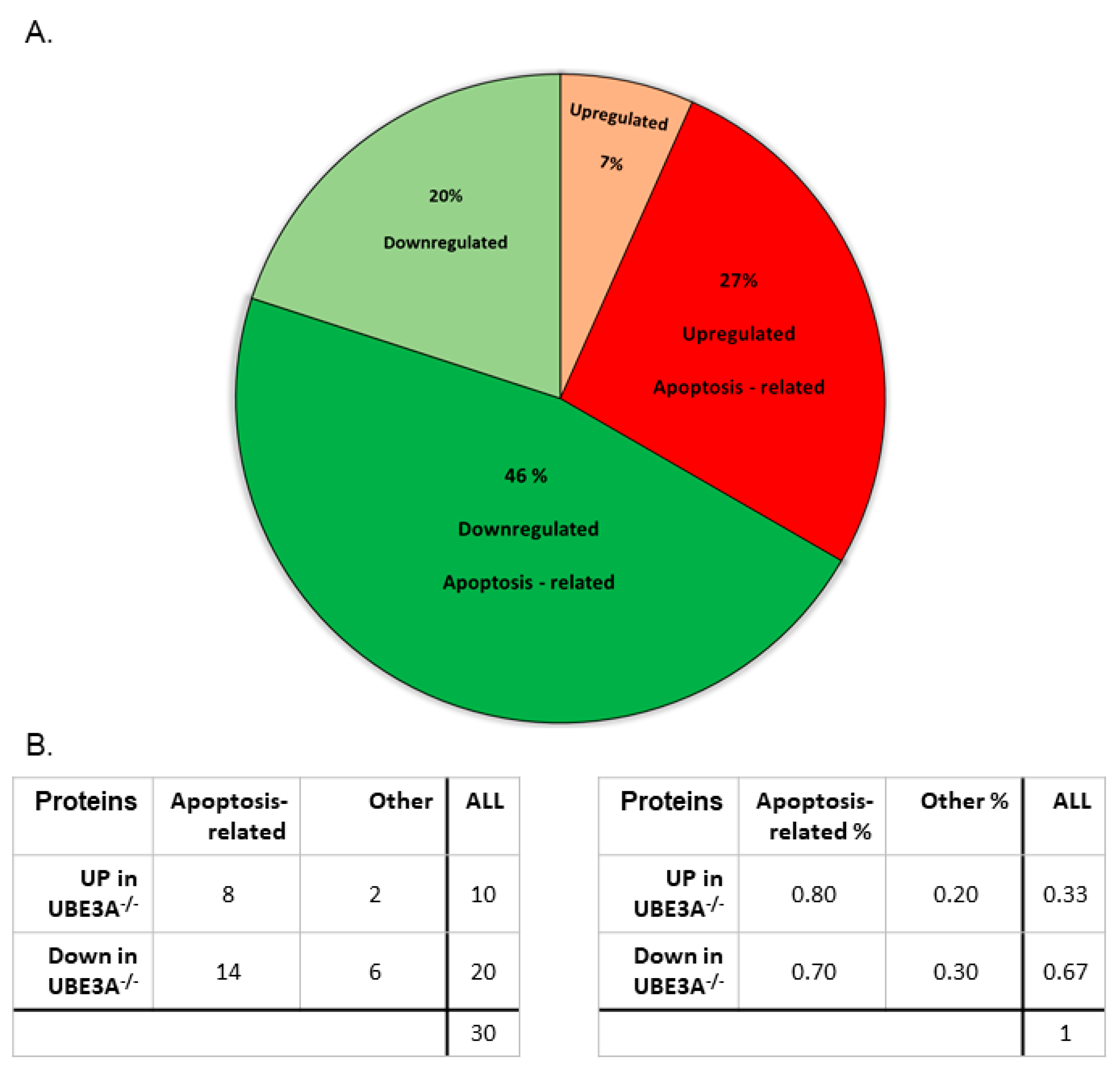
3.6. Proteomic Analysis Supports Altered Apoptotic Processes Triggered by Ube3a Deletion
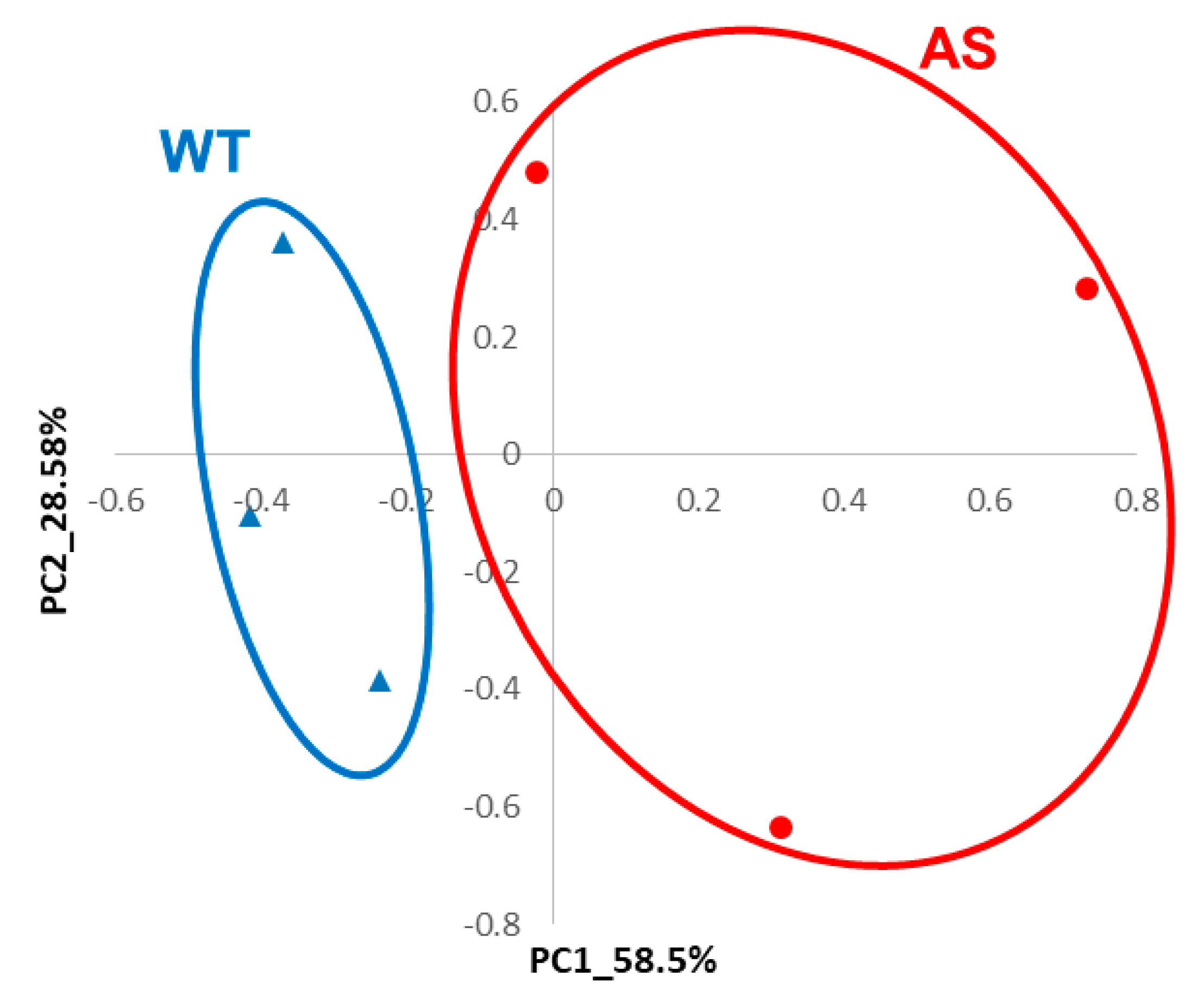
3.7. Mouse Brain RNA-Seq Data Also Reveals Alterations in Apoptotic Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ramamoorthy, S.; Nawaz, Z. E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl. Recept. Signal. 2008, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Sailer, C.; Offensperger, F.; Julier, A.; Kammer, K.-M.; Walker-Gray, R.; Gold, M.G.; Scheffner, M.; Stengel, F. Structural dynamics of the E6AP/UBE3A-E6-p53 enzyme-substrate complex. Nat. Commun. 2018, 9, 4441. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Li, Y.; Chang, W.-T.; Chen, Z.-C.; Cheng, J.-T.; Tsai, C.-C. Ubiquitin-protein ligase E3a (UBE3A) as a new biomarker of cardiac hypertrophy in cell models. J. Food Drug Anal. 2018, 27, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Gustin, R.M.; Bichell, T.J.V.; Bubser, M.; Daily, J.; Filonova, I.; Mrelashvili, D.; Deutch, A.Y.; Colbran, R.J.; Weeber, E.J.; Haas, K.F. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol. Dis. 2010, 39, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamoorthy, S.; Dhananjayan, S.C.; DeMayo, F.J.; Nawaz, Z. Isoform-Specific Degradation of PR-B by E6-AP Is Critical for Normal Mammary Gland Development. Mol. Endocrinol. 2010, 24, 2099–2113. [Google Scholar] [CrossRef] [Green Version]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. Review the HECT Family of E3 Ubiquitin Ligases: Multiple Players in Cancer Development. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef]
- Mishra, A.; Godavarthi, S.K.; Jana, N.R. UBE3A/E6-AP regulates cell proliferation by promoting proteasomal degradation of p27. Neurobiol. Dis. 2009, 36, 26–34. [Google Scholar] [CrossRef]
- Kumar, S.; Talis, A.L.; Howley, P. Identification of HHR23A as a Substrate for E6-associated Protein-mediated Ubiquitination. J. Biol. Chem. 1999, 274, 18785–18792. [Google Scholar] [CrossRef] [Green Version]
- Underbrink, M.P.; Howie, H.L.; Bedard, K.M.; Koop, J.I.; Galloway, D.A. E6 Proteins from Multiple Human Betapapillomavirus Types Degrade Bak and Protect Keratinocytes from Apoptosis after UVB Irradiation. J. Virol. 2008, 82, 10408–10417. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Niebler, M.; Qian, X.; Höfler, D.; Kogosov, V.; Kaewprag, J.; Kaufmann, A.M.; Ly, R.; Böhmer, G.; Zawatzky, R.; Rösl, F.; et al. Post-Translational Control of IL-1β via the Human Papillomavirus Type 16 E6 Oncoprotein: A Novel Mechanism of Innate Immune Escape Mediated by the E3-Ubiquitin Ligase E6-AP and p53. PLoS Pathog. 2013, 9, e1003536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawaz, Z.; Lonard, D.M.; Dennis, A.P.; Smith, C.L.; O’Malley, B.W. Proteasome-dependent degradation of the human estrogen receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Mohsin, S.K.; Gatalica, Z.; Fu, G.; Sharma, P.; Nawaz, Z. Decreased Expression of E6-Associated Protein in Breast and Prostate Carcinomas. Endocrinology 2005, 146, 1707–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, A.; Oh, A.S.; Bowden, E.T.; Lahusen, T.; Lorick, K.L.; Weissman, A.M.; Schlegel, R.; Wellstein, A.; Riegel, A.T. E6AP Mediates Regulated Proteasomal Degradation of the Nuclear Receptor Coactivator Amplified in Breast Cancer 1 in Immortalized Cells. Cancer Res. 2006, 66, 8680–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, D.; Paul, P.J.; Gulati, T.; Deb, S.; Khoo, C.; Russo, A.; Gallo, E.; Blandino, G.; Chan, A.-L.; Takano, E.; et al. E6AP promotes prostate cancer by reducing p27 expression. Oncotarget 2017, 8, 42939–42948. [Google Scholar] [CrossRef]
- Shai, A.; Pitot, H.C.; Lambert, P. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res. 2010, 70, 5064–5073. [Google Scholar] [CrossRef] [Green Version]
- Sell, G.L.; Margolis, S.S. From UBE3A to Angelman syndrome: A substrate perspective. Front. Mol. Neurosci. 2015, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Vatsa, N.; Jana, N.R. UBE3A and Its Link with Autism. Front. Mol. Neurosci. 2018, 11, 448. [Google Scholar] [CrossRef]
- Dindot, S.; Antalffy, B.A.; Bhattacharjee, M.B.; Beaudet, A.L. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2007, 17, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Kaphzan, H.; Buffington, S.A.; Jung, J.I.; Rasband, M.N.; Klann, E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J. Neurosci. 2011, 31, 17637–17648. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.-H.; Armstrong, D.; Albrecht, U.; Atkins, C.; Noebels, J.L.; Eichele, G.; Sweatt, J.D.; Beaudet, A.L. Mutation of the Angelman Ubiquitin Ligase in Mice Causes Increased Cytoplasmic p53 and Deficits of Contextual Learning and Long-Term Potentiation. Neuron 1998, 21, 799–811. [Google Scholar] [CrossRef] [Green Version]
- Kaphzan, H.; Hernandez, P.; Jung, J.I.; Cowansage, K.K.; Deinhardt, K.; Chao, M.V.; Abel, T.; Klann, E. Reversal of impaired hippocampal long-term potentiation and contextual fear memory deficits in Angelman syndrome model mice by ErbB inhibitors. Biol. Psychiatry 2012, 72, 182–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaphzan, H.; Buffington, S.A.; Ramaraj, A.B.; Lingrel, J.B.; Rasband, M.N.; Santini, E.; Klann, E. Genetic reduction of the α1 Subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in angelman syndrome. Cell Rep. 2013, 4, 405–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levav-Cohen, Y.; Wolyniec, K.; Alsheich-Bartok, O.; Chan, A.-L.; Woods, S.J.; Jiang, Y.-H.; Haupt, S.; Haupt, Y. E6AP is required for replicative and oncogene-induced senescence in mouse embryo fibroblasts. Oncogene 2011, 31, 2199–2209. [Google Scholar] [CrossRef] [Green Version]
- Santini, E.; Turner, K.L.; Ramaraj, A.B.; Murphy, M.P.; Klann, E.; Kaphzan, H. Mitochondrial Superoxide Contributes to Hippocampal Synaptic Dysfunction and Memory Deficits in Angelman Syndrome Model Mice. J. Neurosci. 2015, 35, 16213–16220. [Google Scholar] [CrossRef] [Green Version]
- De Zio, D.; Giunta, L.; Corvaro, M.; Ferraro, E.; Cecconi, F. Expanding roles of programmed cell death in mammalian neurodevelopment. Semin. Cell Dev. Biol. 2005, 16, 281–294. [Google Scholar] [CrossRef]
- Lossi, L.; Castagna, C.; Merighi, A. Caspase-3 Mediated Cell Death in the Normal Development of the Mammalian Cerebellum. Int. J. Mol. Sci. 2018, 19, 3999. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Miura, M. Programmed Cell Death in Neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Thomaidou, D.; Mione, M.C.; Cavanagh, J.F.R.; Parnavelas, J.G. Apoptosis and Its Relation to the Cell Cycle in the Developing Cerebral Cortex. J. Neurosci. 1997, 17, 1075–1085. [Google Scholar] [CrossRef]
- Pozueta, J.; Lefort, R.; Ribe, E.; Troy, C.M.; Arancio, O.; Shelanski, M. Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nat. Commun. 2013, 4, 1939. [Google Scholar] [CrossRef] [Green Version]
- Ertürk, A.; Wang, Y.; Sheng, M. Local Pruning of Dendrites and Spines by Caspase-3-Dependent and Proteasome-Limited Mechanisms. J. Neurosci. 2014, 34, 1672–1688. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Ji, Y.; Ding, Y.; Jiang, W.; Sun, Y.; Lu, B.; Nagappan, G. BDNF pro-peptide regulates dendritic spines via caspase-3. Cell Death Dis. 2016, 7, e2264. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Gilbert, J.P.; Huo, Y.; Sharaflari, R.; Nee, M.; Qiao, H.; Man, H.-Y. Faculty Opinions recommendation of The Autism Protein Ube3A/E6AP Remodels Neuronal Dendritic Arborization via Caspase-Dependent Microtubule Destabilization. J. Neurosci. 2018, 38, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Doll, C.A.; Broadie, K. Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Front. Cell. Neurosci. 2014, 8, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiffele, P.; Beg, A.A. Angelman syndrome connections. Nature. 2010, 468, 907–908. [Google Scholar] [CrossRef]
- Bayır, H.; Kagan, V. Bench-to-bedside review: Mitochondrial injury, oxidative stress and apoptosis—There is nothing more practical than a good theory. Crit. Care 2008, 12, 206. [Google Scholar] [CrossRef] [Green Version]
- Atlante, A.; De Bari, L.; Bobba, A.; Marra, E.; Calissano, P.; Passarella, S. Cytochrome c, released from cerebellar granule cells undergoing apoptosis or excytotoxic death, can generate protonmotive force and drive ATP synthesis in isolated mitochondria. J. Neurochem. 2003, 86, 591–604. [Google Scholar] [CrossRef] [Green Version]
- Stojanovski, D.; Johnston, A.J.; Streimann, I.; Hoogenraad, N.J.; Ryan, M.T. Import of nuclear-encoded proteins into mitochondria. Exp. Physiol. 2003, 88, 57–64. [Google Scholar] [CrossRef]
- Siddiqui, M.F.; Elwell, C.; Johnson, S. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism-Open Access 2016, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Jarskog, L.F.; Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 846–858. [Google Scholar] [CrossRef]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.J.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial dysfunction in schizophrenia: Evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulyakova, N.; Andreazza, A.C.; Mills, L.R.; Eubanks, J.H. Mitochondrial Dysfunction in the Pathogenesis of Rett Syndrome: Implications for Mitochondria-Targeted Therapies. Front. Cell. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Cali, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagalinec, M.; Liiv, M.; Hodurova, Z.; Hickey, M.; Vaarmann, A.; Mandel, M.; Zeb, A.; Choubey, V.; Kuum, M.; Safiulina, D.; et al. Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome. PLoS Biol. 2016, 14, e1002511. [Google Scholar] [CrossRef]
- Marazziti, D.; Baroni, S.; Picchetti, M.; Landi, P.; Silvestri, S.; Vatteroni, E.; Catena Dell’Osso, M. Psychiatric disorders and mitochondrial dysfunctions. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 270–275. [Google Scholar]
- Su, H.; Fan, W.; Coskun, P.E.; Vesa, J.; Gold, J.-A.; Jiang, Y.-H.; Potluri, P.; Procaccio, V.; Acab, A.; Weiss, J.H.; et al. Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci. Lett. 2009, 487, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Berkowitz, B.A.; Lenning, J.; Khetarpal, N.; Tran, C.; Wu, J.Y.; Berri, A.M.; Dernay, K.; Haacke, E.M.; Shafie-Khorassani, F.; Podolsky, R.H.; et al. In vivo imaging of prodromal hippocampus CA1 subfield oxidative stress in models of Alzheimer disease and Angelman syndrome. FASEB J. 2017, 31, 4179–4186. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, T.; Schwarzfischer, M.; Skylaki, S.; SchaubergeriD, B.; Hoppe, P.S.; Loeffler, D.; Kokkaliaris, K.; Hastreiter, S.; Skylaki, E.; Filipczyk, A.; et al. Software tools for single-cell tracking and quantification of cellular and molecular properties. Nat. Biotechnol. 2016, 34, 703–706. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 002832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.M.; Baseler, M.; Lane, H.C.; Lempicki, R. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2015, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- Koyavski, L.; Panov, J.; Simchi, L.; Rayi, P.R.; Sharvit, L.; Feuermann, Y.; Kaphzan, H. Sex-Dependent Sensory Phenotypes and Related Transcriptomic Expression Profiles Are Differentially Affected by Angelman Syndrome. Mol. Neurobiol. 2019, 56, 5998–6016. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression with Random Forest. R News 2002, 2, 18–22. [Google Scholar]
- Venables, W.; Ripley, B. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002; ISBN 0-387-95457-0. [Google Scholar]
- Zhang, Q.; Xiao, X. Genome sequence–independent identification of RNA editing sites. Nat. Methods 2015, 12, 347–350. [Google Scholar] [CrossRef] [Green Version]
- Wilks, S.S. The Large-Sample Distribution of the Likelihood Ratio for Testing Composite Hypotheses. Ann. Math. Stat. 1938, 9, 60–62. [Google Scholar] [CrossRef]
- Roehm, N.W.; Rodgers, G.H.; Hatfield, S.M.; Glasebrook, A.L. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Methods 1991, 142, 257–265. [Google Scholar] [CrossRef]
- Carbonari, M. New use for an old reagent: Cell cycle analysis of DNA content using flow cytometry in formamide treated cells. Cytom. Part A 2016, 89, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raisova, M.; Hossini, A.M.; Eberle, J.; Riebeling, C.; Wieder, T.; Sturm, I.; Daniel, P.T.; Orfanos, C.E.; Geilen, C.C. The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J. Investig. Dermatol. 2001, 117, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staedtler, F.; Hartmann, N.; Letzkus, M.; Bongiovanni, S.; Scherer, A.; Marc, P.; Johnson, K.J.; Schumacher, M. Robust and tissue-independent gender-specific transcript biomarkers. Biomarkers 2013, 18, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.Y.; Fu, G.; Ismail, A.; Srinivasan, S.; Cao, X.; Tu, Y.; Lu, S.; Nawaz, Z. Multifunction Steroid Receptor Coactivator, E6-Associated Protein, Is Involved in Development of the Prostate Gland. Mol. Endocrinol. 2006, 20, 544–559. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Li, M.; Zhang, S.; Lu, Y.; Liang, Y.; Zhao, K.; Li, Y. Insulin-like growth factor 1/insulin-like growth factor 1 receptor signaling protects against cell apoptosis through the PI3K/AKT pathway in glioblastoma cells. Exp. Ther. Med. 2018, 16, 1477–1482. [Google Scholar] [CrossRef]
- Fan, J.; Wei, Q.; Liao, J.; Zou, Y.; Song, D.; Xiong, D.; Ma, C.; Hu, X.; Qu, X.; Chen, L.; et al. Noncanonical Wnt signaling plays an important role in modulating canonical Wnt-regulated stemness, proliferation and terminal differentiation of hepatic progenitors. Oncotarget 2017, 8, 27105–27119. [Google Scholar] [CrossRef] [Green Version]
- Baysal, B.E.; Sharma, S.; Hashemikhabir, S.; Janga, S.C. RNA Editing in Pathogenesis of Cancer. Cancer Res. 2017, 77, 3733–3739. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q. Analysis of RNA Editing Sites from RNA-Seq Data Using GIREMI. Methods Mol Biol. 2018, 1751, 101–108. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Gamell, C.; Gulati, T.; Levav-Cohen, Y.; Young, R.J.; Do, H.; Pilling, P.; Takano, E.; Watkins, N.; Fox, S.B.; Russell, P.; et al. Reduced abundance of the E3 ubiquitin ligase E6AP contributes to decreased expression of the INK4/ARF locus in non-small cell lung cancer. Sci. Signal. 2017, 10, eaaf8223. [Google Scholar] [CrossRef] [PubMed]
- Wenric, S.; Shemirani, R. Using Supervised Learning Methods for Gene Selection in RNA-Seq Case-Control Studies. Front. Genet. 2018, 9, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Southwell, D.G.; Paredes, M.F.; Galvao, R.P.; Jones, D.L.; Froemke, R.C.; Sebe, J.Y.; Alfaro-Cervello, C.; Tang, Y.; García-Verdugo, J.M.; Rubenstein, J.L.; et al. Intrinsically determined cell death of developing cortical interneurons. Nature 2012, 491, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daily, J.; Smith, A.G.; Weeber, E.J. Spatial and temporal silencing of the human maternal UBE3A gene. Eur. J. Paediatr. Neurol. 2012, 16, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Matsui, H.; Gavinio, R.; Asano, T.; Uemura, N.; Ito, H.; Taniguchi, Y.; Kobayashi, Y.; Maki, T.; Shen, J.; Takeda, S.; et al. PINK1 and Parkin complementarily protect dopaminergic neurons in vertebrates. Hum. Mol. Genet. 2013, 22, 2423–2434. [Google Scholar] [CrossRef] [Green Version]
- Wolyniec, K.; Levav-Cohen, Y.; Jiang, Y.-H.; Haupt, S.; Haupt, Y. The E6AP E3 ubiquitin ligase regulates the cellular response to oxidative stress. Oncogene 2012, 32, 3510–3519. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.C. Proapoptotic multidomain Bcl-2/Bax-family proteins: Mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006, 13, 1378–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Deng, S.; Liu, H.; Liu, Y.; Yang, Z.; Xing, T.; Jing, B.; Zhang, X. Knockdown of ubiquitin protein ligase E3A affects proliferation and invasion, and induces apoptosis of breast cancer cells through regulation of annexin A2. Mol. Med. Rep. 2012, 12, 1107–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guadagno, J.; Swan, P.; Shaikh, R.; Cregan, S.P. Microglia-derived IL-1β triggers p53-mediated cell cycle arrest and apoptosis in neural precursor cells. Cell Death Dis. 2015, 6, e1779. [Google Scholar] [CrossRef] [Green Version]
- Shan, H.; Bian, Y.; Shu, Z.; Zhang, L.; Zhu, J.; Ding, J.; Lu, M.; Xiao, M.; Hu, G. Fluoxetine protects against IL-1β-induced neuronal apoptosis via downregulation of p53. Neuropharmacology 2016, 107, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Corbett, G.; Roy, A.; Pahan, K. Gemfibrozil, a lipid-lowering drug, upregulates IL-1 receptor antagonist in mouse cortical neurons: Implications for neuronal self-defense. J. Immunol. 2012, 189, 1002–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schizas, N.; Perry, S.; Andersson, B.; Wählby, C.; Kullander, K.; Hailer, N.P. Differential Neuroprotective Effects of Interleukin-1 Receptor Antagonist on Spinal Cord Neurons after Excitotoxic Injury. Neuroimmunomodulation 2018, 24, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Jin, F.; Van Deursen, J. The yin and yang of the Cdkn2a locus in senescence and aging. Cell Cycle 2008, 7, 2795–2802. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, Z.; Lonard, D.M.; Smith, C.L.; Lev-Lehman, E.; Tsai, S.Y.; Tsai, M.-J.; O’Malley, B.W. The Angelman Syndrome-Associated Protein, E6-AP, Is a Coactivator for the Nuclear Hormone Receptor Superfamily. Mol. Cell. Biol. 1999, 19, 1182–1189. [Google Scholar] [CrossRef] [Green Version]
- Pantic, B.; Trevisan, E.; Citta, A.; Rigobello, M.P.; Marin, O.; Bernardi, P.; Salvatori, S.; Rasola, A. Myotonic dystrophy protein kinase (DMPK) prevents ROS-induced cell death by assembling a hexokinase II-Src complex on the mitochondrial surface. Cell Death Dis. 2013, 4, e858. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Farajollahi, S.; Maas, S. Molecular diversity through RNA editing: A balancing act. Trends Genet. 2010, 26, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, C.-P.; Maggi, L.B.J.; Weber, J.D. The Role of RNA Editing in Cancer Development and Metabolic Disorders. Front. Endocrinol. 2018, 9, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, S.S.; Jun, H.-I.; Bahn, J.H.; Azghadi, A.; Ramaswami, G.; Van Nostrand, E.L.; Nguyen, T.B.; Hsiao, Y.-H.E.; Lee, C.; Pratt, G.A.; et al. Faculty Opinions recommendation of Widespread RNA editing dysregulation in brains from autistic individuals. Nat. Neurosci. 2019, 22, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Raabe, C.A.; Voss, R.; Kummerfeld, D.-M.; Brosius, J.; Galiveti, C.R.; Wolters, A.; Seggewiss, J.; Huge, A.; Skryabin, B.V.; Rozhdestvensky, T.S. Ectopic expression of Snord115 in choroid plexus interferes with editing but not splicing of 5-Ht2c receptor pre-mRNA in mice. Sci. Rep. 2019, 9, 4300. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Shiromoto, Y.; Ota, H.; Song, C.; Kossenkov, A.V.; Wickramasinghe, J.; Showe, L.C.; Skordalakes, E.; Tang, H.-Y.; Speicher, D.W.; et al. ADAR1 controls apoptosis of stressed cells by inhibiting Staufen1-mediated mRNA decay. Nat. Struct. Mol. Biol. 2017, 24, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Chen, Y.T.; Chang, Y.F.; Liu, H.; Kuo, Y.P.; Shih, C.T.; Liao, W.C.; Chen, H.W.; Tsai, W.S.; Tan, B.C.M. ADAR1-mediated 3’ UTR editing and expression control of antiapoptosis genes fine-tunes cellular apoptosis response. Cell Death Dis. 2017, 8, e2833. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Han, G.; Li, S.; Song, Y.; Shen, H.; Zhai, Y.; Wang, Y.; Zhang, F.; Dong, N.; Li, T.; et al. Association between the T6459C point mutation of the mitochondrial MT-CO1 gene and susceptibility to sepsis among Chinese Han people. J. Cell. Mol. Med. 2018, 22, 5257–5264. [Google Scholar] [CrossRef]
- Silva-Santos, S.; Van Woerden, G.M.; Bruinsma, C.F.; Mientjes, E.; Jolfaei, M.A.; Distel, B.; Kushner, S.A.; Elgersma, Y. Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Investig. 2015, 125, 2069–2076. [Google Scholar] [CrossRef] [Green Version]
- Dupret, D.; Revest, J.-M.; Koehl, M.; Ichas, F.; De Giorgi, F.; Costet, P.; Abrous, N.; Piazza, P.V. Spatial Relational Memory Requires Hippocampal Adult Neurogenesis. PLoS ONE 2008, 3, e1959. [Google Scholar] [CrossRef] [Green Version]
- Dupret, D.; Fabre, A.; Döbrössy, M.D.; Panatier, A.; Rodríguez, J.J.; Lamarque, S.; Lemaire, V.; Oliet, S.H.R.; Piazza, P.-V.; Abrous, N. Spatial learning depends on both the addition and removal of new hippocampal neurons. PLoS Biol. 2007, 5, e214. [Google Scholar] [CrossRef] [Green Version]
- Van Woerden, G.M.; Harris, K.D.; Hojjati, M.R.; Gustin, R.M.; Qiu, S.; Freire, R.D.A.; Jiang, Y.-H.; Elgersma, Y.; Weeber, E.J. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of αCaMKII inhibitory phosphorylation. Nat. Neurosci. 2007, 10, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, S.; Nawaz, Z. E3 ubiquitin protein ligase, E6-associated protein (E6-AP) regulates PI3K-Akt signaling and prostate cell growth. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1809, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simchi, L.; Panov, J.; Morsy, O.; Feuermann, Y.; Kaphzan, H. Novel Insights into the Role of UBE3A in Regulating Apoptosis and Proliferation. J. Clin. Med. 2020, 9, 1573. https://doi.org/10.3390/jcm9051573
Simchi L, Panov J, Morsy O, Feuermann Y, Kaphzan H. Novel Insights into the Role of UBE3A in Regulating Apoptosis and Proliferation. Journal of Clinical Medicine. 2020; 9(5):1573. https://doi.org/10.3390/jcm9051573
Chicago/Turabian StyleSimchi, Lilach, Julia Panov, Olla Morsy, Yonatan Feuermann, and Hanoch Kaphzan. 2020. "Novel Insights into the Role of UBE3A in Regulating Apoptosis and Proliferation" Journal of Clinical Medicine 9, no. 5: 1573. https://doi.org/10.3390/jcm9051573
APA StyleSimchi, L., Panov, J., Morsy, O., Feuermann, Y., & Kaphzan, H. (2020). Novel Insights into the Role of UBE3A in Regulating Apoptosis and Proliferation. Journal of Clinical Medicine, 9(5), 1573. https://doi.org/10.3390/jcm9051573